

Abstract
Botulinum neurotoxins (BoNTs) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. The natural product toosendanin is traditional Chinese medicine that has reported antibotulinum properties in animal models. To establish what chemical functionalities are necessary for the antibotulinum properties found within toosendanin, a study was initiated with the goal of using function-oriented synthesis (FOS) as a strategy to begin to unravel toosendanin’s powerful anti-botulinum properties. From these studies a new synthetic strategy is put forth allowing access to a 4-acetoxy CD fragment analogue (14) of toosendanin, which was achieved from mesityl oxide and acetylacetone in 14-steps. Animal studies on this fragment are also reported.
Keywords: Botulinum neurotoxins, Toosendanin, Function-oriented synthesis (FOS), 4-Acetoxy CD fragment
1. Introduction
Clostridium botulinum is a rod-shaped, Gram positive, sporulating anaerobic bacillus that is widely distributed in the environment.1 The spores, which are resistant to heat, desiccation, chemicals, radiation and oxygen, facilitate survival for very long periods. Under anaerobic conditions, and in the correct nutritional environment, spore germination and cell division take place. The global distribution of C. botulinum in the environment varies widely. The botulinum neurotoxins (BoNTs) comprise a family of seven immunologically distinct proteins synthesized primarily by strains of the anaerobic bacteria. These toxins (BoNT A–G) are the most lethal poisons known, with BoNT serotype A having a LD50 for a 70 kg human of a mere 0.8 μg by inhalation.2 Historically associated with food poisoning, these proteinaceous toxins inhibit the release of acetylcholine at neuromuscular junctions by cleavage of SNARE proteins, resulting in progressive flaccid paralysis.3–4 It is as a result of this potent neurotoxic activity that BoNTs are of substantial concern as potential bioterrorist weapons.5–7
2. Results and Discussion
We have embarked on a program to identify new small molecule inhibitors of BoNT/A that are non-proteinaceous in nature and that work by a different mechanism other than inhibiting the catalytic light chain of the BoNT/A protease.8 Limonoids, 1, are tetranortriterpenoids with a 4,4,8-trimethylfuranylsteroid skeleton (Fig. 1) derived from euphane or tirucallane triterpenoids, and, in general, have intense bitter taste.9 Meliaceae plants are a rich source of limonoids. Limonoids from the neem tree Melia azadirachta indica and the bead tree M. azedarach have been well studied, mainly because of their marked insect anti-feedant properties.10 Along this same vein of research, M. toosendan, which is native to China and known to contain limonoid constituents, has been found to possess effective antihelmintics.11 A major limonoid constituent found in Melia toosendan is the compound toosendanin, (2, Fig. 1), which appears to have multiple modes of action in insects including damage to midgut tissues, inhibition of esterases, cytochrome P450-aldrin epoxidase and proteinase activities.12
Figure 1.

General structure of a Limonoid 1 and Toosendanin 2.
Toosendanin from a structural diversity standpoint is a fascinating molecule. The core carbon skeleton of toosendanin, is comprised of a cyclopentanoperhydrophenanthrene (3, Fig. 2), a molecular framework that consists of four fused, non-planar rings (labeled A–D) and a densely pack core of 14 stereocenters. Interestingly, this framework is commonly found in all mammalian steroids and hormones. What has intrigued us besides toosendanin’s anti-parasitic properties are two reports in the 1980’s describing toosendanin as also having anti-botulinum properties.13 Strikingly, these findings have remained dormant, as little research has been conducted upon this molecule from a synthetic standpoint.
Figure 2.

Toosendanin’s core ring system 3.
To probe toosendanin’s anti-botulinum properties two tactics were envisioned. The first being a total synthesis of the molecule that ultimately would allow derivatives to be assessed, and thus a means to explore toosendanin’s structure in relationship to its biological activity. A second was what Wender has termed Function-Oriented Synthesis (FOS).14 Because of the complexity of toosendanin and the insurmountable odds of a total synthesis that would provide large quantities of this natural product or closely related derivatives we have set a course on investigating this molecule from a FOS standpoint. The underpinnings of FOS are that the function of a biologically active lead structure can be emulated, tuned, or possibly improved by replacement with simpler scaffolds designed to embrace the key activity-determining structural features of the natural product. Because it is unknown which elements and/or structural features impact toosendanin’s biological activity we have decided to break the molecule into two fragments consisting of the AB and CD rings. In this paper, we report our initial work exploring the CD ring of toosendanin and its potential biological function.
We envisioned any synthesis emulating the CD rings of toosendanin, would need to incorporate the epoxide moiety, shared by rings C and D at the C-14 and C-15 positions. This functionality we hypothesized to be crucial, as this moiety is not observed within any steroidal skeletons, including mammalian or plant (Fig. 1). While the epoxide was deemed critical, the furan ring was not, as we have found through semi-synthesis efforts (Janda et al. unpublished results) that this ring in a reduced chemical state did not alter the molecules biological activity. Furthermore, this functionality was readily susceptible to both oxidation and reduction conditions, which would clearly hamper any practical synthesis of an abbreviated structure if included within our synthetic scheme (Janda et al. unpublished data).
Overall, there are few de novo synthetic efforts that describe the successful syntheses of similar limonoids or key structural fragments. Selected examples are dumsin15 and fragments of epoxyazadiradione.16 Our FOS approach would build on the teachings of Mateos, et al., who has recently described a synthesis of the limonoid fragment 4 in 10 steps with an overall yield of 44% (Fig. 3).17
Figure 3.
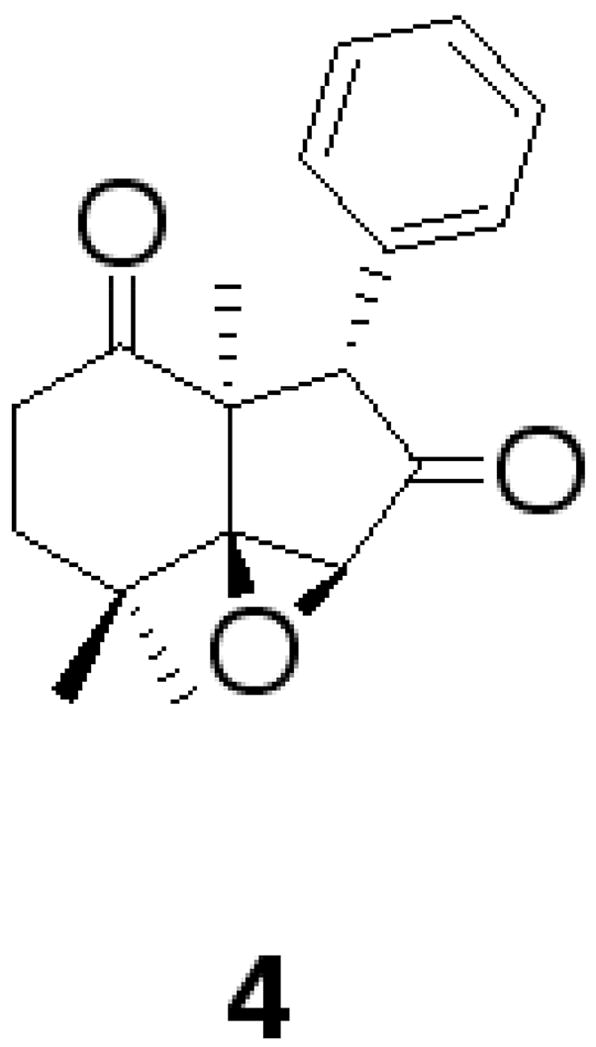
Limonoid fragment synthesized by Mateos, et al. 4.
We envisioned four crucial cyclization steps in the synthesis of a FOS toosendanin CD fragment: the Robinson annulation, an aldol condensation, the Nazarov cyclization, and an epoxidation reaction. Using these series of reactions it was envisioned that at various stages appropriate substituents could be embedded within the CD ring. Importantly, this approach would enable the first synthesis of the CD ring fragment of toosendanin correctly displaying all heteroatom substituents found within the natural product. Finally it is worth noting that a previous paper focusing on the reaction scope within limonoid construction has considered the possibility of building such a desired fragment, however, no compound bearing the α-acetoxy ketone functionality within ring C was reported.18
In developing a synthesis, our starting point featured a Robinson-type annulation of mesityl oxide 5 and acetylacetone 6 using BF3 as a catalyst, which bestowed α-cyclocitral (Scheme 1).16, 18 It is appropriate to note that α-cyclocitral 7 has been a preparatory material for many liminoid derivatives, particularly for SAR studies of insect antifeedant development.16–22 The convenience of starting with 7 is most certainly due to the fact that it can be readily synthesized on a multi-gram scale with inexpensive starting materials. Selective reduction16 of the conjugated carbonyl of diketone 7 using NaBH4/CeCl3 at −40 °C furnished the hydroxyketone 8, which could then be utilized for epoxidation with m-chloroperbenzoic acid affording 9. Aldol condensation of 9 with benzaldehyde provided dihydroxyketone 10 in 89% overall yield from 7.
Scheme 1.

Synthesis of compound 10. Reagents and conditions: (a) BF3·Et2O, 5 °C; (b) NaBH4, CeCl3·7H2O, MeOH, −40 °C; (c) mCPBA, CH2Cl2, rt; 12 h, 98% (d) PhCHO, NaOH, EtOH, rt, 13 h, 96%.
With dihydroxyketone 10, a Nazarov electrocyclization reaction was envisioned. For comparative purposes we note that the Nazarov electrocyclization reaction of a, b, and c (Fig. 4) in the presence of acid grants the corresponding cyclization compounds.18, 22 In lieu of these reports, we attempted the reaction of d with acid for the synthesis of a “non-functionalized” moiety at C3, however, this reaction failed. It is well understood that to accomplish the required cyclization a keto-group at C4, and a phenyl group at C3 are needed; unfortunately these requirements limit diversity, and as stated also dictate the stereochemical outcome.
Figure 4.

Nazarov electrocyclization reaction of a–d.
Thus, the Nazarov electrocyclization reaction of 10 in the presence of acid granted 11a and 11b (inseparable), 11c, and 11d. We were pleased to observe taht the cyclization exclusively formed the cis-fused product,17–18, 21, 23 placing the core ring system in the desired geometry of toosendanin. Reduction of the unsaturated C-ring by catalytic hydrogenation supplied compound 12a and 12b (inseparable), 12c, and 12d respectively (Scheme 2). We attempted the asymmetric reduction of 11a–d with a plethora of reagents [for example, (2S,5S)-(−)-5-benzyl-3-methyl-2-(5-methyl-2-furyl)-4-imidazolidi-none, binaphthyl derivatives, to name a few] that would have provided the correct C3-stereocenter, but, unfortunately the lone reaction that succeeded was palladium-carbon.
Scheme 2.

Synthesis of compounds 12a–d. Reagents and conditions: (a) 10−1 M HClO4/0.5 M Ac2O/AcOEt, rt, 63 h; (b) Pd-alumina, EtOH, rt, 18 h; (c) Pd-carbon, MeOH, rt, 21 h.
The relative stereochemistry of each of these compounds was determined by 2D-ROESY spectrum (Fig. 5). Thus, for 12a’s ROESY correlation was seen between H-4 (geminal with the acetoxy group) and Me-7 (axial); H-3 (geminal with the phenyl group) and Me-3a (the angular methyl group); Me-3a and H-7a. For the case with 12b, we could see a correlation [(sv between H-3 and Me-3a; Me-3a and H-7a; for 12c, H-3 and Me-3a and H-7a; H-4 and Me-7 (axial); H-5 and Me-7 (axial); while for 12d, H-3 and Me-3a; H-7a, Me-3a and H-4 (geminal with the acetoxy group); H-5 and Me-7 (axial)].
Figure 5.

The relative stereochemistry of compounds 12a–d was determined by 2D-ROESY experiments.
The D-ring ketone of 12a and 12b was reduced to sec-alcohol by treatment with 9-BBN (Scheme 3),22 subsequent dehydoxylation of this sec-alcohol with thionychloride in pyridine afforded the desired olefin, which upon hydrolysis of the diacetyl functionalities with potassium carbonate gave dihydroxyolefin 13 in 19% overall yield from 12. Selective protection of dihydroxyolefin 13 using TBSOTf at −78 °C provided the 7-TBS protected compound in 19% yield. The hydroxyl group at C-6 was oxidized by Dess-Martin’s reagent, which on subsequent deprotection of the silyl group with TBAF, followed by acetylation with acetic anhydride in pyridine, and epoxydation of olefin with m-chloroperbenzoic acid granted 14 in 14% overall yield from 13 (Scheme 3).
Scheme 3.

Synthesis of compound 14. Reagents and conditions: (a) 9-BBN, THF, rt, 4 h; (b) SOCl2, Pyridine, CH2Cl2, rt, 12 h; (c) K2CO3, MeOH, rt, 86 h, 19% for three steps; (d) TBSOTf, DIPEA, CH2Cl2, −78 °C, 2.5 h; (e) Dess-Martin reagent, CH2Cl2, rt, 13 h; (f) DMAP, rt, 24 h; (h) m TBAF, THF, rt, 2.5 h; (g) Ac2O, Pyridine, cat. CPBA, CH2Cl2, rt, 6 h, 14% for five steps.
To determine 14’s relative stereochemistry we again looked to the 2D-ROESY spectrum (Fig. 6). We could see a ROESY correlation between H-3 (geminal with the phenyl group) and Me-3a (the angular methyl group), H-4 (geminal with the acetoxy group) and H-6 (equatorial).
Figure 6.

The relative stereochemistry of compound 14 was determined by 2D-ROESY experiment.
To investigate the potential bioactivity of epoxide 14, we utilized the mouse lethality assay (MLA).8d This assay is considered the “gold standard” for the field and is used to assess any new protein or small molecule that may possess anti-botulinum activity. Before the assay was initiated with 14, we first investigated potential toxicity issues with this molecule. Gratifyingly, at a concentration of 2.5 mM this compound showed no propensity to be toxic with any of the animals it was administered, n = 4. Thus, mice were injected iv with 14 followed immediately by ip injection of approximately 10LD50s of BoNT/A. The mice were observed over a 48 hours period and unfortunately all mice succumbed to BoNT/A toxicity. In contrast the natural product toosendanin at this same concentration extended time to death 7.1 hours.
3. Conclusions
The work reported was initiated with a goal of using FOS as a strategy to begin to unravel toosendanins powerful anti-botulinum properties. Thus, a new synthetic approach to a 4-acetoxy CD fragment analogue of Toosendanin was achieved from mesityl oxide and acetylacetone in 14-steps. Unfortunately, this molecule provided no efficacy in vivo. However, we note that our synthetic route should allow access to other CD ring analogs, for example exchange of benzaldehyde to other aldehydes, would allow entry into other unnatural analogues. In addition, an acetyl group functional change would also provide another series of FOS-molecules. These compounds as well as the AB ring synthesis will be reported in due course.
4. Experimental
In general, reagents and solvents were used as purchased without further purification. All flash column chromatography was performed using silica gel 60 (230–400 mesh). Analytical and preparative thin-layer chromatography (TLC) was performed using Merck Kieselgel 60 F254 silica gel plates (0.25, 0.5, or 1 mm). 1H NMR spectra was recorded on Bruker DRX-600 (600 MHz), DRX-500 (500 MHz), or Varian Inova-400 (400 MHz) spectrometers. 13C NMR spectra was recorded on Bruker DRX-600 (150 MHz) spectrometer. All 2D-ROESY experiments (τm = 200ms) were also recorded on Bruker DRX-600 (600 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm) on the δ scale from an internal standard (NMR descriptions: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet). High-resolution mass spectra was recorded on an Agilent ESI-TOF mass spectrometer.
4.1. 1-(5-hydroxy-1,3,3-trimethyl-7-oxabicyclo[4.1.0]heptan-2-yl)ethanone (9)
m-Chloroperbenzoic acid (mCPBA) (77%, 107.5 mg, 0.48 mmol) was added to a solution of 8 (87.3 mg, 0.48 mmol) in CH2Cl2 (2 mL) at room temperature, and the mixture was stirred at room temperature for 12 hours. 5% Na2SO3 aq. was added and the resulting heterogeneous mixture was vigorously stirred. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were washed with 5% Na2CO3 aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with Et2O–hexane (2:1, v/v) to give 9 (93.3 mg, 98%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 4.09 (ddd, J = 10.2, 6.3, 2.4 Hz, 1H), 3.17 (d, J = 2.3 Hz, 1H), 2.49 (s, 1H), 2.29 (s, 3H), 1.78 (dd, J = 13.1, 10.4 Hz, 1H), 1.48–1.41 (m, 1H), 1.47 (s, 3H), 0.94 (s, 3H), 0.92 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 207.2, 66.1, 61.5, 60.5, 60.1, 37.2, 33.7, 32.5, 29.2, 26.9, 24.8; HRMS [MH]+ calcd for C11H19O3, 199.1329; Found, 199.1329.
4.2. (E)-1-(3,4-dihydroxy-2,6,6-trimethylcyclohex-1-enyl)-3-phenylprop-2-en-1-one (10)
Under argon atmosphere, NaOH (52.6 mg, 1.3 mmol) was added to a solution of 9 (130.3 mg, 0.66 mmol) and benzaldehyde (66.8 μL, 0.66 mmol) in EtOH (3 mL) at room temperature, and the mixture was stirred at room temperature for 13 hours. After evaporation of the solvent, the whole was made acidic by adding 1 N HCl under ice cooling and extracted with Et2O. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with AcOEt–hexane (5:1, v/v) to give 10 (180.7 mg, 96%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.59–7.54 (m, 2H), 7.48 (d, J = 16.2 Hz, 1H), 7.43–7.39 (m, 3H), 6.78 (d, J = 16.2 Hz, 1H), 4.04–3.97 (m, 2H), 1.86–1.76 (m, 1H), 1.77 (s, 3H), 1.63–1.53 (m, 1H), 1.23 (s, 3H), 1.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 201.2, 146.6, 144.0, 134.3, 130.9, 129.6, 129.0, 128.6, 127.7, 70.4, 66.7, 40.8, 36.3, 29.7, 29.5, 28.0, 19.1; HRMS [MH]+ calcd for C18H23O3, 287.1642; Found, 287.1648.
4.3. (3aRS,4RS,5RS,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyl-3a,4,5,6,7,7a-hexahydro-1H-indene-4,5-diyl diacetate (11a), (3aRS,4SR,5RS,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyl-3a,4,5,6,7,7a-hexahydro-1H-indene-4,5-diyl diacetate (11b), (3aRS,4RS,5SR,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyl-3a,4,5,6,7,7a-hexahydro-1H-indene-4,5-diyl diacetate (11c), and (3aRS,4SR,5SR,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyl-3a,4,5,6,7,7a-hexahydro-1H-indene-4,5-diyl diacetate (11d)
Under argon atmosphere, 10 (484.6 mg, 1.7 mmol) was dissolved in 48 mL of 10−1 M HClO4/0.5 M Ac2O/AcOEt reagent.18 The solution was allowed to stand at room temperature for 63 hours. Saturated NaHCO3 aq. was added to quench the reaction. The organic layer was separated, and the aqueous phase was extracted with AcOEt. The extract was washed with 5% Na2CO3 aq. and brine, dried over Na2SO4, and evaporated under reduced pressure to leave a solid. The residue was recrystallized from AcOEt–hexane to give a mixture of 11a and 11b (311.3 mg, 50%) as a colorless powder. The mother liquor was purified by p-TLC on SiO2 with AcOEt–hexane (1:4, v/v) to give 11c (109.0 mg, 17%) as a yellow oil and 11d (77.8 mg, 12%) as a colorless oil in the order of elution. 11a: 1H NMR (400 MHz, CDCl3) δ: 7.52–7.43 (m, 5H), 6.05 (s, 1H), 5.73 (d, J = 3.3 Hz, 1H), 4.91 (ddd, J = 11.1, 4.7, 3.4 Hz, 1H), 2.25 (s, 1H), 1.99 (s, 3H), 1.95–1.87 (m, 1H), 1.93 (s, 3H), 1.61–1.53 (m, 1H), 1.51 (s, 3H), 1.28 (s, 3H), 1.01 (s, 3H); 13C NMR (150 MHz, CDCl3) (mixture of 11a and 11b) δ: 208.5, 206.4, 180.7, 179.3, 170.7, 170.2, 170.0, 169.8, 136.3, 134.0, 133.2, 130.2, 130.0, 128.9, 128.2, 127.6, 127.3, 74.6, 71.9, 70.1, 68.4, 64.3, 63.7, 50.9, 50.4, 43.4, 39.2, 34.3, 33.9, 32.7, 32.3, 29.0, 25.9, 25.1, 22.2, 21.1, 20.9, 20.6, 20.0; HRMS [MH]+ calcd for C22H27O5, 371.1853; Found, 371.1854. 11b (data obtained from the spectrum of mixture): 1H NMR (400 MHz, CDCl3) δ: 7.39–7.34 (m, 3H), 7.32–7.28 (m, 2H), 6.08 (s, 1H), 5.50 (d, J = 8.0 Hz, 1H), 5.14 (dt, J = 8.0, 2.1 Hz, 1H), 2.27 (s, 1H), 2.16–2.04 (m, 1H), 1.94 (s, 3H), 1.67 (s, 3H), 1.61–1.53 (m, 1H), 1.34 (s, 3H), 1.29 (s, 3H), 1.17 (s, 3H). 11c: 1H NMR (400 MHz, CDCl3) δ: 7.44–7.37 (m, 5H), 6.27 (s, 1H), 5.65 (d, J = 2.1 Hz, 1H), 5.30 (ddd, J = 12.1, 5.5, 2.3 Hz, 1H), 2.12 (s, 1H), 2.00 (s, 3H), 1.93–1.83 (m, 1H), 1.75 (s, 3H), 1.60 (s, 3H), 1.58–1.51 (m, 1H), 1.40 (s, 3H), 1.27 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 205.5, 174.0, 170.7, 169.5, 134.0, 131.2, 129.8, 128.8, 127.7, 71.7, 68.6, 62.4, 51.6, 36.2, 33.5, 32.7, 28.7, 25.6, 21.0, 20.5; HRMS [MH]+ calcd for C22H27O5, 371.1853; Found, 371.1861. 11d: 1H NMR (400 MHz, CDCl3) δ: 7.44–7.33 (m, 5H), 6.26 (s, 1H), 5.32 (d, J = 3.6 Hz, 1H), 5.05 (ddd, J = 9.3, 7.2, 3.6 Hz, 1H), 2.25 (s, 1H), 2.09–2.02 (m, 1H), 2.07 (s, 3H), 1.71 (s, 3H), 1.55–1.50 (m, 1H), 1.54 (s, 3H), 1.34 (s, 3H), 1.14 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 207.0, 177.5, 169.7, 169.3, 135.3, 132.2, 129.6, 128.6, 127.6, 75.9, 72.1, 63.4, 50.6, 41.6, 33.6, 33.0, 29.7, 28.4, 23.7, 21.2, 20.6; HRMS [MH]+ calcd for C22H27O5, 371.1853; Found, 371.1856.
4.4. (3RS,3aSR,4RS,5RS,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyloctahydro-1H-indene-4,5 -diyl diacetate (12a) and (3RS,3aSR,4SR,5RS,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyloctahydro-1H-indene-4,5 -diyl diacetate (12b)
A solution of 11a and 11b (34.0 mg, 0.09 mmol) in EtOH (2 mL) was hydrogenated in the presence of Pd-alumina (5 wt %, 34.0 mg) at room temperature, and 1 atom for 18 hours. Pd-alumina was filtered off, and the filtrate was evaporated under reduced pressure to leave an oil, which was purified by p-TLC on SiO2 with Et2O–hexane (1:1, v/v) to give a mixture of 12a and 12b (34.2 mg, 100%) as a colorless oil. 12a: 1H NMR (500 MHz, CDCl3) δ: 7.32–7.19 (m, 5H), 5.37 (q, J = 4.0 Hz, 1H), 4.88 (d, J = 4.4 Hz, 1H), 2.99 (dd, J = 13.1, 8.1 Hz, 1H), 2.86 (dd, J = 18.8, 13.2 Hz, 1H), 2.56 (dd, J = 18.8, 8.2 Hz, 1H), 2.16 (s, 1H), 1.98 (s, 3H), 1.78 (dd, J = 15.3, 3.9 Hz, 1H), 1.68–1.61 (m, 1H), 1.67 (s, 3H), 1.35 (s, 3H), 1.32 (s, 3H), 1.24 (s, 3H); 13C NMR (150 MHz, CDCl3) (mixture of 12a and 12b) δ: 214.8, 214.1, 170.5, 170.0, 169.9, 137.5, 136.3, 128.7, 128.2, 128.1, 127.1, 127.0, 71.6, 70.9, 69.2, 68.6, 66.7, 66.5, 50.7, 49.5, 49.0, 47.4, 41.1, 40.9, 40.6, 38.5, 33.7, 32.5, 31.3, 30.3, 29.7, 28.9, 23.2, 23.1, 21.2, 21.0, 20.3; HRMS [MH]+ calcd for C22H29O5, 373.2009; Found, 373.2006. 12b (data obtained from the spectrum of mixture): 1H NMR (500 MHz, CDCl3) δ: 7.32–7.18 (m, 5H), 5.16–5.09 (m, 1H), 5.04 (d, J = 9.5 Hz, 1H), 3.06–2.90 (m, 2H), 2.58–2.51 (m, 1H), 2.05 (d, J = 1.5 Hz, 1H), 1.92 (s, 3H), 1.65–1.61 (m, 2H), 1.59 (s, 3H), 1.40 (s, 3H), 1.25 (s, 3H), 1.19 (s, 3H).
4.5. (3RS,3aSR,4RS,5SR,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyloctahydro-1H-indene-4,5 -diyl diacetate (12c)
A solution of 11c (102.7.0 mg, 0.28 mmol) in MeOH (6 mL) was hydrogenated in the presence of Pd-carbon (10 wt %, 50.5 mg) at room temperature, and 1 atom for 21 hours. Pd-carbon was filtered off, and the filtrate was evaporated under reduced pressure to leave an oil, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:3, v/v) to give 12c (89.9 mg, 87%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.33–7.29 (m, 2H), 7.28–7.24 (m, 1H), 7.14–7.10 (m, 2H), 5.13 (ddd, J = 12.7, 4.4, 2.2 Hz, 1H), 4.71 (d, J = 1.9 Hz, 1H), 3.23–3.18 (m, 1H), 2.82 (dd, J = 18.8, 12.0 Hz, 1H), 2.56 (ddd, J = 18.7, 10.4, 1.0 Hz, 1H), 2.14 (s, 3H), 1.97–1.89 (m, 2H), 1.86 (s, 3H), 1.55 (s, 3H), 1.53 (s, 3H), 1.33 (dd, J = 12.9, 4.4 Hz, 1H), 1.21 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 213.6, 170.5, 169.0, 135.4, 129.0, 128.3, 127.6, 72.8, 68.9, 62.8, 50.7, 49.1, 42.3, 36.0, 33.7, 31.2, 29.4, 26.2, 21.7, 20.9; HRMS [MNa]+ calcd for C22H28O5Na, 395.1829; Found, 395.1829.
4.6. (3RS,3aSR,4SR,5SR,7aSR)-3a,7,7-trimethyl-1-oxo-3-phenyloctahydro-1H-indene-4,5 -diyl diacetate (12d)
A solution of 11d (135.7 mg, 0.37 mmol) in MeOH (6 mL) was hydrogenated in the presence of Pd-carbon (10 wt %, 60.5 mg) at room temperature, and 1 atom for 21 hours. Pd-carbon was filtered off, and the filtrate was evaporated under reduced pressure to leave an oil, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:3, v/v) to give 12d (82.5 mg, 61%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 7.35–7.30 (m, 2H), 7.29–7.20 (m, 3H), 4.87–4.80 (m, 1H), 4.24 (s, 1H), 3.23 (dd, J = 13.9, 8.2 Hz, 1H), 3.04 (dd, J = 17.6, 13.9 Hz, 1H), 2.51 (ddd, J = 17.6, 8.2, 1.6 Hz, 1H), 2.15 (s, 1H), 2.07 (s, 3H), 2.02–1.95 (m, 1H), 1.99 (s, 3H), 1.58–1.48 (m, 1H), 1.56 (s, 3H), 1.28 (s, 3H), 1.19 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 216.7, 169.2, 168.2, 136.3, 128.4, 127.4, 75.5, 72.0, 62.8, 51.3, 46.7, 43.9, 39.3, 34.6, 32.1, 29.7, 28.4, 24.8, 21.7, 21.1; HRMS [MNa]+ calcd for C22H28O5Na, 395.1829; Found, 395.1834.
4.7. (1RS,6RS,7RS,7aSR)-4,4,7a-trimethyl-1-phenyl-2,4,5,6,7,7a-hexahydro-1H-indene-6, 7-diol (13)
To a solution of 12a and 12b (935.0 mg, 2.5 mmol) in THF (20 mL) was slowly added 9-BBN (0.5 M, 10 mL, 5.0 mmol). The reaction mixture was stirred under argon atmosphere at room temperature for 4 hours, and then MeOH (10 mL) was slowly added, stirring for 3 hours. Removal of the solvent afforded a crude product. To a solution of crude product in CH2Cl2 (10 mL) at 0 °C under argon atmosphere were added pyridine (0.8 mL, 10 mmol) and SOCl2 (0.4 mL, 5.0 mmol). The reaction mixture was stirred at room temperature for 12 hours, and then poured into ice-water. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2. The whole was washed with 5% Na2CO3 and brine, dried over Na2SO4. Removal of the solvent afforded a crude product, which was purified by flash chromatography on SiO2 with AcOEt–hexane (1:7, v/v) to give diacetoxyolefin. 1 N aqueous solution of K2CO3 (3 mL) was added to a solution of diacetoxyolefin in MeOH (10 mL) and CH2Cl2 (2 mL) at room temperature, and the mixture was stirred at room temperature for 86 hours. After evaporation of the solvent, the whole was made acidic by adding 2 N HCl under ice cooling and extracted with AcOEt. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with AcOEt–hexane (1:4, v/v) to give 13 (128.6 mg, 19%, 3 steps from mixture of 12a and 12b) as a colorless amorphous. 1H NMR (600 MHz, CDCl3) δ: 7.28–7.16 (m, 5H), 5.65–5.59 (m, 1H), 3.88 (q, J = 3.6 Hz, 1H), 3.14 (dd, J = 7.9, 1.7 Hz, 1H), 3.08 (d, J = 3.6 Hz, 1H), 2.92 (ddd, J = 16.8, 7.9, 1.7 Hz, 1H), 2.43–2.38 (m, 1H), 1.80 (dd, J = 14.6, 3.3 Hz, 1H), 1.51 (s, 3H), 1.33 (s, 3H), 1.27 (dd, J = 14.7, 3.6 Hz, 1H), 1.16 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 154.9, 144.9, 128.4, 128.0, 126.5, 122.2, 72.2, 72.0, 57.0, 55.8, 43.3, 37.3, 33.0, 32.1, 30.9, 23.3; HRMS [MNa]+ calcd for C18H24O2Na, 295.1668; Found, 295.1665.
4.8. (1SR,3RS,3aRS,4RS,7aRS)-4-Acetoxy-1,7a-epoxy-3-phenyl-3a,7,7-trimethyl-1,2,3a,4,7, 7a-hexahydro-3H-inden-5-one (14)
To a solution of 13 (115.9 mg, 0.43 mmol) in CH2Cl2 (5 mL) at −78 °C under argon atmosphere were added diisopropylethylamine (DIPEA) (0.1 mL, 0.86 mmol) and t-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) (98%, 0.2 mL, 0.86 mmol). The reaction mixture was stirred at −78 °C for 2.5 hours. After addition of H2O, the whole was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:9, v/v) to give 7-TBS protected product which was desired (30.6 mg, 19%) as a colorless oil, 6-TBS protected product (69.0 mg, 42%) as a colorless oil, and unreacted 13 (25.8 mg, 22%) as a colorless oil in the order of elution. To a solution of 7-TBS protected product (30.6 mg, 0.08 mmol) in CH2Cl2 (3 mL) at room temperature under argon atmosphere was added Dess-Martin reagent (97%, 41.6 mg, 0.1 mmol). The reaction mixture was stirred at room temperature for 13 hours. After addition of saturated NaHCO3 aq., the whole was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to leave an oil. To a solution of crude oil in THF (2 mL) at room temperature under argon atmosphere was added TBAF (1.0 M, 0.1 mL, 0.12 mmol). The reaction mixture was stirred at room temperature for 2.5 hours. Removal of the solvent afforded a crude product, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:9, v/v) to give 7-hydroxyketone (16.0 mg, 75%, 2 steps from 7-TBS protected product) as a colorless oil. To a solution of 7-hydroxyketone (16.0 mg, 0.06 mmol) in pyridine (0.5 mL) at room temperature were added Ac2O (0.25 mL) and catalytic amount of 4-dimethylaminopyridine (DMAP). The reaction mixture was stirred at room temperature for 24 hours. Removal of the solvent afforded a crude product, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:7, v/v) to give 7-acetoxyketone (17.8 mg, 96%) as a colorless oil. mCPBA (77%, 15.3 mg, 0.07 mmol) was added to a solution of 7-acetoxyketone (17.8 mg, 0.06 mmol) in CH2Cl2 (1.0 mL) at room temperature, and the mixture was stirred at room temperature for 6 hours. 5% Na2SO3 aq. was added and the resulting heterogeneous mixture was vigorously stirred. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were washed with 5% Na2CO3 aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by p-TLC on SiO2 with AcOEt–hexane (1:5, v/v) to give 14 (18.7 mg, 14%, 5 steps from 13) as a colorless amorphous: 1H NMR (600 MHz, CDCl3) δ: 7.30–7.25 (m, 2H), 7.25–7.19 (m, 1H), 7.17–7.12 (m, 2H), 5.07–5.06 (m, 1H), 3.80 (s, 1H), 2.85 (dd, J = 10.5, 8.3 Hz, 1H), 2.59 (d, J = 13.2 Hz, 1H), 2.42 (d, J = 13.2 Hz, 1H), 2.37–2.27 (m, 2H), 1.36 (s, 3H), 1.23 (s, 3H), 1.16 (s, 3H), 1.00 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 202.4, 169.6, 138.0, 128.6, 128.3, 127.0, 76.2, 70.9, 61.2, 51.3, 50.5, 50.0, 36.5, 30.1, 27.4, 24.3, 19.6, 17.3; HRMS [MH]+ calcd for C20H25O4, 329.1747; Found, 329.1750.
Acknowledgments
This work was supported by National Institute of Health (NIH) grant (AI 072358) and The Skaggs Institute for Chemical Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References and notes
- 1.Johnson EA, Bradshaw M. Toxicon. 2001;39:1703. doi: 10.1016/s0041-0101(01)00157-x. [DOI] [PubMed] [Google Scholar]
- 2.Schantz EJ, Johnson EA. Microbiol Rev. 1992;56:80. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson L. Annu Rev Pharmacol Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 4.Dickerson TJ, Janda KD. ACS Chem Biol. 2006;1:359. doi: 10.1021/cb600179d. [DOI] [PubMed] [Google Scholar]
- 5.Madsen JM. Clin Lab Med. 2001;21:593. [PubMed] [Google Scholar]
- 6.Christopher GW, Cieslak TJ, Pavlin JA, Eitzen EM., Jr JAMA. 1997;278:412. [PubMed] [Google Scholar]
- 7.Zilinskas RA. JAMA. 1997;278:418. [PubMed] [Google Scholar]
- 8.(a) Èapková K, Yoneda Y, Dickerson TJ, Janda KD. Bioorg Med Chem Lett. 2007;17:6463. doi: 10.1016/j.bmcl.2007.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boldt GE, Eubanks LM, Janda KD. Chem Commun. 2006:3063. doi: 10.1039/b603099h. [DOI] [PubMed] [Google Scholar]; (c) Boldt GE, Kennedy JP, Janda KD. Org Lett. 2006;8:1729. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc Natl Acad Sci USA. 2007;104:2602. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. ACS Chem Biol. 2007;14:533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Nakatani M. Heterocycles. 1999;50:595. [Google Scholar]
- 10.Carpinella C, Ferrayoli C, Valladares G, Defago M, Palacios S. Biosci Biotechnol Biochem. 2002;66:1731. doi: 10.1271/bbb.66.1731. [DOI] [PubMed] [Google Scholar]
- 11.Xie YS, Fields PG, Isman MB, Chen WK, Zhang X. J Stored Prod Res. 1995;31:259. [Google Scholar]
- 12.(a) Akhtar Y, Isman MB. J Chem Ecol. 2003;29:1853. doi: 10.1023/a:1024802328458. [DOI] [PubMed] [Google Scholar]; (b) Carpinella MC, Defago MT, Valladares G, Palacios SM. J Agric Food Chem. 2003;51:369. doi: 10.1021/jf025811w. [DOI] [PubMed] [Google Scholar]; (c) Zhou JB, Minami Y, Yagi F, Tadera K, Nakatani M. Heterocycles. 1997;45:1781. [Google Scholar]
- 13.(a) Li P, Zou J, Miao W, Ding F, Meng J, Jai G, Li J, Ye H, He X. Zhongcaoyao. 1982;13:28. [Google Scholar]; (b) Zou J, Miao W, Ding F, Meng J, Ye H, Jia G, He X, Sun G, Li P. J Trad Chin Med. 1985;5:29. [PubMed] [Google Scholar]
- 14.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2008;41:40. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 15.Paquette LA, Hong FT. J Org Chem. 2003;68:6905. doi: 10.1021/jo0301346. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Mateos A, Martin de la Nava EM, Pascual Coca G, Rubio Gonzalez R, Ramos Silvo AI, Simmonds MSJ, Blaney WM. J Org Chem. 1998;63:9440. [Google Scholar]
- 17.Fernandez-Mateos A, Martin de la Nava EM, Rabanedo Clemente R, Rubio Gonzalez R, Simmonds MSJ. Tetrahedron. 2005;61:12264. [Google Scholar]
- 18.Fernandez-Mateos A, Martin de la Nava EM, Rubio Gonzalez R. Tetrahedron. 2001;57:1049–1057. [Google Scholar]
- 19.Fernandez-Mateos A, Pascual Coca G, Rubio Gonzalez R, Tapia Hernandez C. J Org Chem. 1996;61:9097. [Google Scholar]
- 20.Fernandez-Mateos A, Lopez Barba AM. J Org Chem. 1995;60:3580. [Google Scholar]
- 21.Fernandez-Mateos A, Lopez Barba A, Pascual Coca G, Rubio Gonzalez R, Tapia Hernandez C. Synthesis. 1997:1381. [Google Scholar]
- 22.Fernandez-Mateos A, Mateos Buron L, Martin de la Nava EM, Rubio Gonzalez R. J Org Chem. 2003;68:3585. doi: 10.1021/jo0205311. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Mateos A, Lopez Barba AM, Martin De La Nava E, Pascual Coca G, Perez Alonso JJ, Ramos Silvo AI. Tetrahedron. 1997;53:14131. [Google Scholar]