

Abstract
The structure of the extracellular, three-domain poliovirus receptor (CD155) complexed with poliovirus (serotype 1) has been determined to 22-Å resolution by means of cryo-electron microscopy and three-dimensional image-reconstruction techniques. Density corresponding to the receptor was isolated in a difference electron density map and fitted with known structures, homologous to those of the three individual CD155 Ig-like domains. The fit was confirmed by the location of carbohydrate moieties in the CD155 glycoprotein, the conserved properties of elbow angles in the structures of cell surface molecules with Ig-like folds, and the concordance with prior results of CD155 and poliovirus mutagenesis. CD155 binds in the poliovirus “canyon” and has a footprint similar to that of the intercellular adhesion molecule-1 receptor on human rhinoviruses. However, the orientation of the long, slender CD155 molecule relative to the poliovirus surface is quite different from the orientation of intercellular adhesion molecule-1 on rhinoviruses. In addition, the residues that provide specificity of recognition differ for the two receptors. The principal feature of receptor binding common to these two picornaviruses is the site in the canyon at which binding occurs. This site may be a trigger for initiation of the subsequent uncoating step required for viral infection.
Poliovirus (PV: family Picornaviridae, genus Enterovirus) is the etiologic agent of poliomyelitis, a human disease of the central nervous system. Effective vaccines against PV have been developed against the three known poliovirus serotypes: PV1, PV2, and PV3 (1). The structure of PV (2), like other picornaviruses such as rhinoviruses (3), consists of an icosahedral protein shell with an external diameter of ≈300 Å that encapsidates a plus strand of RNA of roughly 7,500 bases. The capsid consists of 60 copies each of three surface viral proteins: VP1, VP2, and VP3 (32, 29, and 26 kDa, respectively) and also 60 copies of the internally located VP4 (7 kDa). The three larger proteins adopt an eight-stranded, antiparallel β-barrel fold that is common to many other viral capsid structures (4).
Picornavirus infection is initiated by attachment to specific cell surface molecules. All three PV serotypes recognize the same cellular receptor molecule, the PV receptor (CD155) (5–7). In contrast, at least 78 of the more than 100 human rhinovirus (HRV) serotypes recognize intercellular adhesion molecule-1 (ICAM-1 or CD54) as a cellular receptor, and other picornaviruses recognize a variety of different cell surface molecules (1). Although ICAM-1 is known to be involved in adhesion of lymphocytes to damaged or infected cells, the normal function of CD155 is uncertain.
Rhino- and enteroviruses have a narrow surface depression (“canyon”) that surrounds each of the 12 5-fold vertices. The rhinovirus receptors originally were predicted to be long, narrow molecules that could bind to conserved residues within the canyon (3), the premise being that this might permit a strategy for the virus to escape host immune surveillance because bulkier neutralizing antibodies would be unable to enter the canyon. This prediction turned out to be correct with regard to the shape of the receptor for the major group of rhinoviruses (8, 9) and its site of attachment to the virus (10). However, the rationale of the prediction was questioned when the footprint of a neutralizing antibody was found to extend beyond the rims of the canyon (11), although naturally selected escape mutations that prevented antibody neutralization were located only on the viral surface outside the canyon (12). Genetic and mutational studies have mapped the CD155-binding site into the PV canyon (13–17), as also was done for the ICAM-1-binding site into the HRV canyon (cf. ref. 18).
CD155 and ICAM-1 are membrane-anchored, single-span glycoproteins whose extracellular regions have three and five domains, respectively, with Ig-like folds (Fig. 1). Alternate splicing of the CD155 mRNA yields four different isotypes (α, β, γ, and δ), in which the extracellular domains are identical. CD155-β and -γ, however, are secreted, whereas the membrane-bound CD155-α and -δ serve as PV receptors (6, 7). The amino-terminal domain, D1, in both CD155 (13, 19, 20) and ICAM-1, contains the virus recognition site. Hence, virus attachment occurs at a site on the receptor distal from the plasma membrane, a property that may be important for successful initiation of infection of cells by virus.
Figure 1.

Comparison of the mature structures of ICAM-1, the receptor for the major group of rhinoviruses, with the human PV receptor (hCD155), the monkey PV receptor (mCD155), and the murine poliovirus receptor-related protein 2 (mPRR2). Sites of glycosylation are indicated by shaded circles. The number of amino acids is shown for each domain.
Ig superfamily domains have a β-barrel fold in which all β-strands (labeled A to G) run parallel or antiparallel to the long axis of the domain. The fold of the CD155 D1 domain resembles (see “Model Fitting” in Materials and Methods) that of an Ig variable (V) domain [nomenclature reviewed by Chothia and Jones (21)] (Fig. 2), whereas the fold of the ICAM-1 D1 domain is intermediate (I) between the variable and constant (C) Ig folds. An Ig-like V domain has two extra β-strands, labeled C′ and C", between β-strands C and D. Thus, the D1 domain of CD155 has 32 more residues than the D1 domain of ICAM-1. Furthermore, D1 in CD155 has two potential glycosylation sites, whereas the ICAM-1 D1 is unglycosylated. Absence of carbohydrate in CD155 D1 is known to enhance its binding to PV (16).
Figure 2.
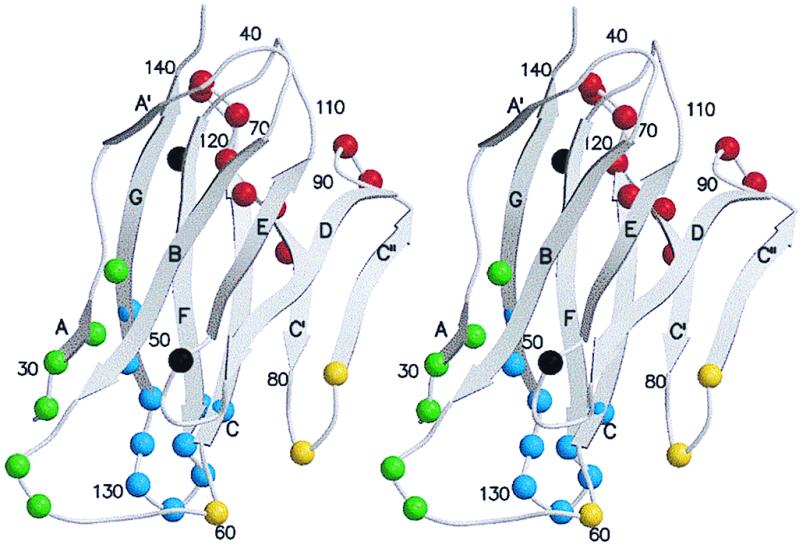
The Cα backbone of domain D1 of CD155 based on its homology to protein zero. Shown is the nomenclature of the β-strands, the two sites of potential glycosylation, and strategically numbered residues. Residues that were found to make contact with the north rim of the canyon are colored green, with the south rim yellow, with the floor blue, and with the additional surface area red. Potential glycosylation sites are colored black.
Cell entry and uncoating are initiated when PV and HRV recognize their respective receptors (22, 23). Purified, soluble CD155 as well as the membrane-anchored receptor convert infectious PV (160S) to altered (“A”) particles (135S) (7). VP4 is absent in A particles (7), and the N terminus of VP1 is externalized (24). Slightly longer incubation leads to the formation of 80S particles, which are devoid of the genomic RNA. It is uncertain, however, whether the 135S and 80S particles are intermediates in the uncoating pathway (25–27). Competition between binding of receptor and a cellular lipid moiety within VP1, in a hydrophobic pocket immediately beneath the canyon floor, has been suggested as the event that initiates uncoating (23).
Cryo-electron microscopy (cryo-EM) and x-ray crystallography have been used to examine interactions of ICAM-1 with two different HRV serotypes (18, 28). Here, we present a similar study of CD155 interaction with PV and demonstrate that the mode of interaction is distinct from that observed in the HRV–ICAM-1 complexes. As predicted by mutagenesis studies (13–17), the footprint of CD155 on PV covers a region of the canyon similar to that found in HRV, but the CD155 molecule binds to PV in a different and more tangential orientation compared with the radial orientation of ICAM-1 bound to HRV.
Materials and Methods
Sample Preparation.
All experiments were carried out with PV1(RIPO), a highly attenuated derivative of PV1(Mahoney) [PV1(M)] from which the cognate internal ribosomal entry site was exchanged with that of HRV2 (29). PV1(RIPO) has a wild-type type 1 capsid, expressing growth properties in HeLa cells at 37°C indistinguishable from that of PV1(M) (29).
A soluble derivative of CD155 was prepared by fusing the coding region of the 337 N-terminal codons of CD155 (including all three extracellular Ig-like domains) to the N-terminal coding region of human placental alkaline phosphatase (AP), using plasmid pAPtag2 (30) to yield plasmid pCD155-AP. The secreted fusion protein, CD155-AP (the calculated molecular mass in its unglycosylated form is 90 kDa), was expressed with this vector in 293 cells (human embryonic kidney cells transformed with adenovirus E1A,B genes) and was purified from cell supernatants (S.M., X.P., and E.W., unpublished results). Incubation of purified PV1(M) with purified CD155-AP reduced infectivity of the virus by about 5 orders of magnitude, whereas purified AP or mouse poliovirus receptor-related protein (mPRR2) had no effect on viral infectivity under the same conditions (S.M., X.P., and E.W., unpublished results).
Cryo-EM.
Purified samples containing 40 μl of 8.75 mg/ml PV1 were mixed with 12 μl of 30 mg/ml CD155-AP at 4°C and incubated for 1 hr. Small (3.5-μl) aliquots of this mixture were adhered to carbon-coated electron microscope (EM) grids and vitrified in liquid ethane as described (31). Control samples of PV (i.e., not incubated with CD155-AP) were prepared similarly for cryo-EM. Electron micrographs were recorded on Kodak SO-163 film in a Philips CM200 FEG microscope (Philips, Eindhoven, the Netherlands), at a nominal magnification of 38,000 and a dose level of ≈20e−/Å2 (Table 1). Although the virus–receptor complexes tended to aggregate, a sufficient number of them were not overlapped severely with neighboring particles and, hence, permitted the computation of a three-dimensional reconstruction. In addition, despite identical particle concentrations in the samples, the virus particles visible in fields far outnumbered the virus–receptor complexes. These observations demonstrated both the instability of the virus–receptor complex and a more hydrophobic character of the virus when associated with receptor, as is observed for A particles (7). Micrographs were digitized on a Zeiss PHODIS microdensitometer at 14-μm intervals, which corresponds to 3.68 Å at the specimen.
Table 1.
EM data collection and processing
| PV1(M)–CD155-AP | PV1(M) | |
|---|---|---|
| Underfocus, μm* | 1.3 ∼ 2.1 | 1.3 ∼ 2.3 |
| No. of micrographs | 16 | 10 |
| No. of particles† | 1,156 (1,699) | 1,081 (1,476) |
| Correlation coefficient‡ | 0.305 (0.040) | 0.310 (0.032) |
| Resolution (Å) | 22Å | 22Å |
Determined from the contrast transfer function of the microscope.
† The number of particles included in each three-dimensional map. The total number of boxed particles is given in parentheses.
‡ Averaged real space correlation coefficient (CC) and SD (in parentheses) for all particles, where cc = ∑(ρ1ρ2−<ρ1><ρ2>)/[∑(ρ12−<ρ1>2)∑(ρ22−<ρ2>2)]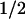 , computed over all radii of the final model.
, computed over all radii of the final model.
An earlier reconstruction of the PV1 A particle (P. R. Chipman, R. R. Rueckert, T.S.B., and M.G.R., unpublished results) served as an initial model for determining the orientation of each projected particle by means of the model-based polar-Fourier transform method (32). Orientation refinement was monitored by means of correlation coefficients computed with real and reciprocal space data (33). The final correlation coefficients were a little lower than those generally observed in reconstructions of other, especially larger, icosahedral particles (cf. ref. 34) and in which particle overlap is not a problem, as was the case with the PV–CD155-AP complex sample. All maps were computed with Fourier–Bessel procedures, and eigenvalue spectra were used to monitor the conditioning of the linear, least-squares equations (35). The resolution of each of the two maps (Table 1) was estimated by splitting the image data into two sets and comparing structure factors obtained in the separate reconstructions. The defocus level was calculated for each image and used to compute the phase-contrast transfer function corrections to include data to about 18-Å resolution in favorable cases (31). However, reasonable phase agreement was retained only to about 22-Å resolution between the two independent reconstructions.
Although the reconstructions were made with respect to a model of known hand, caution was exercised to ensure that the hand was not flipped during refinement. The hand was checked by noting the asymmetric features in the shape and environment of the canyon in the PV and HRV x-ray and EM structures (Fig. 3). A distinctive “landmark” is the “V” shape of the canyon within one icosahedral asymmetric unit (Fig. 3c), in which the base of the V lies clearly displaced to the “east” (see below for definition of north, south, east, and west). All reconstructions of both the HRV and PV samples displayed these same asymmetric features.
Figure 3.

(a) Stereoview of the cryo-EM reconstruction showing the complex of PV1(M) with human CD155. The outline of one geometric icosahedral asymmetric unit is shown. Note that the receptor leans towards the southeast. (b) Stereoview of the cryo-EM reconstruction showing the complex of HRV16 with its ICAM-1 receptor (from ref. 28). The outline of one geometric icosahedral asymmetric unit is shown. Note that the receptor leans towards the southwest. (c) Stereoview of a cryo-EM reconstruction of PV, also showing the geometric icosahedral asymmetric unit. Note the asymmetric shape of the canyon with its most southerly point slightly east of center (arrow) and the smaller peak southwest of the canyon (arrow). These features establish the correct hand of the reconstructions in a and b and are consistent with the x-ray results, where the absolute hand is known. (d) Density (green) representing one CD155 molecule (black) fitted with the Cα backbone structure of the closest homologous structures found in the PDB for each of the three domains. Shown also is the difference map (blue) between the cryo-EM density and the unglycosylated CD155 model to show the sites of glycosylation. The potential glycosylation sites are shown on the CD155 backbone (red).
Difference Map Calculations.
The x-ray crystal structure of PV1(M) [Protein Data Bank (PDB) ID code 2PLV] was used as a reference for scaling the pixel size and the density of the cryo-EM reconstructions. Structure factors were calculated from the x-ray coordinates with a “temperature” factor of 1,000 Å2. These structure factors then were used to compute the reference electron density map to a resolution of 20 Å. The program mama (36) was used to zero out all electron density except for that within an annulus, defined by radii of 110–145 Å, to avoid the impact of the internal RNA and the external receptor in the canyon, neither of which is present in the x-ray structure. The program mave (36) was used to scale the PV–CD155-AP complex and PV EM maps to the x-ray map by using various radial shells to check the results for consistency (28). A difference electron density map then was computed by subtracting the scaled PV map from the scaled PV–CD155-AP map.
A difference map was calculated by subtracting the calculated contribution of the PV and CD155 atomic coordinates from the cryo-EM reconstruction map. This difference map identified the density corresponding to the glycosylation sites (Fig. 3d). Because of the increasing flexibility of the CD155 receptor at progressively larger distances from the virus surface, the scaling of the CD155 model density to the EM reconstruction was performed separately for each of the three Ig-like domains. The difference map showed the sites of three of the carbohydrate moieties (Fig. 3d).
Model Fitting.
In the absence of an atomic structure of CD155, it was necessary to find the closest available homologous models from the PDB for the fitting experiments. A blast (37) search (using the default values for gap penalties and other parameters) of the CD155 amino acid sequence (5) was conducted for each separate domain against all Ig superfamily entries in the PDB. The myelin membrane adhesion molecule—“human protein zero” (PDB ID code 1NEU)—gave the best fit for domain D1 (Fig. 4), which corresponded to an Ig-like V domain. An Ig light chain constant domain, derived from an antiidiotype Fab–Fab complex (PDB ID code 1CIC), was the best fit for domain D2 and corresponded to a constant Ig-like C2 domain. Finally, the insect immune protein (PDB ID code 1BIH) was the best fit for domain D3 and corresponded to an Ig-like I domain. The sequence identity in each instance was about 25%. These homologous structures then were modified by substituting CD155 amino acid side chains in each structure and using the original side chain positions as a guide. The program o (39) was used to visually fit these model domain structures to the cryo-EM density map.
Figure 4.
Sequence alignment of human CD155 domain D1 with protein zero. Secondary structural elements are indicated, and potential glycosylation sites are marked with an *. Residues implicated in binding to PV are marked with an X (15, 16) and a + (14, 17, 38) in the lines above the residue numbers. The color code for residues that are involved in binding to PV is the same as in Fig. 2.
There was considerable uncertainty about the choice of rotational orientation for each domain relative to the long axis of the domain. This problem was solved by noting that the hinge between the V and C domains in Ig superfamily multidomain structures, like CD2 and CD4, tends to bend to keep the surface created by the B, E, and D strands of domain D1 (“B,E,D” surface) facing the F,G surface of domain D2 (28). The elbow angle between pairs of domains (about 165°) was clearly evident in the density map, and this constrained the rotation about the long axis of the molecule. The structure of CD2 (PDB ID code 1HNF) was used as a suitable two-domain, Ig superfamily model and fitted into the cryo-EM density map. A least-squares procedure then was used to superimpose the CD155 homologs of D1 and D2 onto the CD2 Cα coordinates. D3 was fitted to the density by limiting the position of its amino end close to the carboxyl end of D2. The results of the modeling were verified by observing that the difference densities attributed to carbohydrate moieties were juxtaposed to potential glycosylation sites in CD155 (Fig. 3d). Cα coordinates of the CD155 homology model with respect to an icosahedral reference system have been deposited with the PDB (ID code 1DGI).
Results and Discussion
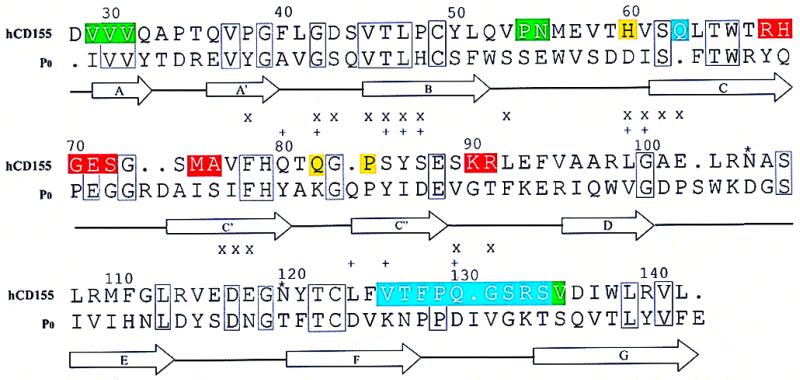
The three-dimensional reconstruction of the PV–CD155-AP complex shows a good fit between the known PV structure and the cryo-EM density (Fig. 3a). Features such as the asymmetric shape of the canyon and the small protrusion on the south side of the canyon are present in both reconstructions. Nevertheless, it is probable that there are small differences in the virus structure, particularly when the differences in its aggregation tendencies are considered. The reconstruction also shows that the receptor is located, as anticipated, in the PV canyon at approximately the analogous site occupied by ICAM-1 when bound to HRV16 or HRV14 (10, 18, 28). However, the orientation of the CD155 molecule, relative to the surface of the virus, is completely different from that adopted by ICAM-1 on HRV (Fig. 3 a and b). For an icosahedral asymmetric unit defined with a 5-fold axis in the “north” and with adjacent 3-, 2-, and 3-fold axes to the “south,” the ICAM-1 molecule leans toward the southwest, whereas CD155 leans toward the southeast. Furthermore, ICAM-1 adopts a more radial orientation with respect to the HRV surface, whereas CD155 is more tangential to the surface of PV, with much of the C,C′,C" surface of domain D1 contacting the viral surface (Fig. 5).
Figure 5.
Stereoview of CD155 (yellow) docked onto PV. VP1, VP2, and VP3 coloring is blue, green, and red, respectively.
The density ascribed to CD155 decreased rapidly as a function of distance from the viral surface, and no significant density was detected in the difference map beyond a 260-Å radius. The mean densities for the PV protein capsid, the RNA, and the D1 domain of CD155 were about the same, indicating that roughly full occupation was obtained for CD155 at each of the 60 independent sites on the virus. The CD155 density by itself, as visualized in the difference map (Fig. 3d), showed densities for domains D1, D2, and D3 of 6, 4, and 2 SDs, respectively, with the largest noise peak being about 1 SD. The rapid decrease in density at progressively higher radii along the length of the receptor shows that there is considerable flexibility at the hinges between domains, which, therefore, allows the more distal domains greater freedom of motion and an ability to adopt different positions relative to the viral surface. Despite this property, domains D1, D2, and D3 were easily docked and fitted into the reconstructed difference density map (Fig. 3d). The elbow angles between pairs of domains were clearly apparent, with one of the potential N-glycosylation sites in D1 and two in domain D2, consistent with the features observed in the difference electron density (Fig. 3d). However, the large AP domain (53 kDa when unglycosylated) fused to the C terminus of the CD155 molecule was not seen in the cryo-EM reconstruction. From this observation, we presume that the fusion protein adopts highly variable orientations relative to the 60 binding sites on the virus surface, and this causes its density to average out upon enforcement of icosahedral symmetry during the three-dimensional reconstruction process. In contrast, the large AP moiety could be clearly seen as “lollipops” on unprocessed, negatively stained samples of the complex at a radius of about 280 Å from the viral center (data not shown). Nevertheless, the presence of the large AP at the C terminus demonstrated what had already been determined by constructing chimeric receptors (13, 19, 20)—that the domain closest to the virus surface must be domain D1 (12.6 kDa).
Domain D1 of human CD155 has potential N-glycosylation sites at residues Asn-105‖ and Asn-120. One of these sites is evident in the difference map (Fig. 3d). The site at Asn-120 lies fairly close to the rim of the canyon, whereas the site at Asn-105 is well outside of the canyon. However, if a different orientation of D1 had been assumed in the fitting procedure, it would have brought Asn-105 into the canyon, causing steric hindrance with the virus surface, further confirming the correct docking of the CD155 homology model into the cryo-EM density. Similarly, the two potential glycosylation sites in monkey CD155 (22) are positioned in the model so as not to cause any steric hindrance with the viral surface. In contrast, mPRR2 (7) has a potential glycosylation site corresponding to Gln-130, within the interface between CD155 and PV, suggesting that glycosylation at this site would contribute to the inability of mPRR2 to bind PV (7).
It may not be mere coincidence that the closest available homologous structure to domain D1 of CD155 is the peripheral nerve myelin protein zero. The presence of CD155 on neurons and of protein zero on the surrounding myelin sheath may indicate a recent evolution from a common precursor. The biological function of CD155 is not clear; however, available evidence suggests that CD155 plays a role in the development of the central nervous system during embryogenesis (M. Gromeier, D. Soleckie, and E.W., unpublished data).
The 115-residue D1 domain of CD155 has an Ig-like V fold and is substantially different from the 84-residue D1 domain of ICAM-1, which has an Ig-like I fold (21). If the β-strands of domain D1 of the CD155 model are superimposed on the equivalent strands of domain D1 in ICAM-1 when docked to HRV, then the additional C′ and C" strands of CD155 collide with the south side of the canyon. This provides a steric restriction on the use of CD155 as an HRV receptor, in addition to differences in the specific site interactions required for binding. Binding of ICAM-1 to the site of the PV receptor has similar steric problems generated by the different elbow angle.
The footprint of CD155 on the surface of PV was established from the atomic model of the PV–CD155 complex by identifying those PV residues with atoms that lie within 4.0 Å from any CD155 atom in the modeled complex (Fig. 6). The viral and receptor surfaces involved in the interface are both in excellent agreement with mutational data (see below) and, hence, validate the accuracy of the model building. Unlike ICAM-1, which contacts primarily the floor and south wall of the HRV canyon, CD155 overlaps the north and south walls as well as floor of the canyon. Indeed, it has been suggested that CD155 may be in contact with residues from two neutralization antigenic sites on either side of the canyon (15, 16). Because the D1 domain of CD155 leans toward the virus surface, rather than being oriented radially like ICAM-1, much of its C,C′,C" face makes additional, extensive contact with the viral surface. In this respect, utilization of the C,C′,C" face by CD155 for interaction with its viral ligand is similar to CD4 with HIV (40, 41). With a 1.4-Å-radius probe as a basis for measurement, the CD155 footprint covers 1,300 Å2 of the PV surface. In comparison, ICAM-1 only has a 900-Å2 footprint on HRVs (28). The larger CD155 footprint can be attributed mostly to the contacts made between the C,C′,C" surface and the virus.
Figure 6.
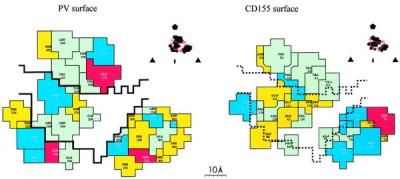
The footprint of the CD155 on the PV surface, defined by those residues on the viral surface that have any atoms within 4 Å of any atom in the receptor. (Inset) One icosahedral asymmetric unit with the footprint outlined and the limits of the canyon. (Left) The footprint on the virus (the canyon has a black outline). (Right) The residues of CD155 in contact with the viral surface. Each residue is colored in accordance with its chemical properties: green, hydrophobic; yellow, hydrophilic; red, acidic; and blue, basic.
The north rim of the canyon is formed by the VP1 BC loop (residues 1,091** to 1,107) and the VP1 βE to αB loop (residues 1,166–1,169). These residues form a hydrophobic surface that interacts with an equally extensive hydrophobic surface (Fig. 6) on CD155 (residues 29–31 at the amino terminus of the βA strand, residues 54 and 55 at the beginning of the BC loop, and residue 135 near the beginning of the βG strand). Both PV loops that form the north rim of the canyon had been implicated correctly by mutagenesis studies as being involved in binding of CD155 (15).
The south rim of the canyon forms a hydrophilic surface by virtue of residues 2,138–2,142 and 2,172 in the “puff” (a large insertion in VP2 between strand βE and helix αB) and residues 1,223–1,226 in the GH loop of VP1. These residues contact the C" strand of CD155, consistent with the mutational analysis of PV (17) and of CD155 (16, 38).
The floor of the canyon is formed primarily by residues in the GH loop of VP1 (1,228–1,235), the GH loop of VP3 (3,180), and the end of the VP1 BC loop (residue 1,109). These interact with residues (126–135) in the FG loop of CD155, consistent with the mutational analysis of the virus (14, 17) and of CD155 (15).
The viral surface in contact with the C,C′,C" surface of CD155 domain D1 is formed by the “knob” (residues 3,058–3,062, an insertion in β-strand B of VP3) and the C-terminal region of VP1 (residues 1,294–1,301). This assignment agrees with studies of CD155 mutants (15), although the involvement of the corresponding viral surface was not implicated in these studies. Contacts in this region are strengthened by complementary electrostatic interactions between Glu-71 in CD155 and Lys-3062 and between Lys-90 and Arg-91 in CD155 and Asp-1298. Analogous complementary interactions were found to be important in determining the specificity of ICAM-1 for the major receptor group of rhinoviruses (18, 28). Thus, the occurrence of some charge complementarity between PV and CD155 also may play a significant role in the recognition process.
Substantial evidence (28) suggests that the structures observed for the HRV–ICAM-1 complexes represent an initial recognition event. Only subsequently is the receptor likely to bind deeper into the canyon and thereby possibly compete out the lipid moiety in the VP1 pocket (23), which then leads to virus destabilization and progressive disassembly and release of the genomic RNA. CD155 binding may follow a similar pathway, as evidenced in the EM results by the substantial loss of particles upon incubation of PV with soluble CD155 (see Materials and Methods). It has been speculated (28) that the natural breathing of picornaviruses (42) might facilitate receptor binding to both the north and south walls of the canyon and, thus, maintain a channel along the 5-fold axis to permit the externalization of VP4, the amino end of VP1, and, eventually, the RNA. For PV, the receptor already appears to be in contact with both walls of the canyon in the initial recognition event. The presence of CD155, therefore, may simply prevent natural breathing in PV and keep pores open as the receptor binds deeper into the canyon.
The markedly different mode of interaction of CD155 with PV compared with ICAM-1 with HRV might seem surprising. Nevertheless, these two receptors share several common features: they both bind into the picornavirus canyon, initiate uncoating, and are long, thin molecules that extend far from the cell surface. The similar location of binding in the canyon suggests that it is the site itself that is important, not the orientation that the bound receptor adopts. This may be required, as suggested originally (3), to hide a part of the site from neutralizing antibodies and to regulate virus stability by competition between the binding of receptor and the lipid-like pocket factor in VP1. The apparent need to have a receptor molecule that is long and extends far from the cell surface may indicate a requirement for the virus to bind to molecules that, by virtue of Brownian motion, are mobile and, hence, promote binding of multiple receptors to unoccupied binding sites on the virus, facilitating cell entry and uncoating.
Acknowledgments
We thank Wei Zhang and Norman Olson for advice in collecting and processing the cryo-EM data, Rob Ashmore for programs, Benoît Hébert for useful discussions, and Sharon Wilder and Cheryl Towell for help in the preparation of the manuscript. This work was supported by National Institutes of Health grants to M.G.R. (AI11219), T.S.B. (GM33050 and AI 45976), R.J.K. (GM56279), and E.W. (AI32100, AI39485, and AI15122) and a National Science Foundation shared equipment grant to T.S.B. (BIR-9112921). We also gratefully acknowledge a Purdue University reinvestment grant in support of structural studies.
Abbreviations
- A particles
altered (135S) particles
- AP
human placental alkaline phosphatase
- C
Ig constant domain
- CD155
poliovirus receptor
- CD155-AP
secreted fusion protein
- cryo-EM
cryo-electron microscopy
- EM
electron microscope (or microscopy)
- HRV
human rhinovirus
- I
Ig intermediate domain
- ICAM-1
intercellular adhesion molecule-1
- mPRR2
mouse poliovirus receptor-related protein 2
- PDB
Protein Data Bank
- PV
poliovirus
- PV1(M)
poliovirus type 1 (Mahoney)
- V
Ig variable domain
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1DGI).
The CD155 amino acid sequence numbering includes a 27-aa signal sequence at the amino end. Some of the publications from the Racaniello laboratory (14, 17, and 38) start counting only after the end of this special sequence.
Amino acids for VP1, VP2, VP3, and VP4 are numbered starting at 1,001, 2,001, 3,001, and 4,001, respectively.
References
- 1.Rueckert R R. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Vol. 1. Philadelphia: Lippincott; 1996. pp. 609–654. [Google Scholar]
- 2.Hogle J M, Chow M, Filman D J. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 3.Rossmann M G, Arnold E, Erickson J W, Frankenberger E A, Griffith J P, Hecht H J, Johnson J E, Kamer G, Luo M, Mosser A G, et al. Nature (London) 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- 4.Rossmann M G, Johnson J E. Annu Rev Biochem. 1989;58:533–573. doi: 10.1146/annurev.bi.58.070189.002533. [DOI] [PubMed] [Google Scholar]
- 5.Mendelsohn C L, Wimmer E, Racaniello V R. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 6.Koike S, Horie H, Ise I, Okitsu A, Yoshida M, Iizuka N, Takeuchi K, Takegami T, Nomoto A. EMBO J. 1990;9:3217–3224. doi: 10.1002/j.1460-2075.1990.tb07520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimmer E, Harber J J, Bibb J A, Gromeier M, Lu H H, Bernhardt G. In: Cellular Receptors for Animal Viruses. Wimmer E, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 101–127. [Google Scholar]
- 8.Staunton D E, Merluzzi V J, Rothlein R, Barton R, Marlin S D, Springer T A. Cell. 1989;56:849–853. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 9.Greve J M, Davis G, Meyer A M, Forte C P, Yost S C, Marlor C W, Kamarck M E, McClelland A. Cell. 1989;56:839–847. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 10.Olson N H, Kolatkar P R, Oliveira M A, Cheng R H, Greve J M, McClelland A, Baker T S, Rossmann M G. Proc Natl Acad Sci USA. 1993;90:507–511. doi: 10.1073/pnas.90.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith T J, Chase E S, Schmidt T J, Olson N H, Baker T S. Nature (London) 1996;383:350–354. doi: 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherry B, Rueckert R. J Virol. 1985;53:137–143. doi: 10.1128/jvi.53.1.137-143.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freistadt M S, Racaniello V R. J Virol. 1991;65:3873–3876. doi: 10.1128/jvi.65.7.3873-3876.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colston E, Racaniello V R. EMBO J. 1994;13:5855–5862. doi: 10.1002/j.1460-2075.1994.tb06930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harber J, Bernhardt G, Lu H H, Sgro J Y, Wimmer E. Virology. 1995;214:559–570. doi: 10.1006/viro.1995.0067. [DOI] [PubMed] [Google Scholar]
- 16.Bernhardt G, Harber J, Zibert A, deCrombrugghe M, Wimmer E. Virology. 1994;203:344–356. doi: 10.1006/viro.1994.1493. [DOI] [PubMed] [Google Scholar]
- 17.Liao S, Racaniello V R. J Virol. 1997;71:9770–9777. doi: 10.1128/jvi.71.12.9770-9777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bella J, Kolatkar P R, Marlor C W, Greve J M, Rossmann M G. Proc Natl Acad Sci USA. 1998;95:4140–4145. doi: 10.1073/pnas.95.8.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koike S, Ise I, Nomoto A. Proc Natl Acad Sci USA. 1991;88:4104–4108. doi: 10.1073/pnas.88.10.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selinka H C, Zibert A, Wimmer E. Proc Natl Acad Sci USA. 1991;88:3598–3602. doi: 10.1073/pnas.88.9.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chothia C, Jones E Y. Annu Rev Biochem. 1997;66:823–862. doi: 10.1146/annurev.biochem.66.1.823. [DOI] [PubMed] [Google Scholar]
- 22.Koike S, Ise I, Sato Y, Mitsui K, Horie H, Umeyama H, Nomoto A. Semin Virol. 1992;3:109–115. [Google Scholar]
- 23.Rossmann M G. Protein Sci. 1994;3:1712–1725. doi: 10.1002/pro.5560031010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fricks C E, Hogle J M. J Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dove A W, Racaniello V R. J Virol. 1997;71:4728–4735. doi: 10.1128/jvi.71.6.4728-4735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arita M, Koike S, Aoki J, Horie H, Nomoto A. J Virol. 1998;72:3578–3586. doi: 10.1128/jvi.72.5.3578-3586.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curry S, Chow M, Hogle J M. J Virol. 1996;70:7125–7131. doi: 10.1128/jvi.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolatkar P R, Bella J, Olson N H, Bator C M, Baker T S, Rossmann M G. EMBO J. 1999;18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gromeier M, Alexander L, Wimmer E. Proc Natl Acad Sci USA. 1996;93:2370–2375. doi: 10.1073/pnas.93.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanagan J G, Leder P. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-t. [DOI] [PubMed] [Google Scholar]
- 31.Baker T S, Olson N H, Fuller S D. Microbiol Mol Biol Rev. 1999;63:862–922. doi: 10.1128/mmbr.63.4.862-922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker T S, Cheng R H. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 33.Dryden K A, Wang G, Yeager M, Nibert M L, Coombs K M, Furlong D B, Fields B N, Baker T S. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao Y, Olson N H, Xu W, Anderson D L, Rossmann M G, Baker T S. Cell. 1998;95:431–437. doi: 10.1016/s0092-8674(00)81773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuller S D, Butcher S J, Cheng R H, Baker T S. J Struct Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 36.Kleywegt G, Jones T A. ESF/CCP4 Newsl. 1993;28:56–59. [Google Scholar]
- 37.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Morrison M E, He Y-J, Wien M W, Hogle J M, Racaniello V R. J Virol. 1994;68:2578–2588. doi: 10.1128/jvi.68.4.2578-2588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Ryu S E, Kwong P D, Truneh A, Porter T G, Arthos J, Rosenberg M, Dai X, Xuong N, Axel R, Sweet R W, et al. Nature (London) 1990;348:419–426. doi: 10.1038/348419a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Yan Y, Garrett T P J, Liu J, Rodgers D W, Garlick R L, Tarr G E, Husain Y, Reinherz E L, Harrison S C. Nature (London) 1990;348:411–418. doi: 10.1038/348411a0. [DOI] [PubMed] [Google Scholar]
- 42.Lewis J K, Bothner B, Smith T J, Siuzdak G. Proc Natl Acad Sci USA. 1998;95:6774–6778. doi: 10.1073/pnas.95.12.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]