

Abstract
The serpins are a family of proteinase inhibitors that play a central role in the control of proteolytic cascades. Their inhibitory mechanism depends on the intramolecular insertion of the reactive loop into β-sheet A after cleavage by the target proteinase. Point mutations within the protein can allow aberrant conformational transitions characterized by β-strand exchange between the reactive loop of one molecule and β-sheet A of another. These loop-sheet polymers result in diseases as varied as cirrhosis, emphysema, angio-oedema, and thrombosis, and we recently have shown that they underlie an early-onset dementia. We report here the biochemical characteristics and crystal structure of a naturally occurring variant (Leu-55–Pro) of the plasma serpin α1-antichymotrypsin trapped as an inactive intermediate. The structure demonstrates a serpin configuration with partial insertion of the reactive loop into β-sheet A. The lower part of the sheet is filled by the last turn of F-helix and the loop that links it to s3A. This conformation matches that of proposed intermediates on the pathway to complex and polymer formation in the serpins. In particular, this intermediate, along with the latent and polymerized conformations, explains the loss of activity of plasma α1-antichymotrypsin associated with chronic obstructive pulmonary disease in patients with the Leu-55–Pro mutation.
Alpha-1-antichymotrypsin is an acute-phase protein and a member of the serine proteinase inhibitor or serpin superfamily. It is synthesized by the liver and bronchial epithelial cells (1), and plasma levels start to rise within 8 hr of an inflammatory response and double within 16 hr (2). The precise role of α1-antichymotrypsin is uncertain but it is thought to act as an anti-inflammatory agent inhibiting chymotrypsin, cathepsin G, mast cell chymase, neutrophil chemotaxis, and superoxide production (3–8). Like all members of the serpin superfamily, α1-antichymotrypsin is characterized by a dominant β-sheet A and a mobile reactive loop that acts as a pseudosubstrate for the cognate proteinase (9, 10). After docking the loop is cleaved by the proteinase at the P1-P1′ bond, and the acyl-enzyme intermediate is trapped as an irreversible complex by insertion of the cleaved loop into the A-sheet (11, 12). The intact reactive loop of α1-antichymotrypsin also can be stably incorporated into the A-sheet in an inactive latent conformation. This conformational transition causes local antiproteinase deficiency, which may be important in the pathogenesis of smoking-induced chronic airflow obstruction (6, 13). Chronic airflow obstruction also has been associated with mutations of α1-antichymotrypsin that result in plasma deficiency by the retention of variant α1-antichymotrypsin within hepatocytes (14–16).
We describe here the biochemical characterization of the Leu-55–Pro mutant of α1-antichymotrypsin, which results in plasma deficiency and chronic obstructive pulmonary disease (15). Our data show that this mutant can adopt not only the native and inactive latent conformations identified in other serpins, but also an inactive, stable, polymerogenic intermediate. This unusual intermediate has been crystallized and the structure has been solved. It provides a satisfying explanation for the unusual biochemistry with partial insertion of the reactive loop into β-sheet A. Surprisingly however, the lower part of the A-sheet is filled by a β-strand derived from residues Asn-163 to Thr-170, which are normally part of the F-helix and the connecting loop to strand 3A. This configuration is likely to approximate an intermediate in the generation of aberrant serpin conformations associated with disease and provides insights into the inhibitory mechanism of active members of the serpin superfamily.
Materials and Methods
Expression and Characterization of Leu-55–Pro α1-Antichymotrypsin.
Leu-55–Pro α1-antichymotrypsin was prepared by PCR mutagenesis and confirmed by complete sequencing of the α1-antichymotrypsin cDNA. Recombinant wild-type and Leu-55–Pro α1-antichymotrypsin were expressed in the pzm-S plasmid in Escherichia coli N4830–1 with a temperature-sensitive promoter as detailed (4, 5). The cells were lysed in a French press, and α1-antichymotrypsin was purified by Q-Sepharose and DNA chromatography as described by Rubin et al. (4). The eluate then was dialyzed into 50 mM Tris, 5 mM EDTA, pH 7.4, loaded onto a heparin-Sepharose column (Amersham Pharmacia XK16/40), equilibrated in the same buffer, and eluted with a 0–0.4 M gradient of KCl. Purity was confirmed by SDS/PAGE, and protein concentration was determined by Lowry assay. Inhibitory activity, association kinetics, nondenaturing and transverse urea gradient PAGE, electron microscopy, heat stability assays, and the assessment of the rate of insertion of a synthetic 12-mer reactive loop peptide (Ac-Ser-Glu-Ala-Ala-Ala-Ser-Thr-Ala-Val-Val-Ile-Ala) into β-sheet A of α1-antichymotrypsin were performed as detailed (6).
Crystallization and Structure Determination.
Leu-55–Pro α1-antichymotrypsin eluted from the final column as two distinct peaks denoted γ and δ. Crystals of δ Leu-55–Pro α1-antichymotrypsin were grown by equilibrating a 1-μl solution of protein (10 mg/ml in 50 mM Tris, 50 mM KCl, pH 7.4) with 2 μl of precipitant as a hanging drop over 1 ml of precipitant [20% (wt/vol) PEG 4000, 0.2 M (NH4)2SO4, 0.1 M NaOAc, pH 4.5] at 18°C for 2–4 weeks. The crystals had space group P21 with unit cell dimensions of a = 41.7 Å, b = 122.1 Å, c = 41.7 Å, β = 101°. Diffraction data were collected at room temperature from a single crystal on station 7.2 at the Daresbury Synchrotron (Warrington, U.K.) by using a Mar345 detector and a wavelength of 1.448 Å. Data were integrated with mosflm (17) and processed with programs from the CCP4 suite (18) (Fig. 1). The structure was solved with amore (19) by using the 2.1-Å resolution structure of cleaved P10 Arg α1-antichymotrypsin (20) as the search model. After initial rigid body refinement the model was further refined by cycles of positional and B factor refinement with the program cns (21) using the maximum-likelihood refinement target (22) followed by model building with xfit (23). A total of 1,036 reflections were set apart for the free R factor evaluation and σA estimation. These reflections also were used for sigmaa weighting of the electron density maps used in model building (24). After seven rounds of refinement and rebuilding, the standard and free R factors had converged at 0.197 and 0.243, respectively, using all data and bulk solvent correction. The final model consisted of residues 26–173, 178–352, and 358–397 and did not contain bound water molecules. Weak density was apparent for residues 86–89, 104–110, 122–125, and 212–214, and for these regions the conformation of the search model was maintained.
Figure 1.

Schematic diagram showing the diffraction data characteristics as a function of resolution. The data completeness drops off sharply beyond 2.5 Å as a result of radiation decay, but the higher-resolution reflections are still useful as indicated by the signal-to-noise ratio. In total 18,805 reflections were measured, yielding 11,373 unique observations between 41.0 and 2.3 Å. The overall multiplicity-weighted Rmerge (43) is 13.1%, increasing to 48.5% in the highest-resolution shell.
Results and Discussion
Biochemical Characterization of Leu-55–Pro α1-Antichymotrypsin.
Leu-55–Pro α1-antichymotrypsin eluted from a heparin-Sepharose column as two distinct peaks, denoted γ and δ, at 0.12 and 0.17 M KCl, respectively (Fig. 2A). The γ peak was consistently smaller than δ, and both peaks contained intact α1-antichymotrypsin when assessed by SDS/PAGE and N-terminal sequencing (1ASNSP-). γ Leu-55–Pro α1-antichymotrypsin eluted at the same KCl concentration as wild-type α1-antichymotrypsin and was 20% active as an inhibitor of bovine α-chymotrypsin compared with the wild-type protein, which was typically 40% active. γ Leu-55–Pro α1-antichymotrypsin migrated as two bands on nondenaturing PAGE with electrophoretic mobilities that are typical of native and latent α1-antichymotrypsin (Fig. 2B). The presence of latent species in addition to native was consistent with the reduced inhibitory activity. This was confirmed by transverse urea gradient PAGE that showed that one component of the γ Leu-55–Pro α1-antichymotrypsin fraction had a similar unfolding profile to wild-type protein whereas the other fraction was resistant to unfolding in up to 8 M urea, as is typical for the latent conformation (Fig. 2C).
Figure 2.

(A) Elution profile of Leu-55–Pro α1-antichymotrypsin from heparin-Sepharose showing a peak (γ) that elutes at the same KCl concentration as wild-type protein (dashed line) and a higher affinity peak (δ). The peak that elutes near the end of the gradient contains dimeric species. (B) 7.5–15% (wt/vol) nondenaturing PAGE of recombinant wild-type α1-antichymotrypsin (lane 1), recombinant γ Leu-55–Pro α1-antichymotrypsin (lane 2), recombinant δ Leu-55–Pro α1-antichymotrypsin (lane 3). Lanes 1 and 3 contain 7.5 μg protein and lane 2 contains 20 μg to give similar band intensities. γ Leu-55–Pro α1-antichymotrypsin migrated as two bands with the electrophoretic mobility of native (N) and latent (L) α1-antichymotrypsin. δ Leu-55–Pro α1-antichymotrypsin had a similar mobility to recombinant wild-type α1-antichymotrypsin with a high molecular mass dimer (D). (C) γ Leu-55–Pro α1-antichymotrypsin (Upper) showed native and latent unfolding transitions on transverse urea gradient PAGE whereas the δ protein (Lower) unfolded with an intermediate profile at approximately 4 M urea. The left of each gel represents 0 M urea and the right 8 M urea. Each gel was loaded with 20 μg protein, which was visualized by silver staining. (d) 7.5–15% (wt/vol) nondenaturing PAGE assessing the thermal stability of wild-type (Top), γ Leu-55–Pro α1-antichymotrypsin (Middle), and δ Leu-55–Pro α1-antichymotrypsin (Bottom). Samples were incubated at 0.3 mg/ml for 30 min at the temperatures indicated, and 7.5 μg protein was loaded in each lane. (E) The native conformation of γ (Upper) and δ (Lower) Leu-55–Pro α1-antichymotrypsin were less stable (as evidenced by the loss of bands on nondenaturing PAGE) than the wild-type protein when incubated under physiological conditions. The samples were incubated at 0.25 mg/ml in 50 mM Tris, 50 mM KCl, pH 7.4 at 41°C for the times indicated and then assessed by 7.5–15% (wt/vol) nondenaturing PAGE. All lanes contain 7.5 μg protein.
The native species of γ Leu-55–Pro α1-antichymotrypsin had association kinetics with bovine α-chymotrypsin (kass = 4.3 ± 0.05 × 105 M−1⋅s−1, Ki = 213 ±4 pM; n = 3) that were similar to those of the wild-type protein (2.3 ± 0.01 × 105 M−1⋅s−1 and 257 ± 3 pM), but it was less stable when assessed by thermal stability and nondenaturing PAGE (Fig. 2D). Native γ Leu-55–Pro α1-antichymotrypsin was also less stable than wild-type α1-antichymotrypsin when incubated at 37°C or 41°C and monitored by inhibitory activity or density of the native band on nondenaturing PAGE (Fig. 2E). This loss of activity was associated with the formation of short chain polymers and aggregates when the protein was visualized by electron microscopy (data not shown). These aggregates dissociated to monomers after heating in 1% (wt/vol) SDS at 100°C for 2 min, which is consistent with polymer formation. The latent component of γ Leu-55–Pro α1-antichymotrypsin remained resistant to heating at 100°C and has been shown previously to be inactive as a proteinase inhibitor (6).
δ Leu-55–Pro α1-antichymotrypsin had a similar electrophoretic mobility on nondenaturing PAGE to recombinant wild-type native α1-antichymotrypsin (Fig. 2B) and was always inactive as an inhibitor of bovine α-chymotrypsin. Inactive variants of α1-antichymotrypsin typically act as substrates and are cleaved at the exposed reactive loop to give a characteristic 4-kDa band shift on SDS/PAGE (25). Incubation of δ Leu-55–Pro α1-antichymotrypsin with bovine α-chymotrypsin in a 2:1 molar ratio of inhibitor to proteinase resulted in nonspecific protein degradation whereas recombinant wild-type protein formed SDS-stable complexes when incubated under identical conditions (data not shown). The conformation of β-sheet A of δ Leu-55–Pro α1-antichymotrypsin was probed with an exogenous 12-mer P14-P3 reactive loop peptide. Peptides incubated in 100-fold molar excess at 37°C annealed to wild-type α1-antichymotrypsin with a t1/2 of 6 hr, but the δ variant accepted a reactive loop peptide much more slowly with a t1/2 of approximately 4 days.
δ Leu-55–Pro α1-antichymotrypsin was more resistant to unfolding in urea than wild-type protein (4 M urea and 1 M urea, respectively; Fig. 2C) but also contained a component with a normal unfolding profile and a high molecular mass species that was resistant to denaturation. The δ Leu-55–Pro α1-antichymotrypsin was more thermodynamically stable than the native component of the γ peak (Fig. 2D). It also had a higher melting temperature than wild-type native α1-antichymotrypsin when assessed by CD at 222 nm, 67.6 ± 0.1°C for wild-type α1-antichymotrypsin and 70.7 ± 0.3°C for the δ Leu-55–Pro mutant, n = 3. Our previous data have shown an inverse correlation between thermal stability of the serpin α1-antitrypsin and the rate of polymerization (26), but this was not the case with δ Leu-55–Pro α1-antichymotrypsin, which readily polymerized when incubated under physiological conditions as assessed by nondenaturing PAGE (Fig. 2E).
Structure of the α1-Antichymotrypsin Intermediate Trapped by the Leu-55–Pro Mutation.
To date, all known serpins have either a native, loop-expelled, labile, active conformation associated with a five-stranded β-sheet A (9, 27) or one of several induced loop inserted, stable, inactive conformations associated with a six-stranded A-sheet. The sixth strand is derived from the reactive loop with or without proteolytic cleavage giving the cleaved (10) and latent (28, 29) forms, respectively. Both the native and latent species were present in the γ Leu-55–Pro α1-antichymotrypsin peak. In contrast the δ peak consisted mainly of a single species that was inactive, was intact on SDS/PAGE, and had stability intermediate between that of native and latent α1-antichymotrypsin. Moreover this species could accept a P14-P3 reactive loop peptide and form polymers, demonstrating that the protein could not be in a latent configuration. This combination of properties suggests a conformation intermediate to the native and latent states. To understand the structural basis of this behavior δ Leu-55–Pro α1-antichymotrypsin was crystallized and the structure was solved at a nominal 2.5-Å resolution (Fig. 1). The protein showed the same overall fold as other members of the serpin superfamily with β-sheets A-C and helices A-I (Fig. 3a). β-sheet A was clearly seen in a six-stranded state, but unlike the latent and cleaved states, the density for s4A was not consistent with full insertion of the reactive loop. There was good density for positions P14-P12 (s4A1) and P10-P3 (s4A2) of s4A, but not for position P11 (Fig. 3b). Instead the density indicated that the reactive loop bends out of the A-sheet at P12 and turns to join s1C. At position P10, the density showed a clear connection to the penultimate turn of the F-helix and the insertion of these and subsequent residues into β-sheet A was supported by the side-chain densities (Fig. 3b).
Figure 3.

(a) Structure of δ Leu-55–Pro α1-antichymotrypsin showing the reactive loop in red, the A-sheet in green, and the F-helix in yellow. Asn-163, whose side chain mimics the peptide plane in position P11 of s4A, is shown in ball-and-stick representation. Residues 353–357 (P6-P2) could not be built and are illustrated as a broken black line. The gate region including s3C and s4C, over which the loop must pass to form the latent conformation, is indicated. The shutter domain underlying the opening of the A-sheet is shown, together with (in black) the five residues at its focus at the s6B-hB junction (Inset). The Leu-55–Pro mutation is shown in red with, in blue, six other mutations that result in serpin polymerization and diseases as diverse as cirrhosis, thrombosis, angio-oedema, and dementia. (b) Cleaved reactive loop of α1-antichymotrypsin in black (20) with final model of δ Leu-55–Pro α1-antichymotrypsin shown in yellow. The sigmaa-weighted 2Fo-Fc (purple) and Fo-Fc (pink) electron density maps are shown before rebuilding the molecular replacement model. The break in s4A (between P12 and P10), the connection to helix F at P10, and the density as the loop leaves the A-sheet at position 12 are clearly visible. Clear Fo-Fc density (pink) at position P7 indicates the presence of Arg-166. (c) β-sheet C from δ Leu-55–Pro α1-antichymotrypsin (white) superimposed on reactive loop cleaved α1-antichymotrypsin (black) showing displacement of the main-chain residues of s1C. (d) Schematic illustration of conformational transitions of the serpins. The active serpin (M) has an exposed reactive center loop (red) similar to that of α1-antitrypsin (30). After activation by heat or destabilizing mutations in the shutter domain (red circle) there is opening of β-sheet A and reactive loop insertion to P12. This state, computationally modeled as a chimera between δ and cleaved α1-antichymotrypsin, is illustrated as M*. The A-sheet can receive the loop of another molecule (black dotted arrow) to form a loop-sheet dimer (P) and then extend to form a chain of polymers. Alternatively if there is sufficient energy to displace s1C the serpin loop can fully insert to form the latent conformation (L) (29). δ Leu-55–Pro α1-antichymotrypsin represents a third conformation in which the open A-sheet is filled by insertion of an unfolded loop of the F-helix (yellow). The figures were prepared with molscript (44).
There is continuous, but weak, density for reactive loop residues 348–352 (P11-P7), which allowed us to include them in the model. However, the lack of prominent side chains in this region leaves some uncertainty in the details of the main-chain conformation. Residues 353–357 (P6-P2) could not be built because of lack of density, and the first interpretable density was for residue Leu-358 (P1). The distance between the Cα atoms of P7 and P1 was 18.3 Å, which implies that the missing five residues must adopt an extended conformation. Residues 359–363 (P1′-P5′) are in a crystal packing interface, which probably explains the better quality density. Density was again weaker for residues 364–366 that connect the reactive loop to s1C. A stereochemical problem around residue 365, indicated by unfavorable dihedral angles, could not be resolved with our data. The Leu-55–Pro mutation is at the N terminus of helix-B. The main-chain dihedral angles in the cleaved structure are compatible with a proline residue, and the main-chain nitrogen is not a hydrogen bonding partner for another protein atom. Moreover the side-chain substitution does not lead to obvious local structural changes that would stabilize the δ form. Thus the effects of the mutation on the structure of the protein are subtle and not clearly defined at this resolution.
Comparison of native α1-antitrypsin (30), native antithrombin with the reactive loop inserted to P14 (29), δ Leu-55–Pro α1-antichymotrypsin with the reactive loop inserted to P12, and the structures of latent antithrombin and PAI-1 (28, 29) shows an interesting correlation of structural changes. In α1-antitrypsin the reactive loop is fully expelled and the A-sheet and s1C are intact. The structure of native antithrombin demonstrates that the initiation of loop insertion to P14 leads to a locally distorted though still five-stranded β-sheet A together with a shift of the reactive loop toward the s4C-s3C turn or gate region but preservation of s1C. The loop of δ Leu-55–Pro α1-antichymotrypsin is inserted to P12 opening the A-sheet to the same extent as in the latent or cleaved structures. The reactive loop has moved even further toward the gate region and s1C has been distorted (Fig. 3c). Finally, full-strand insertion in the latent conformation results from movement of the loop over the gate region and release of s1C (Fig. 3d). This suggests that the transition to full loop insertion is a two-step process. The first is demonstrated in the structure of δ Leu-55–Pro α1-antichymotrypsin with opening of β-sheet A followed by loop insertion up to, but not beyond, P12. The second step will require release of s1C and provides an energy barrier that the serpin must cross for full loop insertion.
The δ form results from a structural transition that stabilizes the P12 inserted state by satisfying the hydrogen bonding potential of β-sheet A. Remarkably, this bonding has been achieved by inserting Asn-163 to Thr-170, which normally form the last turn of the F-helix and the loop before s3A, into positions that correspond to the P10-P3 positions of reactive loop cleaved α1-antichymotrypsin (Fig. 3a). The resulting structure has a near normal hydrogen bonding pattern with the distortion around position P11 compensated by the Asn-163 side-chain carbonyl, which substitutes for the missing main-chain carbonyl. The formation of a six-stranded A-sheet, as usual, stabilizes the molecule and explains the raised melting temperature but it is not as high as the melting temperature of the cleaved and latent forms that have a contiguous s4A (6).
Conformational Transitions and Disease.
The mobility of the reactive loop is necessary for serpin inhibitory function but naturally occurring point mutations can destabilize the protein and result in aberrant conformational transitions and disease. Plotting these dysfunctional mutants on the serpin template shows that they cluster in the proximal (P15-P10) and distal hinge (P6′-P11′) regions of the reactive loop and in the shutter domain below β-sheet A (Fig. 3 a and d). Indeed seven naturally occurring serpin mutants associated with disease cluster in the five amino acids at the focus of the shutter domain, the junction of strand 6B and the B-helix (Fig. 3a). Perturbation of these domains favors sequential intermolecular linkage between the reactive loop of one serpin molecule and the β-sheet A of another (31). These loop-sheet polymers tangle in the endoplasmic reticulum to form inclusion bodies in their cells of origin. Variants of α1-antitrypsin accumulate in hepatocytes to cause liver cirrhosis (32) and mutants of neuroserpin accumulate in neurones, causing a recently described early-onset dementia (33). The consequent lack of circulating protein results in plasma deficiency that is best described with variants of α1-antitrypsin, antithrombin, and C1 inhibitor in association with emphysema, thrombosis, and angio-oedema, respectively (31). Two mutants of α1-antichymotrypsin (Pro-229–Ala and Leu-55–Pro) also have been reported that cause liver retention with hepatocyte inclusions and accompanying plasma deficiency (15, 16). The Leu-55–Pro mutation lies in the shutter domain, and by analogy with the conformationally unstable Mmalton and Siiyama variants of α1-antitrypsin (34, 35), the mutation will predictably increase the flexibility of β-sheet A and favor the formation of loop-sheet polymers in vivo. We show here that this mutant does form polymers at a faster rate than the wild-type protein but also that the A-sheet spontaneously accepts its own reactive loop to form the latent conformation (early eluting γ-peak, Fig. 2 B and C). In addition, the Leu-55–Pro mutation traps a significant proportion of the recombinant α1-antichymotrypsin as the δ intermediate with partial loop insertion.
There is good evidence that 6- and 7-mer peptides corresponding to P14-P8 of the reactive loop can anneal to the upper part of β-sheet A of antithrombin, favoring sheet opening and polymer formation (36). δ Leu-55–Pro α1-antichymotrypsin, which also favors polymer formation, has a partially inserted reactive loop that resembles serpins annealed to small peptides. In both instances the partial insertion at the top of the sheet opens the whole of the gap between strands 3 and 5 such that there is ready acceptance of the reactive loop of a second molecule as s4A2. This exogenous loop must dock in the position that is occupied by the residues connecting s3A and F-helix (30). The biochemical observation that δ Leu-55–Pro α1-antichymotrypsin undergoes even more rapid polymer formation than native α1-antichymotrypsin implies that these residues are readily displaced from the A-sheet by the competing loop of another molecule.
We recently have shown that loop-sheet polymerization is a two-step process with initial opening of β-sheet A followed by exogenous reactive loop insertion. Polymer formation may be described by Eq. 1 (26):
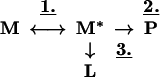 |
1 |
where step 1 represents the conformational change of the serpin to a polymerogenic monomeric form (M*), step 2 represents the formation of polymers (P), and step 3 represents a side pathway that leads to the formation of the latent conformation (L). The Leu-55–Pro mutant of α1-antichymotrypsin favors the spontaneous formation of polymeric and latent protein and hence has some of the proposed properties of the intermediate M*. The mutant has allowed us to trap and crystallize a polymerogenic intermediate that may approximate M* and demonstrates that the initial movement in polymer formation is partial loop insertion and opening of the β-sheet A (Fig. 3d). The second step can either be the acceptance of the reactive loop of another molecule to form polymers (step 2) or full insertion of the endogenous loop to form the latent conformation (step 3). The ratio of the rates of steps 2 and 3 determines the final product: polymeric or latent protein. Unwinding of the F-helix followed by insertion into β-sheet A, as demonstrated in the structure of δ Leu-55–Pro α1-antichymotrypsin, now provides a third possibility (Fig. 3d). That the formation of such partially inserted forms may be general in the serpins is supported by our observations of atypical monomeric L-forms in other members of the family, as for example in pasteurized transfusion concentrate of human antithrombin (37).
Implications for the Serpin Inhibitory Mechanism.
There is now growing evidence that after docking with its target proteinase the reactive loop of the serpin is cleaved and fully inserted into β-sheet A. This swings the proteinase from the upper to the lower pole of the protein (38). Recent studies suggest that this mechanism involves several steps between the formation of the initial Michaelis complex and that of the irreversible final complex (39, 40). The structure of δ Leu-55–Pro α1-antichymotrypsin demonstrates a conformation that fits with the partly inserted loop proposed for the initial Michaelis complex by Mellet et al. (39) with further loop insertion limited by the energy barrier that must be crossed to release s1C. Reactive loop cleavage with the formation of the acyl intermediate then would be required for full loop insertion, distortion of the catalytic triad, and inactivation of the proteinase.
The demonstration that the F-helix–s3A turn can insert into the A-sheet raises the intriguing possibility that such insertion may be a more general feature of the serpin inhibitory or folding mechanisms. There is a caution in this as the insertion may not be feasible to the same extent with plasma α1-antichymotrypsin as Asn-163 is a potential site for glycosylation that would impede strand insertion into the A-sheet. Nevertheless support for the functional basis of this insertion is provided by the striking sequence homology between the reactive loop of α1-antichymotrypsin and the peptide that is inserted as s4A2 in our structure (Fig. 4). Moreover the two residues that insert at position P8 in s4A2, Thr-165 and Thr-351 are highly conserved in the serpin superfamily (41) and there is also conservation of the F-helix 165T-x-G-x-I residues in over 70% of all serpins (42). It is therefore plausible that after docking with the target proteinase the reactive loop inserts into the A-sheet, breaking the stabilizing hydrogen bonds. The F-helix s3A turn then may insert into the A-sheet to shoehorn open the gap between strands 3 and 5 to prepare for acceptance of the reactive loop that is covalently linked to the target proteinase. There is also the possibility that the insertion of the F-helix–s3A loop might occur in an intermediate on the folding pathway of serpins in the endoplasmic reticulum. Active serpins are among the very small number of proteins that fold to a metastable state, instead of their global energy minimum, the latent conformation. A transient insertion of the F-helix–loop region into the bottom of the A-sheet could contribute to a kinetic barrier that blocks full loop insertion thereby preventing the serpin from folding into the lowest energy conformation.
Figure 4.

Structure-based sequence alignment of P9-P1 of the reactive loop (s4A) of α1-antichymotrypsin (above) and the strand of the F-helix (hF) that inserts into the A-sheet in our structure (below). Complete (**) or partial (*) conservation of residues in the reactive loop and hF of α1-antichymotrypsin are shown (conserved) as is the conservation of residues of hF throughout the serpin superfamily (consensus) (41).
Finally, this work also demonstrates how a pathogenic mutation can induce switching of secondary structural characteristics in a protein. Here the loss of a turn from helix F is offset by the adoption of a β-structure by an overlapping sequence of residues with an associated increase in stability. Such switching between helical and β-structure is important as it underpins amyloid fibril formation and the prion hypothesis.
Conclusion
The serpin inhibitory mechanism depends on the flexibility of the reactive loop. We show here that a shutter domain mutation can trap the serpin as an inactive intermediate on the polymerization and complex formation pathway. This conformational intermediate, along with the accompanying transitions to latent and polymerized forms, explains the loss of activity of plasma α1-antichymotrypsin in patients with the Leu-55–Pro mutation in association with chronic obstructive pulmonary disease.
Acknowledgments
We are grateful to Drs. Peter Elliott and Xue Pei, Cambridge Institute for Medical Research, U.K., for their help with crystallization and data collection. This work was supported by the Wellcome Trust and the Medical Research Council (U.K.).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1qmn and r1qmnsf).
References
- 1.Cichy J, Potempa J, Chawla R K, Travis J. J Clin Invest. 1995;95:2729–2733. doi: 10.1172/JCI117975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aronsen K F, Ekelund G, Kindmark C O, Laurell C-B. Scand J Clin Lab Invest Suppl. 124. 1972;29:127–136. doi: 10.3109/00365517209102760. [DOI] [PubMed] [Google Scholar]
- 3.Beatty K, Bieth J, Travis J. J Biol Chem. 1980;255:3931–3934. [PubMed] [Google Scholar]
- 4.Rubin H, Wang Z, Nickbarg E B, McLarney S, Naidoo N, Schoenberger O L, Johnson J L, Cooperman B S. J Biol Chem. 1990;265:1199–1207. [PubMed] [Google Scholar]
- 5.Lomas D A, Stone S R, Llewelyn-Jones C, Keogan M-T, Wang Z M, Rubin H, Carrell R W, Stockley R A. J Biol Chem. 1995;270:23437–23443. doi: 10.1074/jbc.270.40.23437. [DOI] [PubMed] [Google Scholar]
- 6.Chang W-S W, Lomas D A. J Biol Chem. 1998;273:3695–3701. doi: 10.1074/jbc.273.6.3695. [DOI] [PubMed] [Google Scholar]
- 7.Schechter N M, Jordan L M, James A M, Cooperman B S, Wang Z M, Rubin H. J Biol Chem. 1993;268:23626–23633. [PubMed] [Google Scholar]
- 8.Kilpatrick L, Johnson J L, Nickbarg E B, Wang Z, Clifford T F, Banach M, Cooperman B S, Douglas S D, Rubin H. J Immunol. 1991;146:2388–2393. [PubMed] [Google Scholar]
- 9.Wei A, Rubin H, Cooperman B S, Christianson D W. Nat Struct Biol. 1994;1:251–258. doi: 10.1038/nsb0494-251. [DOI] [PubMed] [Google Scholar]
- 10.Baumann U, Huber R, Bode W, Grosse D, Lesjak M, Laurell C-B. J Mol Biol. 1991;218:595–606. doi: 10.1016/0022-2836(91)90704-a. [DOI] [PubMed] [Google Scholar]
- 11.Stratikos E, Gettins P G W. J Biol Chem. 1998;273:15582–15589. doi: 10.1074/jbc.273.25.15582. [DOI] [PubMed] [Google Scholar]
- 12.Wilczynska M, Fa M, Karolin J, Ohlsson P-I, Johansson L B-A, Ny T. Nat Struct Biol. 1997;4:354–357. doi: 10.1038/nsb0597-354. [DOI] [PubMed] [Google Scholar]
- 13.Berman G, Afford S C, Burnett D, Stockley R A. J Biol Chem. 1986;261:14095–14099. [PubMed] [Google Scholar]
- 14.Poller W, Faber J-P, Scholz S, Weidinger S, Bartholome K, Olek K, Eriksson S. Lancet. 1992;339:1538. doi: 10.1016/0140-6736(92)91301-n. [DOI] [PubMed] [Google Scholar]
- 15.Poller W, Faber J-P, Weidinger S, Tief K, Scholz S, Fischer M, Olek K, Kirchgesser M, Heidtmann H-H. Genomics. 1993;17:740–743. doi: 10.1006/geno.1993.1396. [DOI] [PubMed] [Google Scholar]
- 16.Faber J-P, Poller W, Olek K, Baumann U, Carlson J, Lindmark B, Eriksson S. J Hepatol. 1993;18:313–321. doi: 10.1016/s0168-8278(05)80275-2. [DOI] [PubMed] [Google Scholar]
- 17.Leslie A W G. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography. Warrington, U.K.: Daresbury Laboratory; 1992. [Google Scholar]
- 18.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 19.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 20.Lukacs C M, Zhong J Q, Plotnick M I, Rubin H, Cooperman B S, Christianson D W. Nat Struct Biol. 1996;3:888–893. doi: 10.1038/nsb1096-888. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Pannu N S, Read R J. Acta Crystallogr A. 1996;52:659–668. [Google Scholar]
- 23.McRee D E. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 24.Read R J. Methods Enzymol. 1997;278:110–128. doi: 10.1016/s0076-6879(97)77009-5. [DOI] [PubMed] [Google Scholar]
- 25.Plotnick M I, Schechter N, Wang Z M, Liu X, Rubin H. Biochemistry. 1997;36:14601–14608. doi: 10.1021/bi971530j. [DOI] [PubMed] [Google Scholar]
- 26.Dafforn T R, Mahadeva R, Elliott P R, Sivasothy P, Lomas D A. J Biol Chem. 1999;274:9548–9555. doi: 10.1074/jbc.274.14.9548. [DOI] [PubMed] [Google Scholar]
- 27.Elliott P R, Abrahams J-P, Lomas D A. J Mol Biol. 1998;275:419–425. doi: 10.1006/jmbi.1997.1458. [DOI] [PubMed] [Google Scholar]
- 28.Mottonen J, Strand A, Symersky J, Sweet R M, Danley D E, Geoghegan K F, Gerard R D, Goldsmith E J. Nature (London) 1992;355:270–273. doi: 10.1038/355270a0. [DOI] [PubMed] [Google Scholar]
- 29.Carrell R W, Stein P E, Fermi G, Wardell M R. Structure (London) 1994;2:257–270. doi: 10.1016/s0969-2126(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 30.Elliott P R, Lomas D A, Carrell R W, Abrahams J-P. Nat Struct Biol. 1996;3:676–681. doi: 10.1038/nsb0896-676. [DOI] [PubMed] [Google Scholar]
- 31.Stein P E, Carrell R W. Nat Struct Biol. 1995;2:96–113. doi: 10.1038/nsb0295-96. [DOI] [PubMed] [Google Scholar]
- 32.Mahadeva R, Lomas D A. Thorax. 1998;53:501–505. doi: 10.1136/thx.53.6.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis R L, Shrimpton A E, Holohan P D, Bradshaw C, Feiglin D, Sonderegger P, Kinter J, Becker L M, Lacbawan F, Krasnewich D, et al. Nature (London) 1999;401:376–379. doi: 10.1038/43894. [DOI] [PubMed] [Google Scholar]
- 34.Lomas D A, Finch J T, Seyama K, Nukiwa T, Carrell R W. J Biol Chem. 1993;268:15333–15335. [PubMed] [Google Scholar]
- 35.Lomas D A, Elliott P R, Sidhar S K, Foreman R C, Finch J T, Cox D W, Carrell R W. J Biol Chem. 1995;270:16864–16870. doi: 10.1074/jbc.270.28.16864. [DOI] [PubMed] [Google Scholar]
- 36.Fitton H L, Pike R N, Carrell R W, Chang W-S W. Biol Chem. 1997;378:1059–1063. doi: 10.1515/bchm.1997.378.9.1059. [DOI] [PubMed] [Google Scholar]
- 37.Chang W-S W, Harper P L. Thromb Haemostasis. 1997;77:323–328. [PubMed] [Google Scholar]
- 38.Stratikos E, Gettins P G W. Proc Natl Acad Sci USA. 1999;96:4808–4813. doi: 10.1073/pnas.96.9.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellet P, Boudier C, Mely Y, Bieth J G. J Biol Chem. 1998;273:9119–9123. doi: 10.1074/jbc.273.15.9119. [DOI] [PubMed] [Google Scholar]
- 40.Luo Y, Zhou Y, Cooperman B S. J Biol Chem. 1999;274:17733–17741. doi: 10.1074/jbc.274.25.17733. [DOI] [PubMed] [Google Scholar]
- 41.Huber R, Carrell R W. Biochemistry. 1989;28:8951–8966. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 42.Whisstock J, Lesk A M, Carrell R W. Proteins Struct Funct Genet. 1996;26:288–303. doi: 10.1002/(SICI)1097-0134(199611)26:3<288::AID-PROT5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 43.Diederichs K, Karplus A. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 44.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]