

Abstract
During hematopoiesis, myeloid cell leukemia-1 (MCL-1) mediates the survival of bone marrow progenitors and lymphocytes. However, its requirement during myeloid cell differentiation, development, and effector function is less clear. Lineage-specific deletion of MCL-1 in myeloid precursors results in neutropenia due to death during differentiation. The loss of mature neutrophils induced by Mcl-1 deletion was not rescued by genetic deletion of proapoptotic Bim and Puma or by exogenous cytokine treatment. However, blockade of intrinsic apoptosis by lineage-specific deletion of both multidomain proapoptotics Bax and Bak was capable of rescuing the neutropenia associated with Mcl-1 deletion. In the monocytic lineage, despite efficient Mcl-1 deletion, monocytes and macrophages undergo normal development. During the phagocytosis of extracellular bacteria, macrophages concomitantly increase the expression of both MCL-1 and BIM. However, Mcl-1–deficient macrophages exhibit increased sensitivity to death during bacterial phagocytosis that can be abolished by codeletion of Bim. These data suggest that MCL-1 may be necessary to antagonize BIM during macrophage effector responses. Thus, MCL-1 plays selective roles in myeloid development, being required for neutrophil development and setting the threshold for apoptosis during a macrophage effector response.
Introduction
The BCL-2 family is composed of critical cell death regulators that play roles in hematopoietic regulation.1 Proapoptotic BH3-only members respond to death signals and activate the proapoptotic effectors BAX and BAK, which represent an obligate gateway to intrinsic cell death.2 Antiapoptotic members antagonize this process by binding and sequestering BH3-only family members and preventing BAX and BAK activation.3,4 Mcl-1, originally cloned as survival gene induced in a myeloid leukemia cell line, is an antiapoptotic family member whose expression can inhibit cell death in response to a variety of death stimuli.5 Conditional deletion of Mcl-1 has revealed requirements at several stages of hematopoietic and lymphocyte development and its expression is regulated in part by growth factor signaling.6,7 Thus, MCL-1 represents an important antiapoptotic molecule for regulating the survival of hematopoietic populations.
Maintaining regulation of myelopoiesis is essential since myeloid cells are among the shortest lived blood cell lineages. For example, neutrophils and monocytes have half-lives of 8 to 11 hours and 24 to 40 hours, respectively, in circulation leading to the need to replenish with newly produced bone marrow (BM)–derived cells.8 Several studies have implicated various BCL-2 family members in regulating the apoptotic potential of myeloid cells. The antiapoptotic molecule A1-a promotes the survival of neutrophils, as granulocytes lacking A1-a undergo more spontaneous apoptosis than control cells in vitro.9 Furthermore, inhibition of MCL-1 expression by anti-sense approaches led to the spontaneous apoptosis of cultured human neutrophils and macrophages implying an essential role for MCL-1 in promoting their survival.10–12 Furthermore, MCL-1 expression can be induced in a variety of myeloid cell lines in response to growth factor signaling and results in the promotion of survival in these cells.13
Several proapoptotic BH3-only family members have been implicated in maintaining homeostasis during myelopoiesis. Deficiency in Bid has been reported to violate myeloid homeostasis, eventually leading to a fatal malignancy.14 However, Bid-deficient mice do not exhibit overt alterations in myeloid populations until late in life, and the phenotype may be background and/or environment dependent, suggesting that BID may play an indirect role in maintaining myeloid homeostasis.15 Genetic ablation of Puma renders a variety of cell lineages resistant to cellular stress and growth factor withdrawal, but Puma-deficient mice do not exhibit any overt abnormalities in myeloid homeostasis.16,17 In contrast, loss of Bim has been reported to double the number of circulating neutrophils and monocytes and renders them resistant to a variety of death stimuli.18,19 Therefore, evidence indicates that BIM is a critical proapoptotic molecule that modulates the sensitivity of myeloid cells to cell death.
Using a conditional allele of Mcl-1, we have tested the role for MCL-1 in myeloid development and function. These results extend and redefine those of He and colleagues that reported the requirement for MCL-1 in mediating neutrophil but not macrophage survival.20 We demonstrate that myeloid-specific deletion of Mcl-1 blocks neutrophil maturation and induces the accumulation of immature myeloid precursor populations in the BM. Strikingly, neutropenia resulting from Mcl-1 deletion was neither corrected by treatment in vivo or in vitro with exogenous colony stimulation factors nor by combining with lineage-specific deletion of both proapoptotic Bim and Puma, suggesting that neither proapoptotic is essential for mediating cell death in the granulocyte lineage induced by Mcl-1 deletion. However, the death mediated by loss of Mcl-1 in the myeloid lineage is dependent upon the action of the multidomain proapoptotic family members Bax and Bak, as lineage-specific deletion of both was capable of rescuing neutropenia associated with Mcl-1 loss. Despite the normal expression of MCL-1 in wild-type monocytes and macrophages, loss of Mcl-1 does not affect the development of this lineage nor blocks their ability to be recruited to sites of inflammation. However, Mcl-1–deficient macrophages exhibit increased sensitivity to cell death as a result of phagocytosis of bacteria. This hypersensitivity can be abrogated by codeletion of proapoptotic Bim, suggesting that MCL-1 may play a key role in antagonizing BIM during macrophage effector function. Thus, while MCL-1 may be dispensable for macrophage development, it plays a critical role for setting the apoptotic threshold of macrophages carrying out effector function.
Methods
Mice
Mcl-1 conditional and Bax conditional Bak−/− mice (both made in RW4 embryonic stem [ES] cells and maintained on 129/B6 mixed backgrounds) have been described previously.7,21 Mice bearing the conditional allele of Bim (made in RW4 ES cells and maintained on a 129/B6 mixed background) have been previously used to obtain germline Bim-deficient mice.21 Puma-deficient mice (made in E14 ES cells and maintained on a 129/B6 mixed background) have been described previously.16 LysM-Cre mice, obtained from Dr Irmgard Foerster (University of Duesseldorf, Germany) are described previously and have been backcrossed to C57BL/6 more than 15 generations.22 Conditional and LysM-Cre mice were bred to generate mice (129/B6 mixed strain) in which Mcl-1, Bim, Puma, and Bax and Bak could be deleted specifically in the myeloid lineage. All mice used in the present studies were 6 to 12 weeks old and maintained in specific pathogen–free conditions; littermate controls were used as negative controls. All mouse experiments were performed with the approval of the institutional animal care and use committee at St Jude Children's Research Hospital.
Antibody staining and flow cytometry, fluorescence-activated cell sorting, and cytospins
For cell-surface staining, cells were resuspended in 2% fetal calf serum (FCS) in phosphate-buffered saline (PBS), labeled with anti–Gr-1 (BD Biosciences, Franklin Lakes, NJ) and anti–Mac-1 (CD11b; BD Biosciences), and analyzed by flow cytometry (FACSCalibur; BD Biosciences). Cytospins were performed using SuperfrostPlus slides (Erie Scientific, Portsmouth, NH) with a Cyto-Tek (Miles Scientific, Elkhart, IN). Cells were stained with DipQuick staining kit (Jorgensen Laboratories, Loveland, CO). Micrographs were acquired using Nikon ACT software (Nikon, Melville, NY) and a Nikon Eclipse E600 microscope equipped with a Nikon DXM1200 camera and Nikon PLAN APO objectives.
Induction of peritonitis
For neutrophils, 1 mL of sterile 9% bovine milk casein (Sigma-Aldrich, St Louis, MO) in PBS containing 0.9 mM CaCl2 an~d 0.5 mM MgCl2 was intraperitoneally injected. After 16 hours, an additional dose was injected, and 3 hours later, peritoneal cells were collected in PBS plus 0.05% EDTA. For macrophages, 2 mL of 3% Brewer thioglycollate (Sigma-Aldrich) was injected intraperitoneally and incubated for 6 days. Following incubation, the mice were killed and peritoneal cells were collected.
Cytokine treatment
Emergency granulopoiesis was induced as previously described.23 Recombinant human granulocyte colony-stimulating factor (G-CSF; Amgen, Thousand Oaks, CA) was diluted in sterile PBS containing 1% low-endotoxin bovine serum albumin (BSA; Sigma-Aldrich). G-CSF was administered by subcutaneous injection using 250 μg/kg for 5 days. Control mice received an equivalent volume of 1% BSA in PBS in parallel treatments. For G-CSF, macrophage (M)–CSF, and GM-CSF differentiation experiments, BM was cultured with or without 10 ng/mL cytokines (Peprotech, Rocky Hill, NJ). To determine the phenotype of the cultured cells, they were stained for flow cytometry and assessment of cell death using annexin-V–PE, Mac-1 (CD11b)–FITC, and Gr1-APC as well as histologic analyses.
Peripheral blood and tissue isolation and automated blood counts
Peripheral blood was collected in EDTA-coated tubes following retro-orbital puncture. Blood counts were determined with an automated hematology analyzer (Forcyte Hematology Analyzer; Oxford Science, Oxford, CT). BM cells were harvested in Iscove-modified Dulbecco medium (IMDM) containing 10% FCS by flushing with a 27-gauge needle. Spleens were harvested and homogenized in RPMI 1640 containing 10% FCS. BM and spleen cell suspensions were subjected to red blood lysis, washed, and stained as indicated.
Generation of BMDMs
BM was cultured on tissue culture–treated cells in IMDM containing 10% FCS and 20% L929 cell–conditioned media. After 24 hours, nonadherent cells were replated on non–tissue culture–treated plastic dish for 6 days. After 3 days, media is replaced along with any nonadherent cells back to the same dish. On day 7, adherent cells were harvested by Accutase (Innovative Cell Technologies, San Diego, CA) and replated in 6-well non–tissue culture–treated dishes.
Macrophage phagocytosis experiments
Bone marrow–derived macrophages (BMDMs) were plated in 6-well non–tissue-culture–treated dishes at 750 000 cells per well. The concentration of Escherichia coli bacteria (DH5α strain), grown in log phase, was measured by OD600 and by serial dilution plating. E coli, washed in IMDM (no additives), were added at indicated ratios for 2 hours, after which the media is removed, cells were washed with PBS and replaced with IMDM media containing 200 μg/mL gentamicin (Invitrogen, Carlsbad, CA). After 4 hours, the cells were harvested with Accutase, washed, and stained with annexin-V–FITC and propidium iodide.
Immunoblot analysis
Cells were lysed in RIPA buffer with complete protease inhibitors (Sigma-Aldrich) and protein concentrations determined by BCA assay (Thermo Fisher Scientific, Rockford, IL). Proteins were separated by SDS-PAGE, transferred to PVDF membrane (Millipore, Billerica, MA), and immunoblotted with the following antibodies: anti–MCL-1 rabbit polyclonal (Rockland Immunochemical, Gilbertsville, PA), anti-BIM rabbit polyclonal (Calbiochem, San Diego, CA), anti-BAK rabbit polyclonal (Millipore), anti-BAX rabbit polyclonal 651 (gift from the late Dr Stanley Korsmeyer, Harvard Medical School, Boston, MA), anti-actin mouse monoclonal (Millipore), and anti–protein disulfide isomerase (PDI) mouse monoclonal (BD Biosciences). Secondary antibodies were anti-rabbit or anti-mouse horseradish peroxidase–conjugated (Jackson Immunochemical, Bar Harbor, ME) and developed using Western Lightning (PerkinElmer, Waltham, MA).
Cytokine assessment
A multiplex biometric immunoassay kit (Bio-Rad Laboratories, Hercules, CA) was used for cytokine measurement. Mouse sera (50 μL) was incubated with antibody-coupled beads, washed, incubated with biotinylated detection antibody, and detected with streptavidin-phycoerythrin. The serum cytokine concentration was based on a standard curve. Cytokine levels were determined using a multiplex array reader (Luminex 100 xMap Technologies; Bio-Rad).
Results
Selective neutropenia induced by myeloid-specific deletion of Mcl-1
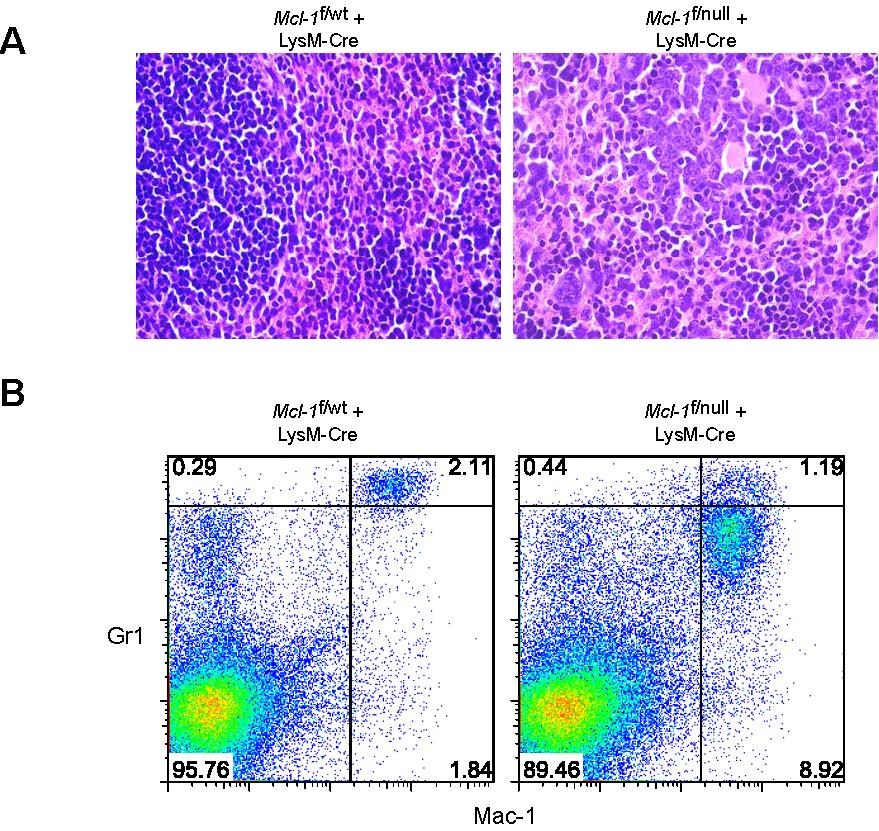
Mcl-1 conditional mice were bred to mice expressing Cre recombinase under the control of the lysozyme M promoter (LysM-Cre) to induce deletion in the myeloid lineage (∼ 83%-98% in mature macrophages and near 100% in the granulocyte lineage).7,22 In Mcl-1–deleted mice, the peripheral blood neutrophils (Mac-1+Gr-1+) were diminished when compared with littermate control mice (Figure 1A). Blood smears from LysM-Cre–deleted animals also exhibited an absence of mature, segmented neutrophils (Figure 1B). Manual differential counts of blood smears demonstrated that circulating neutrophils were virtually absent in Mcl-1–deleted mice and that monocyte numbers were decreased (Table 1). These data indicate that myeloid-specific deletion of Mcl-1 results in the loss of neutrophils, but the monocyte lineage is at least partially spared.
Figure 1.

Myeloid-specific deletion of Mcl-1 results in neutropenia, but spares macrophages. (A) Flow cytometric analysis of peripheral blood from littermate control (Mcl-1f/wt, Mcl-1f/null, Mcl-1f/wt plus LysM-Cre) and myeloid lineage specific Mcl-1–deleted mice (Mcl-1f/f plus LysM-Cre) stained for Mac-1 (CD11b) and Gr1. Gate (Mac-1+Gr1high) indicates the percentage of mature segmented neutrophils in white blood cells. (B) Representative Wright-Giemsa–stained blood smears from littermate control and Mcl-1–deleted mice (20× objective). Insets depict expanded 100× objective view. (C,D) Stained cytospins of peritoneal exudate from littermate control (Mcl-1f/wt) and Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre) (C) harvested 16 hours after intraperitoneal injection of casein to induce neutrophil recruitment (images taken with 20× objective) or (D) harvested 5 days after intraperitoneal injection of thioglycollate to induce macrophage recruitment (images taken with 40× objective). (E) MCL-1 protein levels as determined by immunoblot analysis of BMDMs from control mice (Mcl-1f/wt and Mcl-1f/wt plus LysM-Cre) or from Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre). Blots were probed for actin expression to serve as a loading control.
Table 1.
Differential blood smear counts from myeloid-deleted mice
| Genotype | Peripheral WBCs, % | ||||
|---|---|---|---|---|---|
| Lymphocytes | Neutrophils | Bands | Monocytes | Eosinophils | |
| Littemate control | 78 ± 11 | 14 ± 8 | 0 ± 0 | 7 ± 3 | 1 ± 1 |
| Mcl-1f/null plus LysM-Cre | 96 ± 2 | 0 ± 0* | 1 ± 1 | 2 ± 2 | 1 ± 1 |
| Bimf/f plus LysM-Cre | 83 ± 4 | 14 ± 5 | 0 ± 0 | 2 ± 1 | 1 ± 1 |
| Mcl-1f/nullBimf/f plus LysM-Cre | 97 ± 1 | 0 ± 0* | 0 ± 0 | 2 ± 1 | 1 ± 2 |
| Pumanull | 84 ± 5 | 9 ± 2 | 1 ± 0 | 6 ± 2 | 2 ± 1 |
| Mcl-1f/null Pumanull plus LysM-Cre | 90 ± 5 | 0 ± 1* | 0 ± 0 | 6 ± 6 | 3 ± 2 |
| Mcl-1f/null Bimf/fPumanull plus LysM-Cre | 95 ± 3 | 2 ± 1* | 0 ± 0 | 3 ± 2 | 0 ± 0 |
| Baxf/fBaknull plus LysM-Cre | 75 ± 10 | 14 ± 7 | 1 ± 2 | 10 ± 5 | 0 ± 1 |
| Mcl-1f/f Baxf/f Baknull plus LysM-Cre | 91 ± 4 | 7 ± 3 | 0 ± 1 | 1 ± 1 | 0 ± 0 |
Compilation of average manual differential blood cell counts for cohorts of age-matched mice of indicated genotypes. Each data set is composed of at least 3 mice per group. Error represents SEM.
P < .01 by 2-tailed t test when compared with littermate control population.
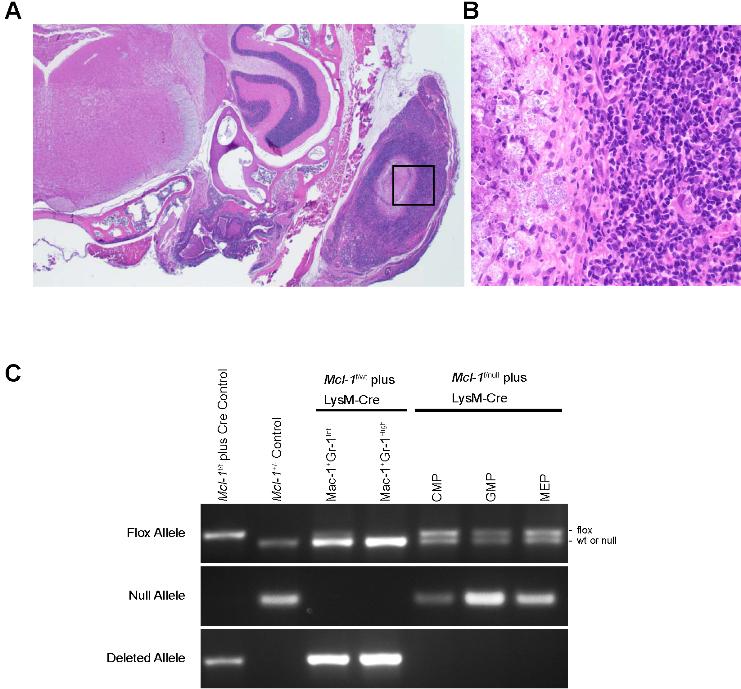
Despite being housed in a barrier facility in a sterilized microisolator cages, an increased incidence of infections were observed in mice with myeloid-specific Mcl-1 deletion. Local expansion resulted in severe dermatitis, rhinitis, adenitis, cellulitis, and otitis with extensive necrosis of subcutaneous tissue, resorption of underlying bone, and bacterial colonization (Figure S1A,B, available on the Blood website; see the Supplemental Materials link at the top of the online article). The inflammatory response was composed of lymphocytes, plasma cells, macrophages, and eosinophils, but neutrophils were conspicuously absent from the inflammatory milieu.
To recruit inflammatory cells to the peritoneal space, casein (neutrophils) or thioglycollate (macrophages) were injected and the peritoneal exudate was collected. Cells collected 16 hours after casein stimulation were composed primarily of activated neutrophils, but in Mcl-1–deleted mice there was a profound deficiency of neutrophils (Figure 1C). In contrast, peritoneal cells collected 5 days after thioglycollate challenge of both littermate control and Mcl-1–deleted mice exhibited robust macrophage recruitment (Figure 1D). There were no overt differences between littermate control and Mcl-1–deleted mice in the recruitment of activated macrophages to the peritoneum (data not shown). To confirm that the macrophage lineage was efficiently deleting Mcl-1, BMDMs were analyzed for MCL-1 expression. Whereas wild-type macrophages express MCL-1, LysM-Cre–mediated deletion results in viable, Mcl-1–deficient macrophages (Figure 1E). These data indicate that myeloid deletion of Mcl-1 results in the loss of neutrophils, but that terminal differentiation of the monocyte/macrophage lineage does not require MCL-1.
Block in granulocyte maturation in LysM-Cre–deleted mice
Myeloid development occurs in the BM and is highly regulated to constantly replace damaged or eliminated myeloid cells. Strikingly, the total cellularity of Mcl-1–deleted mice BM was reduced when compared with littermate controls (Figure 2A). Mice in which Mcl-1 was deleted by LysM-Cre exhibited a dramatic increase in the size of BM cells as measure by increased forward scatter by flow cytometry (data not shown). When the BM was analyzed, myeloid cells expressing Mac-1+Gr1High were detected in littermate control mice (Figure 2B). However, in the Mcl-1–deleted BM, this population was lost and replaced by an expanded population of immature myeloid precursors as evidenced by the Mac-1+Gr1Intermediate cells (Figure 2B).
Figure 2.

Deletion of Mcl-1 blocks the development of mature neutrophils and induces the accumulation of myeloid precursors. (A) Graph of the average number of BM cells from littermate control of Mcl-1f/null plus LysM-Cre mice. ■ represents the total BM number from hind limbs; red bars, the total number of mature neutrophils (Mac-1+ Gr1High). Graph represents average numbers of more than 5 mice analyzed, and error bars denote SEM. *P < .01 by 2-tailed t test when compared with littermate control population. (B) Representative flow cytometric analysis of BM from littermate control and Mcl-1–deleted mice stained for Mac-1 and Gr1. Top right quadrant contains mature neutrophils; bottom right quadrant, immature myeloid precursors. Numbers indicate the percentage of cells from the total BM. (C) Stained cytospins of BM aspirates from littermate control (Mcl-1f/wt) and Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre). Images taken with a 40× objective. (D) Flow cytometric analysis of BM from littermate control and Mcl-1–deleted mice stained for Gr1 and annexin-V to detect apoptotic cells. Top right quadrant contains myeloid lineage cells that are undergoing cell death. Numbers indicate the percentage of total BM cells.
Myeloid lineages arise from the common myeloid progenitor (CMP) and granulocyte monocyte progenitors (GMPs) sequentially.24 It has been reported that LysM-Cre mice can exhibit some deletion in earlier hematopoietic progenitor populations.25 However, in Mcl-1f/null plus LysM-Cre animals, deletion of Mcl-1 in sorted CMP, GMP, and megakaryocyte-erythroid progenitor (MEP) populations was undetectable indicating that the failure to develop mature neutrophils is not due to deletion in early myeloid progenitors, which are MCL-1–dependent (Figure S1C).6
In littermate control mice, the predominant cells present in the BM are band neutrophils with thin ring-shaped nuclei and faint cytoplasm and mature, segmented neutrophils (Figure 2C). However, in LysM-Cre–deleted mice, mature, segmented neutrophils were virtually absent; instead, there was an expansion in myelocyte precursors and metamyelocytes (Table 2; Figure 2C). To address whether the increase in myeloid precursors was due to the death of differentiating cells, BM was stained for Gr1 and then assayed for apoptosis by staining with Annexin-V. The immature myeloid precursors (Gr1Intermediate) present in the BM of Mcl-1–deleted mice display an increased rate of cell death as evidenced by the increase in annexin-V+Gr-1intermediate cells (Figure 2D). These data demonstrate that deletion of Mcl-1 by LysM-Cre results in blockade of normal myeloid differentiation due to the loss of myeloid cells by apoptosis and results in a subsequent expansion of immature precursors.
Table 2.
Myeloid bone marrow differentials from LysM-Cre–deleted mice
| Genotype | BM myeloid cells, % |
|||
|---|---|---|---|---|
| Myelocyte precursor | Metamyelocyte | Band cell | Segmented neutrophil | |
| Littemate control | 17 ± 0 | 13 ± 1 | 22 ± 5 | 48 ± 4 |
| Mcl-1f/null plus LysM-Cre | 59 ± 4 | 28 ± 4 | 11 ± 4 | 1 ± 1* |
| Bimf/f plus LysM-Cre | 13 ± 2 | 7 ± 2 | 15 ± 4 | 65 ± 1 |
| Mcl-1f/nullBimf/f plus LysM-Cre | 63 ± 7 | 26 ± 8 | 10 ± 2 | 0 ± 0* |
| Pumanull | 25 ± 6 | 16 ± 2 | 11 ± 3 | 48 ± 7 |
| Mcl-1f/nullPumanull plus LysM-Cre | 66 ± 5 | 26 ± 8 | 9 ± 2 | 1 ± 1* |
| Mcl-1f/null Bimf/f Pumanull plus LysM-Cre | 74 ± 1 | 22 ± 2 | 5 ± 2 | 1 ± 1* |
| Baxf/fBaknull plus LysM-Cre | 20 ± 6 | 11 ± 2 | 15 ± 2 | 54 ± 7 |
| Mcl-1f/f Baxf/f Baknull plus LysM-Cre | 15 ± 2 | 9 ± 1 | 14 ± 2 | 63 ± 4 |
Compilation of average manual differential myeloid BM counts for cohorts of age-matched mice of indicated genotypes. Each data set is composed of at least 3 mice per group. Error represents SEM.
P < .01 by 2-tailed t test when compared with littermate control population.
The expansion of myeloid precursor populations was also associated with splenomegaly due to increased extramedullary hematopoiesis. Total splenic cell number was increased (average, 14.2 ± 1.8 × 107; n = 10) from that of littermate control mice (average, 7.7 ± 0.6 × 107; n = 12). The splenomegaly is accompanied by the observation of increased myeloid forms in the splenic red pulp in Mcl-1–deleted mice, but is absent in littermate controls (Figure S2A). Flow cytometric analysis of the spleen of Mcl-1–deleted mice demonstrated an increase in the Mac-1+Gr-1Intermediate population indicative of more immature cells similar to those present in the BM (Figure S2B). Therefore, the failure to generate mature neutrophils seems to be due to the death of myeloid precursors in the BM at the stage of transition to mature, segmented neutrophils.
MCL-1 and cytokine-mediated granulopoiesis
Colony-stimulating factors promote the production and survival of myeloid lineages. For example, G-CSF is essential for regulating the production of neutrophils from the BM by stimulating their mobilization into the circulation.26,27 As a result, G-CSF treatment is used clinically to treat neutropenia resulting from either myelosuppressive treatments or types of congenital neutropenia.28,29 Thus, we tested whether the neutropenia observed in Mcl-1–deficient mice could be overcome by daily administration of exogenous G-CSF.23 Blood was analyzed after treatment for effects on the numbers and phenotype of circulating neutrophils. Whereas treatment of littermate control and Mcl-1–deleted mice with vehicle control did not lead to alterations in blood neutrophils (Mac-1+Gr-1+), treatment with G-CSF induced a robust induction in the percentage of neutrophils in control mice (Figure 3A). In contrast, there was no increase in the percentage of circulating neutrophils in LysM-Cre–deleted mice receiving G-CSF treatment. These data indicate that the induction of emergency granulopoiesis requires MCL-1 expression to promote the survival of the neutrophils, and that treatment with exogenous G-CSF cannot overcome this requirement.
Figure 3.

Growth factor signaling cannot rescue neutropenia in Mcl-1–deleted mice. (A) Representative flow cytometric analysis of blood from littermate control (Mcl-1f/wt plus LysM-Cre) and Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre) treated with G-CSF (250 μg/kg) or vehicle control for 5 days. Top right quadrant represents Mac-1 and Gr1 double-positive neutrophils. (B) Percentage of apoptotic mature and immature neutrophils (Mac-1+, Gr1(high and Intermediate), annexin-V+) from littermate control (Mcl-1f/wt plus LysM-Cre) and Mcl-1–deleted (Mcl-1f/null plus LysM-Cre) BM cells cultured in the presence or absence of GM-CSF (10 ng/mL). Data presented are the average of 3 independent experiments, and the error bars represent the SEM. (C) Flow cytometric analysis of BM from littermate control (Mcl-1f/wt plus LysM-Cre) and Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre) cultured in GM-CSF (10 ng/mL) for 1 day. Samples were stained with Mac-1 and Gr1. Top right quadrant designated as mature neutrophils; bottom right quadrant, immature neutrophils. (D) MCL-1 protein levels as determined by immunoblot analysis of BM cells from control mice (Mcl-1f/wt and Mcl-1f/wt plus LysM-Cre) or Mcl-1–deleted mice (Mcl-1f/null plus LysM-Cre) cultured with or without GM-CSF (10 ng/mL) at indicated time points. Blots were probed for PDI expression to serve as a loading control. (E) In vitro myeloid differentiation of Mcl-1f/wt littermate control (■) or Mcl-1f/null plus LysM-Cre–deleted mice (red bar) cultured in G-CSF, M-CSF, or GM-CSF (10 ng/mL). After 48 hours, cells were harvested, cytospun, and subjected to manual BM differential for myeloid precursors and neutrophils. Graphs represent the average of 3 individual experiments; error bars denote SEM.
GM-CSF has been shown to promote the survival of neutrophils by both inducing MCL-1 transcription and by extending the half-life of MCL-1 protein.13,30 Furthermore, it has been reported that GM-CSF treatment can render Mcl-1–deficient neutrophils resistant to apoptosis, leading to the hypothesis that GM-CSF promotes survival in a MCL-1–independent manner.20 To test this hypothesis, BM from littermate control or Mcl-1–deleted mice was cultured in the presence of GM-CSF and monitored for the survival and differentiation status of the cultures. Whereas Mcl-1–deleted cells were extremely sensitive to death induced by culture without growth factors, GM-CSF treatment promoted the survival of both Mcl-1–deleted and littermate control cells over the course of 2 days in culture (Figure 3B). However, the phenotype of the surviving Mcl-1–deleted cells was different from the GM-CSF–cultured littermate control cells. Whereas littermate control cells were Mac-1+Gr1High and exhibited segmented nuclei, the Mcl-1–deleted cells were Mac-1+Gr-1intermediate and only reached the metamyelocyte and band stages of development (Figure 3B,D,E). Culture of BM cells with GM-CSF promotes MCL-1 expression in littermate control cells, and even in GM-CSF–treated cultures of Mcl-1f/null plus LysM-Cre BM we observed cells that have failed to delete Mcl-1 (Figure 3D). These cells may represent cells at a developmental stage prior to efficient LysM-Cre expression or the outgrowth of nondeleted cells in the cultures.
Strikingly, the phenotype of the GM-CSF–cultured Mcl-1–deleted cells is similar to the phenotype observed in the BM of Mcl-1–deleted animals, suggesting that a similar failure to progress may be occurring in vivo. Indeed, serum from LysM-Cre–deleted mice exhibited increased amounts of G-CSF (6937 ± 2008 pg/mL; n = 5) and GM-CSF (255.3 ± 43 pg/mL; n = 5) when compared with littermate control (G-CSF, 185.3 ± 84 pg/mL; GM-CSF, 50 ± 10.6 pg/mL; n = 6) mice in a multiplex serum cytokine assay. These data demonstrate that while GM-CSF can promote the survival of Mcl-1–deleted cells, these cells fail to differentiate into mature, segmented neutrophils either in vitro or in vivo.
To determine the ability of Mcl-1–deleted BM cells to differentiate in response to colony-stimulating factor stimulation, BM from littermate controls of LysM-Cre–deleted mice were cultured in the presence of growth factors. After 2 days of culture, the resulting BM cells were harvested and subjected to histologic examination to determine the potential of the cells to differentiate into neutrophil lineage cells by manual differential cell counts. When cultured in either G-CSF, M-CSF, or GM-CSF, Mcl-1–deleted BM cells failed to give rise to mature, segmented neutrophils (Figure 3E). These data suggest that even when cultured with exogenous colony-stimulating factors, Mcl-1–deleted BM cells are incapable of generating mature neutrophils.
Mcl-1 deficiency induces Bim- and Puma-independent apoptosis in the neutrophil lineage
Proapoptotic BIM has been implicated as a critical regulator of myeloid cell homeostasis because germline ablation increases the number of circulating myeloid cells and renders neutrophils and macrophages resistant to cell death induced by a variety of stimuli.18,19 Whereas Puma-deficient mice do not demonstrate overt alterations in myeloid homeostasis, cells from these mice are resistant to a variety of death-inducing stimuli, including growth factor withdrawal and genotoxic stress.16,17 Furthermore, lymphocytes from mice deficient in both Bim and Puma exhibit an even more dramatic resistance to a variety of death stimuli.31 Because MCL-1 is capable of directly binding and antagonizing both proapoptotic BIM and PUMA, we hypothesized that MCL-1 may act to regulate the susceptibility of myeloid cells to cell death by antagonizing BIM and/or PUMA effector function.7,32,33 Therefore, we sought to identify whether loss of MCL-1 expression results in the induction of cell death in the neutrophil lineage through a BIM- and/or PUMA-dependent manner.
LysM-Cre–expressing mice conditional for Mcl-1 were bred to mice expressing a conditional allele of Bim that flanks exons 1 to 4 with LoxP sites (Figure S3). Cre-mediated recombination results in deletion of exons 1 to 4 and results in a null allele21 (Figure S3). Analyses of protein lysates from Bim-deleted BMDMs demonstrate that LysM-Cre can delete the conditional Bim allele (Figure 4A). Surprisingly, myeloid deletion of Bim did not result in a dramatic increase in circulating myeloid cells (Table 1). These data are distinct from data obtained in mice bearing a germline mutant Bim allele, implying that loss of Bim in BM progenitor populations may contribute more to the myeloid expansion than the loss of Bim in the late stages of myelopoiesis.18 Therefore, conditional LysM-Cre mice are ideal to test whether loss of Mcl-1 at the terminal stages of myeloid development induces Bim-dependent cell death.
Figure 4.

Neutropenia resulting from Mcl-1 deletion cannot be rescued by loss of Bim and Puma. (A) MCL-1 and BIM protein levels as determined by immunoblot analysis of BMDMs generated from control mice (wild-type and Mcl-1f/null) or mice in which Mcl-1 and/or Bim could be deleted by LysM-Cre (Mcl-1f/null plus LysM-Cre, Bimf/f plus LysM-Cre, and Mcl-1f/nullBimf/f plus LysM-Cre). Blots were probed for actin expression to serve as a loading control. (B) Representative flow cytometric analysis of blood from mice of indicated genetic backgrounds stained for Mac-1 (CD11b) and Gr1. Gate (Mac-1+Gr1high) contains mature segmented neutrophils; numbers represent the percentage of white blood cells. (C) Representative flow cytometric analysis of bone marrow from mice of indicated genetic backgrounds. Top right quadrant is designated as mature neutrophils; bottom right quadrant, immature neutrophils. (D) Graph of the average number of total BM cells from the hind limbs of mice of indicated genotypes. Graph represents average numbers of at least 3 mice analyzed per genotype; error bars denote SEM. (E) Graph denotes the average total number of mature BM neutrophils (Mac-1+ Gr1High) from mice of indicated genotypes. Graph represents average numbers of at least 3 mice analyzed per genotype; error bars denote SEM. *P < .01 by 2-tailed t test when compared with littermate control population.
When genetic deletion of Mcl-1 was combined with deletion of Bim or ablation of Puma, mice still exhibited reduced populations of peripheral blood neutrophils (Mac-1+Gr-1+) when compared with littermate controls (Figure 4B; Table 1). These data suggest that loss of either BIM or PUMA expression is insufficient to rescue Mcl-1 deletion in the neutrophil lineage. To test whether loss of both Bim and Puma could rescue Mcl-1 deletion–induced neutropenia, germline Puma-deficient mice were intercrossed with mice conditional for Mcl-1 and Bim. Surprisingly, even the loss of both Bim and Puma were unable to rescue the peripheral neutropenia induced by LysM-Cre–mediated deletion of Mcl-1 (Figure 4B; Table 1). BM analyses confirmed that loss of Bim, Puma, or both together cannot rescue the defects in myeloid development observed due to deletion of Mcl-1 alone (Figure 4C-E; Table 2).
These data imply that despite the recognized role for BIM in regulating myeloid homeostasis, loss of BIM expression at the late stages of myelopoiesis is insufficient to rescue neutrophils from loss of antiapoptotic Mcl-1. Furthermore, even combining Bim deletion with Puma deficiency was incapable of rescuing the effects of Mcl-1 deletion in the myeloid lineage. These data suggest that either other BH3-only molecules beyond BIM and PUMA are operating to induce the developmental death in the absence of MCL-1, or that MCL-1 may be acting directly by inhibiting the BAX and BAK multidomain proapoptotics.34
Neutropenia induced by Mcl-1 deletion can be rescued by codeletion of both Bax and Bak
BAX and BAK represent an obligate gateway for regulating intrinsic cell death.2,35 While in most cases germline ablation of both Bax and Bak results in embryonic lethality, viable mice lacking both genes demonstrate dysregulation of cellular homeostasis in a variety of tissues, including expansion of hematopoietic progenitor populations and accumulation of large numbers of peripheral granulocytes and lymphocytes.35 To overcome the embryonic lethality, a conditional allele of Bax was bred to Bak-deficient mice.21 We bred these mice to Mcl-1–conditional mice expressing LysM-Cre to generate Mcl-1f/fBaxf/fBaknull LysM-Cre mice. Both Mcl-1 and Bax can be efficiently deleted by LysM-Cre as demonstrated by immunoblot analysis (Figure 5A). These mice allowed deletion of Mcl-1 and Bax specifically in the late stages of myelopoiesis on a Bak-deficient background to determine if ablation of Mcl-1 induces death of granulocyte precursors through a BAX- and BAK-dependent pathway. Myeloid-specific ablation of Bax and Bak resulted in minor alterations in mouse BM cellularity and peripheral blood myeloid cells (Figure 5D,E; Table 1). Furthermore, BM differential analyses from Bax and Bak myeloid-deleted mice demonstrate that loss of the multidomain proapoptotics does not overtly perturb myeloid precursor populations when compared with littermate control animals (Table 2). Thus, multidomain proapoptotic deletion during the late stages of myeloid differentiation does not lead to myeloid expansion similar to that observed in germline-deficient mice.35 These data suggest that when deleted after the precursor stages, a BAX- and BAK-independent mechanism may exist to control the homeostasis of mature myeloid lineages.
Figure 5.

Deletion of both Bax and Bak rescues neutropenia induced by myeloid-specific deletion of Mcl-1. (A) MCL-1, BAX, and BAK protein levels as determined by immunoblot analysis of BMDMs generated from control mice (Mcl-1f/wt, Mcl-1f/wt plus LysM-Cre, Mcl-1f/null plus LysM-Cre, and Mcl-1f/wtBaxf/wtBakhet plus LysM-Cre) or mice in which Bax and Bak or Mcl-1, Bax, and Bak could be deleted by LysM-Cre (Baxf/fBaknull plus LysM-Cre and Mcl-1f/fBaxf/fBaknull plus LysM-Cre). Blots were probed for actin expression to serve as a loading control. (B) Flow cytometric analysis of blood from control mice and mice in which Mcl-1, Bax, and Bak were deleted in the myeloid lineage (Mcl-1f/fBaxf/fBaknull plus LysM-Cre) stained for Mac-1 (CD11b) and Gr1. Top right quadrant (Mac-1+Gr1high) contains mature segmented neutrophils. (C) Flow cytometric analysis of BM from control mice and mice in which Mcl-1, Bax, and Bak have been deleted by LysM-Cre. Top right quadrant designated as mature neutrophils; bottom right quadrant, immature neutrophils. (D) Graph of the average number of total BM cells from the hind limbs of mice of indicated genotypes. Graph represents average numbers of at least 3 mice analyzed per genotype; error bars denote SEM. (E) Graph denotes the average total number of mature BM neutrophils (Mac-1+ Gr1High) from mice of indicated genotypes. Graph represents average numbers of at least 3 mice analyzed per genotype; error bars denote SEM. (F) Stained cytospins of peritoneal cells harvested from control and Mcl-1, Bax, and Bak LysM-Cre–deleted mice harvested 16 hours after casein injection. Images taken with 40× objective.
The genetic ablation of either Bax or Bak alone was incapable of rescuing neutrophil survival in the absence of Mcl-1, suggesting a functional redundancy between the multidomain proapoptotics in countering cell death induced by Mcl-1 deletion. However, when the blood of Mcl-1f/fBaxf/fBaknull LysM-Cre mice was compared with that of littermate control mice, a restoration of normal numbers of circulating neutrophils and monocytes was observed (Figure 5B; Table 1). Whereas Mcl-1–deleted mice exhibit an accumulation of immature cells in their BM, Mcl-1f/fBaxf/fBaknull LysM-Cre mouse myeloid development appears normal (Figure 5C-E; Table 2). Furthermore, 16 hours after elicitation of peritonitis with casein, large numbers of mature neutrophils were observed in Mcl-1f/fBaxf/fBaknull LysM-Cre mice (Figure 5F). These data demonstrate that neutropenia resulting from loss of Mcl-1 expression can be rescued by ablation of both Bax and Bak. Thus, these data suggest that loss of MCL-1 in neutrophil precursors results in death through the intrinsic death pathway, requiring at least one multidomain proapoptotic molecule.
MCL-1–regulated apoptosis induction by effector function in macrophages
The phagocytosis and destruction of pyogenic bacteria by macrophages is an important host defense mechanism used to limit infection. Stimulation of Toll-like receptors (TLRs) on macrophages has been reported to induce the expression of proapoptotic BIM, but this induction is insufficient to induce macrophage cell death.36 However, the process of engulfing bacteria can induce apoptosis, suggesting that phagocytosis leads to an additional stimuli that promotes cell death.37–39 Such apoptosis induced by macrophage ingestion of pyogenic bacteria is strongly reduced in Bim-deficient macrophages, implying that BIM is the primary regulator of phagocytosis-induced cell death.36
Despite the expression of MCL-1 in wild-type macrophages, normal macrophage populations were detected in stimulated peritoneal cells or from in vitro BMDMs from LysM-Cre–deleted animals (Figure 1). Therefore, we sought to address whether the lack of MCL-1 expression led to any functional consequences in the macrophage effector function. BMDMs from littermate control or LysM-Cre–deleted mice were cultured with various ratios of live E coli bacteria to macrophages for 2 hours, after which extracellular bacteria were eliminated by gentamicin treatment. Both wild-type and Mcl-1–deleted macrophages were capable of engulfing E coli and inducing reactive oxygen species production, indicating that loss of MCL-1 expression does not adversely perturb either of these processes (data not shown). The macrophages were incubated for an additional 4 hours, after which cell death was assessed by annexin-V and propidium iodide staining. While culture of either control or Mcl-1–deficient macrophages did not induce appreciable cell death, treatment of the Mcl-1–deficient macrophages with bacteria resulted in a dramatic increase in cell death when compared with littermate control macrophages (Figure 6A,B). These data demonstrate that Mcl-1–deficiency renders macrophages more sensitive to cell death induced by phagocytosis of pyogenic bacteria.
Figure 6.

Loss of Mcl-1 in macrophages induces BIM-dependent death induced by phagocytosis. (A) Representative experiment of littermate control (Mcl-1f/wt), Mcl-1–deficient (Mcl-1f/null plus LysM-Cre), and Mcl-1– and Bim-deleted (Mcl-1f/nullBimf/f plus LysM-Cre) BMDMs cultured with indicated ratios of E coli bacteria to macrophages for 2 hours. At 4 hours later, the amount of death was determined by flow cytometric analysis for propidium iodide–positive cells. (B) Compilation of 3 independent experiments performed as described in panel A. Data depicted are average fold of death induced over macrophages cultured without bacteria for various bacteria-to-macrophage ratios. Error bars represent SEM. (C) MCL-1 and BIM protein levels as determined by immunoblot analysis of BMDMs generated from wild-type mice cultured with E coli bacteria (25:1 bacteria-to-macrophage ratio) for indicated time period. Blots were probed for actin expression to serve as a loading control.
To examine why MCL-1 deficiency would render macrophages more sensitive to apoptosis, we assessed the expression of MCL-1 in macrophages cultured with bacteria. When wild-type macrophages engulf extracellular bacteria, MCL-1 protein expression is induced (Figure 6C). Such an induction of MCL-1 protein occurs concomitantly with the induction of proapoptotic BIM expression (Figure 6C). Thus, when Mcl-1 is deleted, the macrophages may not be able to counter the increases in BIM expression and therefore undergo cell death. To test this hypothesis, we generated BMDMs from mice in which both Mcl-1 and Bim were deleted and tested their sensitivity cell death induced by engulfing bacteria. Whereas macrophages from mice lacking Mcl-1 were hypersensitive to phagocytosis-induced apoptosis, macrophages deficient in both Mcl-1 and Bim were markedly more resistant (Figure 6A,B). Therefore, these data suggest MCL-1 levels may be increased to buffer the induction of BIM expression during phagocytosis and upon MCL-1 elimination a BIM-dependent death of the macrophage is promoted.
Discussion
Myeloid cells represent the first line of defense of the host from pathogens by rapidly responding and initiating the immune response. Their production is tightly regulated, as these cell types are the most short-lived blood cells requiring constant replacement from BM progenitor populations. By deleting Mcl-1 in the myeloid lineage, we demonstrate a selective role for MCL-1; it is the obligate survival molecule for neutrophil development, but is not required for protection during monocyte development. Our results redefine and extend the findings of He and colleagues that also identified MCL-1 as an essential survival molecule of neutrophils.20 We demonstrate that LysM-Cre–mediated deletion of Mcl-1 blocks terminal granulocyte development, preventing the maturation of myeloid precursors to the mature, segmented neutrophil. As a result, the BM of mice in which Mcl-1 has been deleted in the myeloid lineage contains an expansion of myeloid cells and exhibits increased apoptosis of immature granulocyte precursors.
Antiapoptotic MCL-1 plays critical roles in the survival of hematopoietic cells; maintaining the survival of developing lymphocytes and early hematopoietic progenitors.6,7 Furthermore, whereas this study examined the role for MCL-1 in lineage-committed myeloid populations, it has been previously demonstrated that MCL-1 is required for the survival of myeloid progenitors. When sorted CMP or GMP populations were subjected to in vitro deletion of Mcl-1 by retroviral Cre-recombinase transfection, both underwent rapid cell death despite the presence of exogenous growth factors.6 These data indicate that MCL-1 plays an obligate role during the early stages of myelopoiesis, where it promotes the survival of the progenitor populations. However, later during granulopoiesis, MCL-1 is required to promote survival during the differentiation of mature neutrophils but appears dispensable for monocyte/macrophage differentiation.
Growth factor signaling has been implicated in regulating hematopoietic development. In myeloid cells, G-CSF has been shown to promote the production of new granulocytes. However, we demonstrate in Mcl-1–deleted mice that the deficiency in neutrophils cannot be rescued by treatment of mice with exogenous G-CSF. Such a failure to overcome the defect is likely due to a failure to generate mature neutrophils from the BM. Furthermore, culture of BM with GM-CSF has been reported to promote the survival of myeloid cells. Indeed, a previous report suggested that growth factor signaling may bypass the need for MCL-1 in mediating neutrophil survival.20 Although GM-CSF protects both wild-type and Mcl-1–deleted BM cells from death, we demonstrate that Mcl-1–deficient cells failed to differentiate into mature, segmented neutrophils. These data demonstrate that whereas the survival of immature BM myeloid precursors such as metamyelocytes and band cells can be enhanced by GM-CSF, the final maturation to segmented neutrophils is still dependent on MCL-1. These data are further supported by the observation that myeloid-specific deletion of Mcl-1 results in increased serum levels of a variety of myeloid growth factors, including GM-CSF and G-CSF. Despite the elevated levels of these growth factors, the neutropenia induced by Mcl-1 deletion is still observed.
The proapoptotics responsible for the death of neutrophils upon Mcl-1 deletion are still unclear. BIM has been reported to play an important role in regulating the homeostasis of both monocyte and neutrophil lineages, but codeletion of both Mcl-1 and Bim was unable to rescue neutrophil survival. Although PUMA has not been implicated in regulating myeloid homeostasis, its deletion renders cells resistant to a variety of cellular stresses, and combining Puma and Bim deficiency has been shown to render lymphocytes dramatically resistant to cell death.31 However, neither Puma ablation nor codeletion of both Puma and Bim was capable or rescuing the effects of myeloid specific Mcl-1 deletion on neutrophil maturation. However, the loss of Mcl-1 in neutrophils does induce apoptosis through the intrinsic death pathway, as the loss of both multidomain proapoptotics Bax and Bak was capable of rescuing Mcl-1–deficient neutrophils. These data suggest that potentially several BH3-only molecules may be operating to induce cell death downstream of Mcl-1 deletion, or that loss of MCL-1 may directly induce the activation of BAX and/or BAK.
Unlike the defects observed in the granulocyte lineage, monocytes are found in circulation and are efficiently recruited to sites of inflammation in Mcl-1–deleted mice. Previously, He and colleagues also demonstrated that MCL-1 is dispensable for monocyte development; however, they did not observe any functional consequences to the loss of MCL-1.20 In contrast, our data indicate that MCL-1 plays a previously unrecognized role in promoting the survival of macrophages carrying out effector function. Our data suggest that during phagocytosis, both MCL-1 and BIM are induced. In the absence of MCL-1, BIM-dependent death occurs, leading to the aberrant apoptosis of the macrophage. Thus, Mcl-1–deficient macrophages may exhibit increased sensitivity to cell death induced by ingestion of bacteria due to their failure to counter BIM induction. Indeed, it has been proposed that the BIM induction observed in neutrophils, under situations in which no cell death is observed, may be due to the antagonism of concomitantly up-regulated MCL-1.40 Our data support this model, as codeletion of both Bim and Mcl-1 effectively restores the death of macrophages to that of littermate controls.
Macrophages cultured with pyogenic bacteria ingest these bacteria to eliminate the pathogens. One consequence of phagocytosis is that it can activate the intrinsic apoptotic pathway and result in the death of the macrophage.36,38,39 This phagocytosis-induced apoptosis has important clinical ramifications because the death of macrophages plays a vital role in the pathogenesis of sepsis. Experimentally, it has been demonstrated that either preventing the death of myeloid cells by overexpression of antiapoptotic BCL-2 or inhibiting effector caspases can be an effective treatment for preventing bacterial sepsis.41,42 It is still unclear whether preventing the death of the macrophage prevents the destructive effects of sepsis. Does it act directly by enhancing bacterial clearance, or do apoptotic macrophages release mediators of inflammation that promote the toxic effects of sepsis? In either case, death of the macrophage could contribute to the onset of sepsis and subsequent lethality.
These studies redefined the role of antiapoptotic MCL-1 in myeloid homeostasis. During the terminal maturation of neutrophils, MCL-1 is needed to promote survival during the transition to mature, segmented neutrophils. Deficiency in Mcl-1 results in the death of granulocyte precursors during development by inducing BAX- and BAK-dependent apoptosis, which cannot be overcome by treatment with exogenous myeloid growth factors. In the monocyte/macrophage lineage, MCL-1 has a more selective role. Although MCL-1 is dispensable for the differentiation of mature macrophages, it plays an important role in regulating the susceptibility of macrophages to death induced by effector function.
Supplementary Material
Acknowledgments
This work is dedicated to the late Stanley Korsmeyer, in whose laboratory some of these experiments were initiated. We would like to thank Ms K. Bisanz and Mr Brian Koss for technical expertise, T. Root for animal husbandry assistance, Ms J. Rogers for multiplex cytokine analyses, and Dr P. Murray (St Jude Children's Research Hospital) for helpful discussions.
This work is supported by the Pew Scholars Program in the Biomedical Sciences, the American Lebanese Syrian Associated Charities (ALSAC), and the National Institutes of Health National Cancer Institute.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.A.S. performed experiments and analyzed the data; K.B. analyzed and collected pathology and histology data; O.T. and J.K.F. generated new reagents; G.P.Z. provided reagents and discussed data; and J.T.O. designed research, performed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joseph T. Opferman, Department of Biochemistry, St Jude Children's Research Hospital, 262 Danny Thomas Pl D-4063C, Memphis, TN 38105; e-mail: joseph.opferman@stjude.org.
References
- 1.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 2.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 4.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 5.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 7.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 8.Lord BI, Woolford LB, Molineux G. Kinetics of neutrophil production in normal and neutropenic animals during the response to filgrastim (r-metHu G-CSF) or filgrastim SD/01 (PEG-r-metHu G-CSF). Clin Cancer Res. 2001;7:2085–2090. [PubMed] [Google Scholar]
- 9.Hamasaki A, Sendo F, Nakayama K, Ishida N, Negishi I, Hatakeyama S. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J Exp Med. 1998;188:1985–1992. doi: 10.1084/jem.188.11.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis. J Leukoc Biol. 2000;68:158–166. [PubMed] [Google Scholar]
- 11.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J Exp Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moulding DA, Giles RV, Spiller DG, White MR, Tidd DM, Edwards SW. Apoptosis is rapidly triggered by antisense depletion of MCL-1 in differentiating U937 cells. Blood. 2000;96:1756–1763. [PubMed] [Google Scholar]
- 13.Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem. 2004;279:26915–26921. doi: 10.1074/jbc.M313875200. [DOI] [PubMed] [Google Scholar]
- 14.Zinkel SS, Ong CC, Ferguson DO, et al. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 2003;17:229–239. doi: 10.1101/gad.1045603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufmann T, Tai L, Ekert PG, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 17.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 18.Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 19.Villunger A, Scott C, Bouillet P, Strasser A. Essential role for the BH3-only protein Bim but redundant roles for Bax, Bcl-2, and Bcl-w in the control of granulocyte survival. Blood. 2003;101:2393–2400. doi: 10.1182/blood-2002-07-2132. [DOI] [PubMed] [Google Scholar]
- 20.Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 23.Panopoulos AD, Zhang L, Snow JW, et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 25.Ye M, Iwasaki H, Laiosa CV, et al. Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity. 2003;19:689–699. doi: 10.1016/s1074-7613(03)00299-1. [DOI] [PubMed] [Google Scholar]
- 26.Lieschke GJ, Grail D, Hodgson G, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 27.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 1996;5:491–501. doi: 10.1016/s1074-7613(00)80504-x. [DOI] [PubMed] [Google Scholar]
- 28.Vadhan-Raj S, Jeha SS, Buescher S, et al. Stimulation of myelopoiesis in a patient with congenital neutropenia: biology and nature of response to recombinant human granulocyte-macrophage colony-stimulating factor. Blood. 1990;75:858–864. [PubMed] [Google Scholar]
- 29.Klein C, Grudzien M, Appaswamy G, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 30.Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. 1998;92:2495–2502. [PubMed] [Google Scholar]
- 31.Erlacher M, Labi V, Manzl C, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 34.Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirschnek S, Ying S, Fischer SF, et al. Phagocytosis-induced apoptosis in macrophages is mediated by up-regulation and activation of the Bcl-2 homology domain 3-only protein Bim. J Immunol. 2005;174:671–679. doi: 10.4049/jimmunol.174.2.671. [DOI] [PubMed] [Google Scholar]
- 37.Baran J, Guzik K, Hryniewicz W, Ernst M, Flad HD, Pryjma J. Apoptosis of monocytes and prolonged survival of granulocytes as a result of phagocytosis of bacteria. Infect Immun. 1996;64:4242–4248. doi: 10.1128/iai.64.10.4242-4248.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hacker H, Furmann C, Wagner H, Hacker G. Caspase-9/-3 activation and apoptosis are induced in mouse macrophages upon ingestion and digestion of Escherichia coli bacteria. J Immunol. 2002;169:3172–3179. doi: 10.4049/jimmunol.169.6.3172. [DOI] [PubMed] [Google Scholar]
- 39.Albee L, Shi B, Perlman H. Aspartic protease and caspase 3/7 activation are central for macrophage apoptosis following infection with Escherichia coli. J Leukoc Biol. 2007;81:229–237. doi: 10.1189/jlb.0506358. [DOI] [PubMed] [Google Scholar]
- 40.Bauer A, Kirschnek S, Hacker G. Inhibition of apoptosis can be accompanied by increased Bim levels in T lymphocytes and neutrophil granulocytes. Cell Death Differ. 2007;14:1714–1716. doi: 10.1038/sj.cdd.4402185. [DOI] [PubMed] [Google Scholar]
- 41.Iwata A, Stevenson VM, Minard A, et al. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J Immunol. 2003;171:3136–3141. doi: 10.4049/jimmunol.171.6.3136. [DOI] [PubMed] [Google Scholar]
- 42.Hotchkiss RS, Chang KC, Swanson PE, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}