

Abstract
The PMR1 mRNA endonuclease catalyzes the selective decay of a limited number of mRNAs. It participates in multiple complexes, including one containing c-Src, its activating kinase, and one containing its substrate mRNA. This study used tandem affinity purification (TAP) chromatography to identify proteins in HeLa cell S100 associated with the mature 60-kDa form of Xenopus PMR1 (xPMR60). Unexpectedly, this identified a number of cytoskeleton-associated proteins, most notably the Ena family proteins mammalian Enabled (Mena) and vasodilator-stimulated phosphoprotein (VASP). These are regulators of actin dynamics that distribute throughout the cytoplasm and concentrate along the leading edge of the cell. xPMR60 interacts with Mena and VASP in vivo, overexpression of Mena has no impact on mRNA decay, and Mena and VASP are recovered together with xPMR60 in each of the major complexes of PMR1-mRNA decay. In a wound-healing experiment induced expression of active xPMR60 in stably transfected cells resulted in a twofold increase in cell motility compared with uninduced cells or cells expressing inactive xPMR60°. Under these conditions xPMR60 colocalizes with VASP along one edge of the cell.
Keywords: mRNA decay, cytoskeleton, PMR1, Mena, VASP, c-Src
INTRODUCTION
It is generally accepted that all mRNAs are regulated to some degree by changes in their rate of turnover, and a recent review (Garneau et al. 2007) summarizes much of what is known about the basic biochemical mechanisms of eukaryotic mRNA decay. In both yeast and vertebrates most mRNAs decay by shortening of the poly(A) tail, followed by 3′-5′ exonuclease-mediated degradation of the mRNA body by the exosome, or removal of the 5′ cap and 5′-3′ degradation by Xrn1. Decapping and 5′-3′ decay is the predominant mechanism in yeast, and the identification of concentrated foci containing proteins involved in this process (P bodies) led to the notion that these are the principle sites of mRNA decay. While there is good evidence that P bodies can serve a similar function in mammalian cells (Franks and Lykke-Andersen 2007), the exosome is notably absent from P bodies, and recent work from our laboratory (Murray and Schoenberg 2007) and others (Mullen and Marzluff 2008) showing that unstable mRNAs decay from both ends is inconsistent with a primary role for P bodies in mRNA decay. Instead, P bodies more likely function primarily as storage sites for nontranslating mRNA (Eulalio et al. 2007; Parker and Sheth 2007).
In addition to these exonuclease-mediated processes, a limited number of mRNAs undergo endonuclease-mediated mRNA decay. Examples of this include the mRNAs encoding Xenopus albumin (Pastori et al. 1991; Yang and Schoenberg 2004), insulin-like growth factor II (van Dijk et al. 1998), transferrin receptor (Binder et al. 1994), and some endoplasmic reticulum-associated proteins (Hollien and Weissman 2006). Endonuclease-mediated mRNA decay differs fundamentally from the exonuclease process in that it targets only a subpopulation of the transcriptome. The selectivity of this process is determined by the formation of a complex between the endonuclease and its translating substrate mRNA (Yang and Schoenberg 2004; Hollien and Weissman 2006). To date, three mRNA endonucleases have been identified and characterized: PMR1 (Chernokalskaya et al. 1998), G3BP (Gallouzi et al. 1998), and IRE1 (Hollien and Weissman 2006).
PMR1 was identified as a ribonuclease activity with selectivity for albumin mRNA that appeared at the same time as the estrogen induced destabilization of serum protein mRNAs in Xenopus liver (Pastori et al. 1991). It was subsequently purified (Dompenciel et al. 1995), and the sequence of peptide fragments and cDNAs showed this to be a member of the peroxidase gene family (Chernokalskaya et al. 1998). Identification of the gene encoding PMR1 proved elusive; the sequence was similar overall to the major peroxidases (myeloperoxidase, lactoperoxidase, thyroperoxidase, eosinophil peroxidase) but differed from these at regions critical for its function in mRNA decay. A combined bioinformatics and biochemical analysis recently identified this as the peroxidasin-like protein (data not shown). We previously referred to the mature 60-kDa form of Xenopus PMR1 as PMR60, but from this point forward it will be denoted xPMR60 to distinguish it from the mammalian forms of the enzyme.
Much of our understanding of PMR1-mediated decay comes from the use of transfected mammalian cell systems that recapitulate properties of this protein seen in experiments with whole animals and cultured hepatocytes. In both frog liver and transfected mammalian cells, xPMR60 is found primarily in two complexes: the functional ∼680-kDa polysome-bound mRNP containing the endonuclease and its substrate mRNA (complex I), and a ∼140-kDa complex that is devoid of target mRNA (complex II) (Yang and Schoenberg 2004). The principal polysome targeting domain, which lies in the C terminus of xPMR60 and its mammalian ortholog, contains an SH2 phosphotyrosine motif, and tyrosine phosphorylation of this site is required for its targeting to polysomes, for its participation in a ∼680-kDa complex with its substrate mRNA, and for endonuclease-mediated mRNA decay (Yang et al. 2004). Recent work identified c-Src as the kinase that catalyzes this key activating step in endonuclease-mediated mRNA decay (Peng and Schoenberg 2007).
In this study, we sought to identify xPMR60-interacting proteins from HeLa cells using S100 as the starting material for selection by tandem affinity purification (TAP). Unexpectedly, this identified a number of the cytoskeleton-associated proteins including two members of the enabled (Ena) family of proteins, mammalian enabled (Mena), and vasodilator-stimulated phosphoprotein (VASP). The Ena proteins, Mena, VASP, and the related protein Evl, regulate actin dynamics and coordinate signal transduction through the actin cytoskeleton (Krause et al. 2003; Kwiatkowski et al. 2003). They have three major functional domains. The N-terminal EVH1 domain binds to proline-rich peptides containing FPPPP (FP4) sequence elements that are present in zyxin, vinculin, and the ActA protein of Listeria monocytogenes but not to APPPP (AP4) repeats. The central portion of the protein is a proline-rich sequence that binds to WW and SH3 domains, including the tyrosine kinases c-Abl and c-Src (Gertler et al. 1995), and the C-terminal EVH2 domain contains binding sites for both F- and G-actin. Mena differs from VASP by the presence of a repeating LERER sequence, and unlike VASP, is expressed in multiple alternatively spliced forms. While these proteins are present in the cytoplasm they can concentrate at the leading edge of the cell, where they function to drive forward movement. We present data showing the interaction of xPMR60 with the Ena/VASP proteins is an inherent aspect of PMR1 mRNA decay. Furthermore, in an assay for cell motility both xPMR60 and VASP colocalize along the edge of the cell opposite the majority of actin stress fibers, that catalytically active xPMR60 stimulates cell motility whereas an inactive form of the enzyme does not.
RESULTS
Recovery of cytoskeleton-associated proteins with xPMR60°-TAP
A stable line of tetracycline-regulated HeLa cells was generated that expresses catalytically inactive xPMR60 (xPMR60°) with a C-terminal tandem affinity (TAP) tag (xPMR60°-TAP) (Yang and Schoenberg 2004). These were grown in 1-L suspension cultures, and S100 prepared from postnuclear extract was processed through TAP chromatography on IgG-Sepharose and calmodulin agarose (Puig et al. 2001). Figure 1 shows a Coomassie blue-stained SDS-polyacrylamide gel of proteins eluted with ethylenediaminetetraacetic acid (EDTA) from the calmodulin agarose column electrophoresed next to a control preparation from nontransfected HeLa cells. Tryptic fragments from each of the indicated bands were analyzed by automated matrix-assisted laser desorption/ionization (MALDI) and liquid chromatography–mass spectrometry (LC-MS)/MS, and proteins that matched to at least 3 peptides are indicated on the left. Unexpectedly, these all are associated with the actin cytoskeleton, including β-actin, α-actinin, IQGAP (which is involved in actin assembly through the Arp2/3 complex) (Le Clainche et al. 2007), calmodulin (which interacts with IQGAP) (Li and Sacks 2003), and myosin alkali light chain 6, a nonskeletal myosin. We also recovered two isoforms of the mammalian ortholog of the Drosophila Enabled protein (Mena) and the closely related VASP.
FIGURE 1.

Tandem affinity purification of xPMR60°-associated proteins. S100 preparations from untransfected HeLa cells (lane 1) or HeLa cells that stably express xPMR60°-TAP (lane 2) were selected first on IgG-Sepharose, and protein recovered by Tev protease cleavage was then bound to calmodulin agarose. Proteins eluted with EDTA were separated on a 4%–20% gradient SDS-polyacrylamide gel and identified by staining with Coomassie blue. Individual bands were cored from the gel and analyzed by MALDI and LC-MS/MS. At least three peptides were identified for each of the proteins indicated on the gel.
Because cytoskeleton-associated RNA-binding proteins (e.g., ZBP1) can control the subcellular distribution and translation of specific mRNAs (Farina et al. 2003; Huttelmaier et al. 2005), two different actin disruptors were employed to test whether Mena and VASP were recovered by the fortuitous association of xPMR60° with one or more unidentified RNA- or actin-binding proteins. Latrunculin A (Lat A) is a marine toxin that binds stoichiometrically to monomeric actin, and cytochalasin D (CD) interferes with the binding of capping proteins to barbed ends of actin filaments. These effectively disrupt the actin cytoskeleton without altering the cytoplasmic distribution of xPMR60° in Cos-1 cells (data not shown). In the experiment in Figure 2A, Cos-1 cells expressing xPMR60°-TAP or the same protein with an inactivated tyrosine phosphorylation site (Y650F) were treated with or without Lat A for 1 h before lysis and recovery of cytoplasmic xPMR60°-containing complexes on IgG-Sepharose. Proteins bound to IgG-Sepharose were recovered by cleaving the TAP tag with Tev protease, the effectiveness of which is seen by the change in the SDS-polyacrylamide gel mobility of xPMR60° by Western blotting with a monoclonal antibody to the N-terminal myc tag. Lat A had no impact on the recovery of Mena with xPMR60° or with the Y650F mutant form of the protein. Although both forms of Mena were recovered in Figure 1, coprecipitation with xPMR60° recovered more of the 75-kDa form. The reason for this is not known; however, it may reflect differences in the phosphorylation state of Mena (Howe et al. 2002). Similar results were seen when protein immunoprecipitated with antibody to the N-terminal myc tag on xPMR60° was analyzed by Western blotting with a polyclonal antibody to VASP that crossreacts with Mena (Fig. 2C).
FIGURE 2.

xPMR60° binding to Mena and VASP is independent of RNA and the integrity of the actin cytoskeleton. (A) Cos-1 cells were transfected with plasmids expressing xPMR60°-TAP or Y650F-TAP and treated without (−) or with (+) Latunculin A (Lat A). Cytoplasmic extracts were bound onto IgG-Sepharose and input protein, and protein recovered by Tev protease cleavage of the bound complexes was analyzed by Western blotting with antibodies to the myc tag on xPMR60° and Mena. (B) Cos-1 cells expressing myc-tagged GFP or xPMR60° were treated without (−) or with (+) cytochalasin D (CD) prior to lysis. Epitope-tagged proteins were recovered from cytoplasmic extracts using immobilized myc antibody, and input and bound complexes were analyzed by Western blotting with antibodies to the myc tag (top panels) or Mena (bottom panels). (C) Extracts from cells treated as in B but without CD addition were analyzed by Western blotting with antibody to the myc tag on xPMR60° and GFP or a rabbit polyclonal antibody to VASP that also cross-reacts with Mena. (D) Cos-1 cells were transfected as in B, and cytoplasmic extracts were treated with RNase A before immunoprecipitation with immobilized myc monoclonal antibody. Recovered proteins were analyzed by Western blotting with antibodies to the myc tag and Mena (left panels), and the effectiveness of the RNase digestion was determined by native agarose gel electrophoresis of RNA recovered from the same samples (right panel).
The cytoskeleton-independent binding of xPMR60° to Mena was confirmed in Figure 2B, where Cos-1 cells expressing myc-tagged GFP or xPMR60° were treated with CD for 30 min before lysis and immunoprecipitation with antibody to the myc tag. The input protein samples show that CD has no effect on the overall expression of either GFP or xPMR60°, but it reduced the amount of 80-kDa Mena without affecting the 75-kDa protein (Fig. 2B, cf. lanes 1,2 and lanes 3,4). Nevertheless, Mena was recovered with xPMR60° but not GFP, and CD had no impact on its recovery. Together the data in Figure 2, A and B, indicate that integrity of the actin cytoskeleton is not required for the recovery of Mena by xPMR60°. Also, similar results obtained with xPMR60° and the nonphosphorylatable Y650F form of the protein indicates that the Mena binding is independent of tyrosine phosphorylation.
The functional unit of PMR1-mediated mRNA decay is an mRNP complex containing the endonuclease and its substrate mRNA (Yang and Schoenberg 2004), and RNase A digestion disrupts the xPMR60-recovery of proteins that depend on RNA for their interaction (Yang et al. 2006). The results in Figure 2D show that treating cytoplasmic extracts from Cos-1 cells expressing GFP or xPMR60° with RNase A prior to immunoprecipitation has no impact on Mena recovery, thus indicating that Mena either binds directly to xPMR60° or these proteins interact through a shared intermediary.
mRNA decay is unaffected by changing the distribution or amount of the Ena/VASP proteins
The EVH1 domains of Mena, VASP, and Evl bind tightly to FPPPP (FP4) repeats (Krause et al. 2003), and previous work took advantage of this to examine the function of these proteins in actin dynamics by coupling FP4 repeats to EGFP and a sequence that targets the fusion protein to outer mitochondrial membrane (EGFP-FP4-mito) (Bear et al. 2000; Scott et al. 2006). EVH1 domain binding is lost when FP4 repeats are replaced with AP4, and a matching fusion protein bearing these repeats serves as control for this experiment. To determine whether the Ena family proteins bind to xPMR60° in vivo, each of these EGFP fusion proteins was cotransfected into Cos-1 cells with a plasmid expressing xPMR60° (Fig. 3A), and their distribution was examined by immunofluorescence microscopy. In cells expressing EGFP-FP4-mito, approximately half of the mitochondria stain for EGFP and xPMR60° (insert), but no costaining was observed in cells expressing EGFP-AP4-mito. This selective relocalization confirms that the Ena/VASP proteins interact in vivo with xPMR60°.
FIGURE 3.

Tyrosine phosphorylation and xPMR60-mediated mRNA decay are unaffected by proteins that change the distribution of the Ena/VASP proteins and by increased expression of Mena. (A) Cos-1 cells transfected with plasmids expressing myc-xPMR60° and EGFP-FP4-mito (left panel) or EGFP-AP4-mito (right panel) were fixed, stained with a monoclonal antibody to the myc tag on xPMR60°, and visualized with AlexaFluor 594-coupled rabbit anti-mouse IgG (red) and EGFP fluorescence. Bar, 10 μM; insets, 2.5× magnification of the areas in the white boxes. (B) U2OS cells were cotransfected with equal amounts of plasmid expressing xPMR60° and EGFP-FP4-mito (FP4-mito) or EGFP-AP4-mito (AP4-mito) and myc-GFP. In the top panels, cytoplasmic extracts (input) were analyzed by Western blotting with antibodies to the myc tag on xPMR60° and GFP (left panel) or GFP to detect the EGFP fusion proteins (right panel). In the bottom panel protein recovered by immunoprecipitation with immobilized myc antibody was analyzed by Western blotting with antibody to the myc tag on xPMR60° and phosphotyrosine monoclonal antibody (PY20). (C) Triplicate cultures of U2OS cells were each transfected with plasmids expressing albumin (substrate) and luciferase (control) mRNAs. Cells in lanes 4–6 were cotransfected with plasmids expressing GFP and empty vector (pcDNA3), cells in lanes 7–9 were cotransfected with plasmids expressing catalytically active xPMR60 and empty vector (pcDNA3), cells in lanes 10–12 were cotransfected with plasmids expressing xPMR60 and EGFP-AP4-mito, and cells in lanes 13–15 were cotransfected with plasmids expressing xPMR60 and EGFP-FP4-mito. Total RNA isolated 24 h after transfection was analyzed by RNase protection assay using a mixed probe for albumin and luciferase mRNA. Lane 1 (M) contains end labeled Hinf ΦX174 restriction fragments, lane 2 contains 1:20 of the undigested probe, lane 3 is the same sample after RNase digestion. The mean of the amount of albumin mRNA normalized to luciferase mRNA in each set of triplicates determined by PhosphorImager analysis is indicated beneath the autoradiogram. (D) Triplicate cultures of U2OS cells were transfected as in C except a plasmid expressing EGFP-Mena was substituted for plasmids expressing EGFP-FP4/AP4-mito fusion proteins. The expression of xPMR60 and myc-GFP and EGFP-Mena were monitored by Western blotting as in B. (E) The impact of EGFP-Mena on xPMR60-catalyzed decay of albumin mRNA was determined as in C by RNase protection assay.
Next, we examined the impact of each of these proteins on tyrosine phosphorylation and xPMR60-catalyzed mRNA decay. In Figure 3B, U2OS cells were cotransfected with equal quantities of plasmids expressing xPMR60° and EGFP-FP4-mito or EGFP-AP4-mito, and protein recovered by immunoprecipitation with antibody to the myc tag on xPMR60° was analyzed by Western blotting with a phosphotyrosine monoclonal antibody (PY20). Neither of the EGFP fusion proteins altered the overall tyrosine phosphorylation of xPMR60°. This experiment was repeated in Figure 3C with the addition of plasmids expressing substrate (albumin) and firefly luciferase (control) mRNA. Triplicate cultures were transfected with each of these plasmids and GFP + empty vector (Fig. 3C, pcDNA3, lanes 4–6), catalytically active xPMR60 + empty vector (Fig. 3C, pcDNA3, lanes 7–9), xPMR60 + EGFP-AP4-mito (Fig. 3C, lanes 10–12), or xPMR60 + EGFP-FP4-mito (Fig. 3C, lanes 13–15). RNA recovered 24 h later was analyzed by RNase protection assay, and changes in albumin mRNA were normalized by PhosphorImager analysis to the luciferase control. Approximately 60% of albumin mRNA was degraded in cultures expressing xPMR60 compared with cells transfected with GFP (Fig. 3C, cf. lanes 4–6 and lanes 7–9), and this was unaffected by either of the EGFP fusion proteins (Fig. 3C, lanes 10–15).
The experiment in Figure 3, D and E, examined the impact of increased Mena expression on mRNA decay. Triplicate cultures were transfected with plasmids expressing myc-tagged GFP or catalytically active xPMR60 and empty vector (pcDNA3) or a plasmid expressing an EGFP-Mena fusion protein, and the expression of each of these proteins was monitored by Western blotting (Fig. 3D). Note that xPMR60 and GFP expression were detected with antibody to the myc tag on each of these proteins, and EGFP-Mena expression was detected with an antibody to GFP that recognizes EGFP, albeit weakly. Increasing the amount of Mena in the cell had no impact on the selective decay of albumin mRNA when assayed by RNase protection assay (Fig. 3E). Taken together, the results in Figure 3 suggest that interaction with the Ena/VASP proteins is an inherent aspect of xPMR60 mRNA decay.
The binding of Mena depends on the overall integrity of xPMR60°
Previous work identified four key functional domains of xPMR60°: a catalytic domain in the central portion of the protein (Chernokalskaya et al. 1998), a C-terminal polysome-targeting domain (Yang and Schoenberg 2004) containing the requisite tyrosine phosphorylation site (Y650) (Yang et al. 2004), a TIA-1-binding domain that targets xPMR60° to stress granules in stressed cells (Yang et al. 2006), and a binding site for c-Src that is required for phosphorylation at Y650 (Peng and Schoenberg 2007) (Fig. 4A, top diagram). The c-Src-binding site contains two repeats of PPXXP, and mutating either of these to PAXXA blocks the interaction with c-Src and tyrosine phosphorylation.
FIGURE 4.

The recovery of Mena is sensitive to the overall integrity of xPMR60° and is not through c-Src. (A) The organization of xPMR60° is shown with the N-terminal TIA-1 binding domain in gray, the PPXXP c-Src binding sites and Y650 indicated in black. Beneath this are the deletion constructs that were used to identify the Mena-binding domain. (B) Cos-1 cells were transfected with the plasmids diagrammed in A together with a plasmid expressing GFP with an N-terminal myc tag. In lane 1, cells were transfected with the GFP expression plasmid and empty vector. Cytoplasmic extracts were immunoprecipitated with myc antibody and recovered protein was analyzed by Western blot with antibodies to the myc tag on GFP and the xPMR60° deletion constructs and to Mena. (C) Cos-1 cells were transfected with plasmids expressing xPMR60° in which the proline residues in the c-Src binding sites at 262 and 265 (P262,265A) or 270 and 273 (P270,273A) were changed to alanine. Cytoplasmic extract was immunoprecipitated with antibody to the myc tag on each of the proteins and input and bound complexes were analyzed by Western blot with antibodies to the myc tag and to Mena.
xPMR60 has neither FP4 or similar proline-rich peptides nor a Lim-3-like domain (Boeda et al. 2007), so it is unlikely that the Ena/VASP proteins bind through their EVH1 domains. We used Western blotting of protein recovered with the battery of N- and C-terminal deletion constructs shown in Figure 4A to determine whether the Ena/VASP proteins bind to one of the previously mapped motifs. Removing the N-terminal 50 amino acids containing the TIA-1-interacting domain (Δ50N) had no impact on the recovery of Mena (Fig. 4B, lane 3); however, Mena binding was lost when 100 amino acids were removed from the N terminus (Fig. 4B, lane 4), and with the exception of Δ100C, with all of the other deletions. The absence of any clear evidence linking one of the mapped domains to Mena binding suggests that binding is dependent on the overall integrity of xPMR60°.
To determine whether Mena was recovered through the shared binding of the Ena/VASP proteins and xPMR60° to c-Src (see below), we examined its recovery with forms of xPMR60° in which the PPXXP c-Src-binding motifs at amino acids 262–265 and 270–273 were individually changed to PAXXA (Fig. 4C; Peng and Schoenberg 2007). Neither of these mutations affected the recovery of Mena by xPMR60°, confirming both that Mena does not bind to these motifs and that the interaction of xPMR60° with Mena is independent of the interaction of xPMR60 with c-Src.
Mena interacts with c-Src and xPMR60°
The identification of adjacent binding sites for c-Src and Mena on xPMR60° suggested that Mena might interact with both proteins. This was first examined using U2OS cells, which like most transformed cell lines have elevated levels of c-Src (Peng and Schoenberg 2007). In the experiment in Figure 5A, cytoplasmic extract from untransfected cells was immunoprecipitated with antibodies to GFP (control) or c-Src, and the recovered proteins were analyzed by Western blotting with antibodies to c-Src and Mena. Mena was recovered by immunoprecipitation with antibody to c-Src but not with antibody to GFP, indicating that these proteins interact in the absence of xPMR60°. Next, U2OS cells were transfected with myc-tagged GFP or xPMR60°, and protein recovered by immunoprecipitation with myc antibody was analyzed by Western blotting with antibodies to the myc tag on xPMR60°, c-Src, and Mena (Fig. 5B). Neither Mena nor c-Src was recovered with GFP, and both proteins were recovered with xPMR60°. Since the interaction of c-Src with Mena is independent of xPMR60°, we asked whether the interaction of xPMR60° with Mena is independent of c-Src. In the experiment in Figure 5C, c-Src-deficient Cos-1 cells (Peng and Schoenberg 2007) were transfected with plasmids expressing GFP (Fig. 5C, lanes 1,6) or xPMR60° and either empty vector, kinase inactive (KI-Src), constitutively active (CA-Src), or wild-type (WT-Src) forms of c-Src. Western blotting of the input samples showed each has equivalent amounts of Mena, and that c-Src was only detected in cells that were transfected with one of the indicated expression plasmids (left panels), and equivalent amounts of xPMR60° were recovered from each of the transfectants by immunoprecipitation with myc antibody (Fig. 5C, lanes 7–10). Importantly, the recovery of Mena was unaffected by either of the active forms of c-Src or the inactive form of c-Src. In studying the interaction of c-Src with xPMR60°, we noted that less CA-Src was reproducibly recovered than KI-Src or WT-Src. We suspect this results from destabilization of the complex after phosphorylation of xPMR60°. Taken together, the data in Figure 5 indicate that xPMR60° and c-Src bind independently to the Ena/VASP proteins.
FIGURE 5.

c-Src binds independently to Mena, and c-Src and Mena are recovered with immunoprecipitated xPMR60°. (A) U2OS cell cytoplasmic extracts were immunoprecipitated with antibodies to GFP or c-Src and the input (lanes 1 and 2) and bound (lanes 3 and 4) fractions were analyzed by Western blotting with antibodies to c-Src and Mena. The open dot in the upper panels (○) is a band that crossreacts with the c-Src antibody. (B) U2OS cells were transfected with plasmids expressing myc-tagged GFP or xPMR60°. Total cytoplasmic extract (input) and complexes recovered by immunoprecipitation with a myc antibody were analyzed by Western blotting with antibodies to the myc tag, Mena, and c-Src. (C) A line of Cos-1 cells with <20% of the amount of endogenous c-Src as the U2OS cells (Peng and Schoenberg 2007) was transfected with myc-tagged GFP or xPMR60° plus pUSE (the empty c-Src expression vector, lanes 1,2,6,7), or plasmids expressing xPMR60° and kinase-inactive c-Src (KI-Src, lanes 3,8), constitutively active c-Src (CA-Src, lanes 4,9), or wild-type c-Src (WT-Src, lanes 5,10). The left panels are Western blots showing expression of GFP, xPMR60°, c-Src, and Mena in the input samples and the right panels are Western blots of complexes recovered by immunoprecipitation with a monoclonal antibody to the myc tag.
Catalytically active xPMR60 stimulates motility but not cell growth
While the preceding data indicate that binding to the Ena/VASP proteins is an inherent property of xPMR60, they do not indicate the function of this interaction. Because the Ena/VASP proteins function in controlling cell motility we asked whether xPMR60 might somehow be linked to this process through its interaction with these proteins. To address this we developed lines of U2OS cells expressing active xPMR60 or inactive xPMR60°-TAP under a tetracycline-inducible promoter and determined the impact of expressing each of these proteins on cell motility using a “wound-healing” assay. In the wound-healing assay an almost confluent monolayer is scored, and the same field is photographed over a period of time to measure the rate at which cells move to fill the gap. By adding mitomycin C at a concentration that blocks replication but does not kill the cells, changes in the rate of wound healing provide a measure of cell motility.
In the experiment in Figure 6, cultures of individual clonal lines expressing active (xPMR60) or inactive (xPMR60°) PMR1 were cultured for 16 h in the presence or absence of doxycycline. At this point mitomycin C was added, the monolayer was scored, and a selected field of each culture was photographed (0 h). The same field in each of triplicate cultures was photographed over the next 26 h, and the mean of multiple measurements of each field was used to determine the percent change in the width of the gap as a function of time. These results are graphed in Figure 6A, and photographs of representative fields taken at 0, 12, and 26 h after the start of the experiment are shown in Figure 6B.
FIGURE 6.

Motility is increased in cells expressing catalytically active xPMR60 but not in cells expressing xPMR60°. (A) Triplicate cultures of tetracycline-inducible U2OS cells expressing xPMR60 (line #5) or xPMR60°-TAP (line #1) were treated without (−Dox) or with 1 μg/mL doxycycline (+Dox) for 16 h to induce protein expression. At t = 0, a scratch was made through each monolayer, mitomycin C was added to block replication, and registration marks were placed on the bottom of each well to define photographic fields for each dish. The same field of triplicate cultures was photographed under phase contrast at the indicated times, and the distance across the gap was determined using image analysis software. Multiple measurements were made for each field, and the mean ± standard deviation of the percent of the gap closed was plotted as a function of time. (B) Representative photographs taken at the start of the experiment, and after 12 and 26 h are shown for a single dish of cells of each of the treatment groups in A. Each time point tracks the same field of cells throughout the experiment. (C) The cells from each of the treatment groups were collected and pooled at the end of the experiment, and protein expression was determined by Western blotting using monoclonal antibody to the myc tag on xPMR60 and xPMR60°-TAP, and a rabbit polyclonal antibody to GAPDH. (D) Two different lines of xPMR60-expressing cells (#1, circles, and #5, squares), and a matching line of tetracycline-inducible myc-GFP-expressing cells (triangles) were cultured in medium without (−Dox, open symbols, dotted lines) or with 1 μg/mL doxycycline (+Dox, solid symbols and lines). Triplicate cultures were harvested at the indicated times, and the number of cells in each well was determined by Coulter counter. The results are plotted as the log of the mean ± standard deviation for each treatment group as a function of time.
xPMR60 expression was undetectable in either cell line cultured without doxycycline (Fig. 6C), and the rate of wound healing was identical for each of these and for a control line of cells expressing myc-tagged GFP under the same inducible promoter (not shown). The active and inactive forms of xPMR60 were induced to the same degree following addition of doxycycline (Fig. 6C), but their expression had dramatically different effects on cell motility. Whereas cells expressing inactive xPMR60°-TAP moved into the wound at the same rate as each of the uninduced cell lines, expression of active xPMR60 resulted in a twofold increase in motility. This was confirmed in three separate experiments using two different clonal lines.
To determine whether xPMR60 affected the rate at which these cells replicate, we performed growth curves using triplicate cultures of two clonal lines of xPMR60-expressing cells that were cultured with and without doxycycline and compared these results with the growth of a clonal line of cells expressing myc-tagged GFP (Fig. 6D). All three lines grew at the same rate regardless of the expression of xPMR60 or GFP, thus confirming that the catalytically active xPMR60 stimulates cell motility and not cell growth.
Immunofluorescence colocalizes xPMR60 and VASP to the leading edge in migrating cells
In the same experiment we also examined the subcellular distribution VASP, xPMR60, and the actin cytoskeleton. xPMR60-expressing cells were plated on coverslips, and induced, scored and handled as described above. These were washed and fixed at intervals over the next 24 h and stained with a mouse monoclonal antibody to the myc tag on xPMR60, a rabbit monoclonal antibody to VASP (different from the one used to detect VASP by Western blotting), and AlexaFluor488-tagged phalloidin (Fig. 7). Prior to scoring the monolayer, VASP was distributed throughout the cytoplasm, and a portion was also seen to concentrate inside the plasma membrane, regardless of whether xPMR60 was expressed (Fig. 7, t = 0 h, top panels). In doxycycline-treated cells, the staining pattern for VASP and xPMR60 was almost superimposable (Fig. 7, t = 0 h, +Dox).
FIGURE 7.

Immunofluorescence localization of xPMR60 and VASP in migrating cells. A parallel set of cultures was grown on coverslips and treated as described in Fig. 6A. These were fixed at the time the monolayer was scored (t = 0) and 12 h later, and stained with a mouse anti-myc monoclonal antibody, a rabbit anti-VASP monoclonal antibody, and AlexaFlour 488-tagged phalloidin to visualize stress fibers. Color separations of the area in each of the white boxes are shown enlarged 1.5-fold on the right side of each micrograph.
After scoring the monolayer, the distribution of VASP and stress fibers changed in a manner consistent with the induction of cell motility, with VASP now concentrating along one edge and actin stress fibers concentrating on the opposite side of the cell (Fig. 7, t = 12 h, −Dox). In doxycycline-treated cells, the change in the subcellular distribution of xPMR60 at this time was particularly striking. Whereas at the beginning of the experiment xPMR60 and VASP distributed throughout the cytoplasm, both of these proteins now concentrated along one edge of the cell, again opposite most stress fibers (Fig. 7, t = 12 h, +Dox). The pattern was similar at 24 h (data not shown). Phalloidin staining was reproducibly greater in xPMR60-expressing cells, a result that is consistent with the twofold increase in motility following induction of xPMR60.
Mena and VASP bind to the functional complex of xPMR60 with its substrate mRNA
The functional unit of endonuclease decay is a ∼680-kDa mRNP (termed Complex I) that contains xPMR60 and its substrate mRNA (Yang and Schoenberg 2004), which can be released from polysomes by treating cells with puromycin prior to lysis or adding EDTA to cytoplasmic extracts. The recovery of Mena and VASP with this complex would provide compelling evidence for a role for these in localizing mRNA decay. In the experiment in Figure 8, cytoplasmic extract from U2OS cells expressing xPMR60°-TAP was treated with EDTA and sedimented on a 10%–40% glycerol gradient. Individual fractions were analyzed directly (input) or applied to IgG-Sepharose and eluted by cleaving the TAP tag with Tev protease (bound). In agreement with the notion that binding to the Ena/VASP proteins is an intrinsic feature of PMR1 mRNA decay, Mena and VASP were both recovered by xPMR60°-TAP in the heaviest fractions of Complex I. In separate experiments each of these proteins was recovered with xPMR60 from fractions at the top of the gradient, and we suspect this reflects in vitro binding of proteins that dissociated during centrifugation.
FIGURE 8.

Glycerol gradient analysis of xPMR60° bound to Mena and VASP. Cytoplasmic extracts from U2OS cells expressing xPMR60°-TAP were treated with EDTA to dissociate bound ribosomes and centrifuged on a 10%–40% glycerol gradient. Individual fractions were analyzed directly (input) or after recovery of xPMR60°-containing complexes by Tev protease cleavage of proteins bound onto IgG-Sepharose (bound). xPMR60° was identified by Western blotting with monoclonal antibody to the myc tag, and Mena and VASP were identified by Western blotting with the VASP polyclonal antibody used in Fig. 3C that detects both proteins. The locations of Complex I and Complex II are indicated at the top, and the arrow indicates the direction of sedimentation. The size standards indicated on the bottom were determined by the sedimentation of gel filtration standards on a parallel gradient.
DISCUSSION
The recovery of Mena and VASP by TAP with xPMR60°-TAP was unexpected. Mena, VASP, and the related protein Evl distribute throughout the cytoplasm and concentrate along the leading edge of the cell, where they regulate dynamic changes in the actin cytoskeleton (Krause et al. 2003), and our first thought was that this was an artifact of xPMR60° binding to RNA and/or the cytoskeleton. Each of these possibilities was ruled out by experiments with two different cytoskeleton disruptors (latruncuin A and cytochalasin D) and by RNase digestion prior to immunoprecipitation (Fig. 2). Ideally, one would use RNAi to knock down the Ena family proteins and study the effects of this on xPMR60°. However, the overlapping function of Mena, VASP, and Evl makes it impractical to target any one of these by RNAi, and attempts to replicate xPMR60-mediated mRNA decay in mouse embryo fibroblasts that lack Mena and VASP and Evl (MVD7 cells [Bear et al. 2000[]) were limited by low transfection efficiency and poor protein expression in these cells.
We therefore turned to other means to determine the relationship between xPMR60, Mena, and VASP. Direct evidence for the in vivo interaction of these proteins came from coexpressing xPMR60° with an EGFP fusion protein containing AP4 or FP4 repeats and a mitochondrial targeting sequence (Fig. 3A, EGFP-FP4-mito, EGFP-AP4-mito). The EVH1 domain of the Ena family proteins binds tightly to FP4 (but not AP4) repeats, and whereas no mitochondrial staining for xPMR60° was observed for cells expressing the AP4 fusion protein, approximately half of the mitochondria stained for xPMR60° in cells expressing the FP4 fusion protein. As effective as EGFP-FP4-mito was at relocating xPMR60, neither it nor EGFP-AP4-mito impacted on tyrosine phosphorylation of xPMR60 or xPMR60 degradation of albumin mRNA (Fig. 3C). Increasing the amount of cellular Mena by cotransfection with a plasmid expressing an EGFP fusion protein also had no impact on albumin mRNA decay (Fig. 3E). These results and the recovery of c-Src and Mena together with immunoprecipitated xPMR60° (Fig. 5B) suggest that the interaction with the Ena/VASP proteins is an integral part of PMR1-mediated mRNA decay.
Further evidence for this was seen by experiments using immunofluorescence to monitor the distribution of xPMR60 and VASP in a wound-healing assay performed with cells that stably express xPMR60 under a tetracycline-inducible promoter. The wound-healing assay measures the motility of cells that are induced to migrate by scoring a confluent monolayer. In nonmigrating cells, VASP distributes throughout the cytoplasm and concentrates inside the plasma membrane. Under these conditions the distribution of xPMR60 overlaps with that of VASP (Fig. 7, top right panels). The distribution of VASP changes in migrating cells, where it concentrates along the side opposite the majority of actin stress fibers. The distribution of xPMR60 also changes, concentrating with VASP along the edge opposite most actin stress fibers (Fig. 7, bottom right panels). Together with the recovery of Mena and VASP with the functional unit of PMR1 mRNA decay (Fig. 8), these data point to their interaction being an integral part of endonuclease decay, with the Ena/VASP proteins controlling the distribution of PMR1 within the cell.
Lastly, we show that expression of catalytically active xPMR60 results in a twofold increase in cellular motility without affecting the rate of cell growth (Fig. 6). We noted previously that the activation of PMR1 by c-Src makes this specialized decay pathway a potential endpoint for this kinase in cancer (Peng and Schoenberg 2007). Increased motility is a hallmark of the invasive growth of cancer cells, and our results raise the possibility that PMR1 brought to the leading edge by the Ena/VASP proteins may contribute to this by increasing the localized decay of specific mRNAs. We have just begun work using “RIP-Chip” (Baroni et al. 2008) to identify mRNAs recovered with xPMR60°-TAP, and while this work is still in its initial stages, one of the confirmed targets, integrin beta-like 1 (ITGBL1) mRNA, is a likely candidate.
MATERIALS AND METHODS
Plasmid constructs
The preparation of plasmids expressing catalytically inactive xPMR60°, Y650F, P262,265A, and P270,273A mutants, and serial xPMR60 deletions were described previously (Yang and Schoenberg 2004; Peng and Schoenberg 2007). The empty vector pUSEamp and plasmids encoding wild-type (WT-Src), kinase-inactive (KI-Src), dominant-negative (DN-Src), or constitutively active (CA-Src) forms were from Upstate Biotechnology. To construct plasmid pTracer-Tet-Myc- xPMR60°-TAP, the sequence containing tetracycline-controlled transactivator (tTA) and myc-xPMR60°-TAP was amplified by primer (5′-TGCAGGGCCCTCAGGTTGACTTCCCCGC-3′ and 5′-ACTGGATATCACGAGGCCCTTTCGTC-3′) and then inserted into pTracer-CMV vector (Invitrogen) between ApaI and EcoRV sites. pcDNA3.1-FP4-mito, pcDNA3.1-AP4-mito, and pcDNA3.1(-)FL-Mena were obtained from Dr. Alpha S. Yap (The University of Queensland).
Antibodies
Monoclonal antibodies to the myc tag (9E10), myc antibody-couple agarose beads, antibodies to c-Src (SRC2 and B-12) and GFP (B2), HRP-coupled rabbit anti-mouse IgG, and mouse anti-rabbit IgG were purchased from Santa Cruz Biotechnology. Phosphotyrosine monoclonal antibody (PY20) was obtained from BD Biosciences. The rabbit antibodies to Mena and VASP used for immunoprecipitations were a generous gift from Frank Gertler (MIT). The VASP antibody used for immunofluorescence was purchased from Cell Signaling Technologies.
Cell culture
HeLa S3 (Tet-off) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS) and 4 mM glutamine. Cos-1 cells were cultured in DMEM plus 10% FBS and 2 mM glutamine. U2OS cells were cultured in McCoy's 5A medium plus 10% FBS. Unless otherwise indicated, cells were collected 40 h after transfection. In the experiments in Figures 2 and 3, the cells were treated with 1 μg/mL latrunculin A (Sigma) for 30 min or 2 μM cytochalasin D (Sigma) for 1 h at 37°C before fixing and staining or harvesting for Western blot analysis.
For the wound healing experiments in Figure 6, A and B, 2.5 × 105 U2OS cells were plated in each well of a 6-well dish and grown for 24 h, at which time 1 μg/mL doxycycline was added to half of the cultures. A total of 0.4 μg/mL mitomycin C was added 16 h later to block replication, and 30 min later the cell monolayer was scratched with a 1000 μL pipette tip. The plates were washed 3× with phosphate-buffered saline (PBS) to remove nonadherent cells, and fresh medium (+ doxycycline, if required, and mitomycin C) was added to the cells. Cells were visualized under phase contrast on an Olympus IX81 microscope at 0, 2, 4, 6, 8, 12, and 26 h post-scratch. Orientation marks placed on the bottom of each well were used to identify and photograph the same field at each time point. At 26 h, cells were harvested and total protein was isolated for Western blotting. To determine the rate at which the cells closed the gap, the width of the gap on each plate at the various time points was determined using the MicroSuite Biological Suite software. “Percent closed” was defined as 0% at the 0-h time point and percent closed was calculated for other time points using the formula:
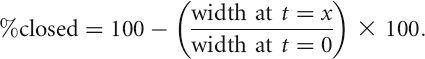 |
For each set of triplicate plates at each time point, an average percent closed and standard deviation was calculated and data was plotted using Excel.
To determine the impact of xPMR60 on the rate of cell growth 2 × 104 cells of xPMR60-expressing U2OS lines #1 and #5 and a matching line of cells expressing myc-GFP (#2) were seeded in each well of 12-well dishes. The next day half of the cultures received 1 μg/mL doxycycline to induce each of these proteins, and at the indicated times triplicate cultures were trypsinized and adjusted to 1 mL. One-half milliliter of the cell suspension was diluted into 10 mL of an isotonic solution and counted using a Coulter Counter. The log of the number of cells/well ± standard deviation is plotted as a function of time in Figure 6D. Selective induction of each protein was confirmed as in Figure 6C by Western blots of cells harvested at the 96 h time point (data not shown).
TAP purification and identification of xPMR60°-interacting proteins
HeLa S3 (Tet-Off) cells (Clontech) were transfected with pTracer-myc-xPMR60°-TAP and stable transfectants were selected by growth in 100 μg/mL Zeocin. xPMR60° and its associated proteins were purified as described by Puig et al. (2001). Cells were suspended in lysis buffer (10 mM HEPES at pH 7.6, 3 mM MgCl2, 10 mM KCl, 5% glycerol, 0.5% NP-40, 25 μl/mL protease inhibitor mixture [Sigma], and 1 mM PMSF) and broken using 30 strokes of a Dounce homogenizer (A pestle). Nuclei were removed by centrifugation at 2000g for 15 min, and the remaining supernatant was centrifuged at 100,000g for 1 h at 4°C. The resultant supernatants were incubated with IgG Sepharose 6 Fast Flow (Amersham Bioscience) (400 μl) at 4°C, the beads were washed three times with IPP150 (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, and 0.1% NP-40), once with TEV buffer (IPP150 containing 0.5 mM EDTA and 1 mM dithiothreitol), and incubated in 1 mL of TEV buffer containing 100 U of TEV protease (Invitrogen) at 4°C for 2 h. Supernatants were diluted to 4 mL with calmodulin binding buffer (10 mM β-mercaptoethanol, 10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, 0.1% NP40), 300 μL of Calmodulin Affinity Resin (Stratagene) was added, followed by a 2-h incubation at 4°C. After washing to remove unbound material the remaining proteins were eluted five times (300 μL each) with calmodulin elution buffer (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.1% NP40, 1 mM magnesium acetate, 1 mM imidazole, 2 mM EDTA, and 10 mM β-mercaptoethanol). The eluted proteins were concentrated by trichloroacetic acid (TCA) precipitation and separated on a 4%–20% SDS-polyacrylamide gel. This was stained with Coomassie blue, and the individual bands were excised and identified by MALDI and LC-MS/MS at the W.M. Keck Biotechnology Resource Facility at Yale University.
Preparation of cell extracts
Cytoplasmic extracts were prepared as described previously (Yang and Schoenberg 2004). Briefly, cells were washed twice with ice-cold PBS, then suspended in cell lysis buffer (10 mM HEPES-KOH at pH 7.5, 10 mM KCl, 5 mM MgCl2, 50 mM NaF, 0.5% Nonidet P-40, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 25 μL/mL protease inhibitor mixture [Sigma], and 10 μL/mL phosphatase inhibitors [Sigma]). After incubation for 15 min on ice, the cells were homogenized with 30 strokes of a Dounce homogenizer (A pestle), and the homogenate was centrifuged for 15 min at 2000g.
Glycerol gradient analysis of xPMR60 complexes
Linear gradients of 10%–40% (v/v) glycerol were prepared in buffer containing 10 mM HEPES-KOH at pH 7.5 and 2 mM DTT. Postmitochondrial extracts were adjusted to 50 mM EDTA before applying to the gradients and sedimentation for 20 h at 83,000g in a Sorvall TH641 rotor. Molecular size markers containing a mixture of thyroglobulin (MW = 669,000), ferritin (MW = 440,000), catalase (MW = 232,000), lactate dehydrogenase (MW = 140,000), and bovine serum abumin (MW = 67,000) were fractionated on a parallel gradient. Individual fractions were analyzed directly by Western blotting or after recovery of xPMR60°-containing complexes on IgG-Sepharose. Proteins bound to IgG-Sepharose were recovered by cleaving the TAP tag with Tev protease and analyzed by Western blotting as described in Yang and Schoenberg (2004).
Confocal microscopy
For the experiment in Figure 3A, 5 × 104 Cos-1 cells were seeded onto glass coverslips (18-mm diameter, Fisher) that were previously treated with 10 μg/mL fibronectin at 37°C for 20 min and washed once with PBS. Coverslips in 12-well plates were transfected with 480 ng of EGFP-FP4-Mito or EGFP-AP4-Mito and 80 ng of myc-xPMR60° plasmid DNA. Cells were fixed in 4% paraformaldehyde in PBS for 1 h and permeabilized in 0.2% Triton X-100 in PBS for 15 min at 25°C. After blocking for 1 h in 5% normal serum in PBS, cells were incubated overnight at 4°C with myc monoclonal antibody, washed three times with PBS, and incubated with rabbit anti-mouse IgG antibody Alexa Fluor 594 (Molecular Probes) for 1 h at 25°C. Finally, cells were washed three times with PBS and incubated with 1 μg/mL of DAPI (4, 6-diamidine-2-phenylindole dihydrochloride, Roche) for 15 min at 25°C. Cells were washed with PBS and mounted with ProLong (Molecular Probes), following the manufacturer's protocol, and examined using a Zeiss 510 confocal microscope.
To study the colocalization of xPMR60 with VASP, 8.3 × 104 U2OS cells stably transfected with myc-xPMR60 under a tetracycline-regulated promoter were plated onto polylysine-treated glass coverslips. Twenty-four hours later, half of the cells were treated with 1 μg/mL doxycycline to induce expression of xPMR, and 16 h later the cell monolayer was scratched with a 1000-mL pipet tip. The cells were washed 3× with PBS to remove dislodged cells, and the growth medium was replaced. Cells were fixed at 0, 4, 8, 12, and 24 h after scratching by treating with 2% paraformaldehyde, 0.05% glutaraldehyde for 10 min at 25°C, then 0.05% glutaraldehyde for 10 min at 25°C. Cells were blocked for 1 h in 5% normal goat serum in PBS at 25°C and then incubated overnight at 4°C with a mouse anti-myc monoclonal antibody (Santa Cruz Biotechnology) and a rabbit anti-VASP monoclonal antibody (Cell Signaling Technologies). Cells were washed 3 times with PBS and incubated for 3 h with goat anti-mouse IgG Alexa Fluor 555, goat anti-rabbit IgG Alexa Fluor 647, and phalloidin Alexa Fluor 488 (all from Molecular Probes) at 25°C. Cells were washed 3 times in PBS, mounted with ProLong Gold (Molecular Probes), and examined using a Zeiss 510 confocal microscope.
Immunoprecipitation and Western blotting
For immunoprecipitation with myc antibody, cell lysates were incubated with monoclonal antibody-coupled beads on a rocking platform for 3 h at 4°C. For immunoprecipitation with c-Src monoclonal antibody or anti-GFP, cell lysates were incubated with antibody for 3 h at 4°C followed by 20 μL of protein A agarose (Santa Cruz Biotechnology) and incubation for 1 h on a rocking platform at 4°C. The beads were washed four times with IPP150 buffer and suspended in SDS-PAGE sample buffer. For Western blot analysis, the immunoprecipitates were separated on a 10% SDS-polyacrylamide gel and electroblotted onto Immobilon-P membrane (Millipore). The membrane was blocked for 1 h at room temperature in 5% nonfat dry milk in TBST buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 0.1% Tween 20), then incubated with the primary antibody overnight at 4°C, washed, and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. Blots were developed with SuperSignal west pico chemiluminescent substrate (Pierce).
RNase protection assay
U2OS were transiently transfected with plasmids expressing albumin and luciferase mRNA plus plasmid expressing myc-GFP or a plasmid expressing catalytically active myc-PMR60, and pcDNA3.1-FP4-mito or pcDNA3.1-AP4-mito (Fig. 3C) or with pcDNA3.1(-)FL-Mena (Fig. 3D,E). Total RNA was isolated 24 h later using TRIzol reagent (Invitrogen). The antisense albumin riboprobe was prepared with the MAXIscript In Vitro Transcription kit (Ambion) from the T7 promoter of a pcRII-Topo plasmid containing exons 14 and 15 of Xenopus albumin cDNA. The antisense luciferase riboprobe was transcribed from the T3 promoter from a pBluescript(SK) plasmid containing the first 153 nucleotides of firefly luciferase cDNA. Ribonuclease protection assay was done as described previously (Yang and Schoenberg 2004) with 5 μg of total RNA hybridized to 600 pg of each riboprobe using the Ribonuclease Protection Assay III kit (Ambion). Protected probe was separated on a denaturing 6% polyacrylamide-urea gel and quantified by PhosphorImager (GE Healthcare) analysis.
ACKNOWLEDGMENTS
We thank Frank Gertler and Nancy Kedersha for their helpful advice and suggestions. We also thank Frank Gertler and Alpha Yap for providing antibodies and expression plasmids. This work was supported by PHS grant R01 GM038277 (to D.R.S.) from the National Institute of General Medical Science. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health. M.S. is supported by a postdoctoral fellowship from the American Heart Association, Great Rivers Division, and support for core facilities was provided by PHS grant P30 CA16058 from the National Cancer Institute to The Ohio State University Comprehensive Cancer Center.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1206209.
REFERENCES
- Baroni T.E., Chittur S.V., George A.D., Tenenbaum S.A. Advances in RIP-Chip analysis: RNA-binding protein immunoprecipitation-microarray profiling. Methods Mol. Biol. 2008;419:93–108. doi: 10.1007/978-1-59745-033-1_6. [DOI] [PubMed] [Google Scholar]
- Bear J.E., Loureiro J.J., Libova I., Fassler R., Wehland J., Gertler F.B. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Binder R., Horowitz J.A., Basilion J.P., Koeller D.M., Klausner R.D., Harford J.B. Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3′ UTR and does not involve poly(A) tail shortening. EMBO J. 1994;13:1969–1980. doi: 10.1002/j.1460-2075.1994.tb06466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeda B., Briggs D.C., Higgins T., Garvalov B.K., Fadden A.J., McDonald N.Q., Way M. Tes, a specific Mena interacting partner, breaks the rules for EVH1 binding. Mol. Cell. 2007;28:1071–1082. doi: 10.1016/j.molcel.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Chernokalskaya E., DuBell A.N., Cunningham K.S., Hanson M.N., Dompenciel R.E., Schoenberg D.R. A polysomal ribonuclease involved in the destabilization of albumin mRNA is a novel member of the peroxidase gene family. RNA. 1998;4:1537–1548. doi: 10.1017/s1355838298980451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompenciel R.E., Garnepudi V.R., Schoenberg D.R. Purification and characterization of an estrogen-regulated Xenopus liver polysomal nuclease involved in the selective destabilization of albumin mRNA. J. Biol. Chem. 1995;270:6108–6118. doi: 10.1074/jbc.270.11.6108. [DOI] [PubMed] [Google Scholar]
- Eulalio A., Behm-Ansmant I., Schweizer D., Izaurralde E. P-body formation is a consequence, not the cause of RNA-mediated gene silencing. Mol. Cell. Biol. 2007;27:3970–3981. doi: 10.1128/MCB.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina K.L., Huttelmaier S., Musunuru K., Darnell R., Singer R.H. Two ZBP1 KH domains facilitate β-actin mRNA localization, granule formation, and cytoskeletal attachment. J. Cell Biol. 2003;160:77–87. doi: 10.1083/jcb.200206003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks T.M., Lykke-Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes & Dev. 2007;21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi I.E., Parker F., Chebli K., Maurier F., Labourier E., Barlat I., Capony J.P., Tocque B., Tazi J. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: A potential link between signal transduction and RNA stability. Mol. Cell. Biol. 1998;18:3956–3965. doi: 10.1128/mcb.18.7.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gertler F.B., Comer A.R., Juang J.L., Ahern S.M., Clark M.J., Liebl E.C., Hoffmann F.M. enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes & Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Howe A.K., Hogan B.P., Juliano R.L. Regulation of vasodilator-stimulated phosphoprotein phosphorylation and interaction with Abl by protein kinase A and cell adhesion. J. Biol. Chem. 2002;277:38121–38126. doi: 10.1074/jbc.M205379200. [DOI] [PubMed] [Google Scholar]
- Huttelmaier S., Zenklusen D., Lederer M., Dictenberg J., Lorenz M., Meng X.H., Bassell G.J., Condeelis J., Singer R.H. Spatial regulation of β-actin translation by Src-dependent phosphorylation of ZBP1. Nature. 2005;438:512–515. doi: 10.1038/nature04115. [DOI] [PubMed] [Google Scholar]
- Krause M., Dent E.W., Bear J.E., Loureiro J.J., Gertler F.B. Ena/VASP proteins: Regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski A.V., Gertler F.B., Loureiro J.J. Function and regulation of Ena/VASP proteins. Trends Cell Biol. 2003;13:386–392. doi: 10.1016/s0962-8924(03)00130-2. [DOI] [PubMed] [Google Scholar]
- Le Clainche C., Schlaepfer D., Ferrari A., Klingauf M., Grohmanova K., Veligodskiy A., Didry D., Le D., Egile C., Carlier M.F., et al. IQGAP1 stimulates actin assembly through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 2007;282:426–435. doi: 10.1074/jbc.M607711200. [DOI] [PubMed] [Google Scholar]
- Li Z., Sacks D.B. Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 2003;278:4347–4352. doi: 10.1074/jbc.M208579200. [DOI] [PubMed] [Google Scholar]
- Mullen T.E., Marzluff W.F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes & Dev. 2008;22:50–65. doi: 10.1101/gad.1622708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray E.L., Schoenberg D.R. A + U-rich instability elements differentially activate 5′-3′ and 3′-5′ mRNA decay. Mol. Cell. Biol. 2007;27:2791–2799. doi: 10.1128/MCB.01445-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R., Sheth U. P bodies and the control of mRNA translation and degradation. Mol. Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Pastori R.L., Moskaitis J.E., Schoenberg D.R. Estrogen-induced ribonuclease activity in Xenopus liver. Biochemistry. 1991;30:10490–10498. doi: 10.1021/bi00107a018. [DOI] [PubMed] [Google Scholar]
- Peng Y., Schoenberg D.R. c-Src activates endonuclease-mediated mRNA decay. Mol. Cell. 2007;25:779–787. doi: 10.1016/j.molcel.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Liu X., Schoenberg D.R. The 90-kDa heat shock protein stabilizes the polysomal ribonuclease 1 mRNA endonuclease to degradation by the 26S proteasome. Mol. Biol. Cell. 2008;19:546–552. doi: 10.1091/mbc.E07-08-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Scott J.A., Shewan A.M., den Elzen N.R., Loureiro J.J., Gertler F.B., Yap A. Ena/VASP proteins can regulate distinct modes of actin organization at cadherin-adhesive contacts. Mol. Biol. Cell. 2006;17:1085–1095. doi: 10.1091/mbc.E05-07-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk E.L., Sussenbach J.S., Holthuizen P.E. Identification of RNA sequences and structures involved in site-specific cleavage of IGF-II mRNAs. RNA. 1998;4:1632–1635. doi: 10.1017/s1355838298981316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Schoenberg D.R. Endonuclease-mediated mRNA decay involves the selective targeting of PMR1 to polyribosome-bound substrate mRNA. Mol. Cell. 2004;14:435–445. doi: 10.1016/j.molcel.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Yang F., Peng Y., Schoenberg D.R. Endonuclease-mediated mRNA decay requires tyrosine phosphorylation of polysomal ribonuclease 1 (PMR1) for the targeting and degradation of polyribosome-bound substrate mRNA. J. Biol. Chem. 2004;279:48993–49002. doi: 10.1074/jbc.M409776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Peng Y., Murray E.L., Otsuka Y., Kedersha N., Schoenberg D.R. Polysome-bound endonuclease PMR1 Is targeted to stress granules via stress-specific binding to TIA-1. Mol. Cell. Biol. 2006;26:8803–8813. doi: 10.1128/MCB.00090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]