

Abstract
Evidence is presented which supports the conclusion that the hormetic dose–response model is the most common and fundamental in the biological and biomedical sciences, being highly generalizable across biological model, endpoint measured and chemical class and physical agent. The paper provides a broad spectrum of applications of the hormesis concept for clinical medicine including anxiety, seizure, memory, stroke, cancer chemotherapy, dermatological processes such as hair growth, osteoporosis, ocular diseases, including retinal detachment, statin effects on cardiovascular function and tumour development, benign prostate enlargement, male sexual behaviours/dysfunctions, and prion diseases.
Keywords: biphasic, dose–response, history of medicine, hormetic, hormesis, J-shaped, U-shaped, anxiolytic, seizure, memory, stroke, prion, prostate, biophosphonates, statins, erectile dysfunction, protein folding, retinal detachment, tumor cell proliferation
Introduction
The following paper will make the case that the hormesis concept is the most fundamental dose–response in the biomedical and toxicological sciences. Over the past decade there has been a remarkable surgence of interest in hormesis as a result of more significance being given to low dose effects and the use of more powerful study designs which have permitted the detection of the hormetic biphasic dose response in the low dose zone. This paper will establish the occurrence of hormesis within the biomedical literature, its quantitative features, mechanistic foundations, and applications to the field of clinical pharmacology. The hormetic dose–response challenges long-standing beliefs about the nature of the dose–response in a low dose zone and has the potential to affect significantly the design of pre-clinical studies and clinical trials as well as strategies for optimal patient dosing in the treatment of numerous diseases. A detailed historical assessment of how and why the hormetic dose response became marginalized in the biomedical literature was published by Calabrese in 2005 [1].
The hormetic dose–response relationship
We created an hormesis database for articles published in the peer-reviewed literature using a priori evaluative criteria in order to assess more systematically and objectively this concept. The criteria take into account the strength of the study design features, magnitude of the low dose stimulation, statistical significance and reproducibility of the findings. To date there are approximately 8000 dose–responses within this relational retrieval database. A detailed description of the database was published in 2005 [2].
The hormetic dose–response may be reliably described as a being a stimulation in the low dose zone, followed by an inhibitory response at higher doses. The magnitude of the stimulatory response at maximum is typically modest, being only about 30–60% above that of the control response (Figure 1). The strong majority of stimulatory responses are less than twice the control value. This is the most distinguishing characteristic of the hormetic dose–response, being its most consistent and reliable feature. The width of the stimulatory response is typically within 100-fold of the zero equivalent point, that is, the dose where the response changes from stimulation to inhibition, (i.e. the threshold value). In a small proportion (i.e. <2%) of the hormetic dose responses analyzed to date, a very broad stimulatory dose–response range has been noted, exceeding 1000-fold. The implications of having a wide stimulatory zone may be clinically significant. For example, the stimulatory zone defines the therapeutic window. It may also define an adverse effect window, as in the case when low doses of anti-tumour drugs stimulate tumour growth [3]. It is also important to recognize that the hormetic stimulatory zone is graphically contiguous with the pharmacologic/toxicologic threshold. This indicates that there is the distinct possibility of a desired therapeutic dose being a toxic dose to some individuals due to extensive interindividual variation.
Figure 1.
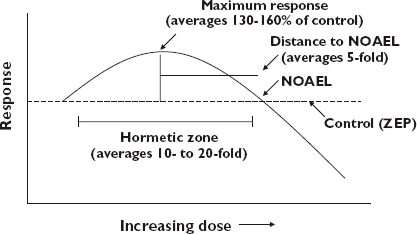
Dose–response curve showing the quantitative features of hormesis
The hormetic dose–response can occur (i) as a direct stimulatory response, (ii) after an initial disruption in homeostasis followed by the modest overcompensation response, or (iii) as a response to an ‘adapting’ or ‘pre-conditioning’ dose that is followed by a more massive challenging dose. The relationship of hormesis to the adaptive response was explored by Davies et al. [4] who defined the optimal condition for a transient hydrogen peroxide adaptation as measured by cell viability in the yeast model S. carerviase. In a critical first step, the authors determined the effect of hydrogen peroxide employing up to nine concentrations and a variety of cell densities. Of particular note was the observation of an hormetic-like biphasic dose-response in which low hydrogen peroxide concentrations (≤0.4 mm) enhanced cell colony growth by approximately 30%. The hydrogen peroxide-induced toxicity started to occur between 0.5 and 0.8 mm. Based on these findings an adapting or preconditioning dose was selected to be one in the low dose stimulatory/hormetic zone. The yeast cell that received the adapting dose in the hormetic zone followed by the challenging (i.e. cell killing) dose not only showed the adaptive response but also displayed a percent viability that exceeded the original control value by approximately 20–50%.
Since the quantitative features of hormetic dose–responses are similar regardless of the biological model, gender, endpoint measured and inducing agent as well as whether it occurs via direct or overcompensation stimulation, it suggests that such features maybe a quantitative index of the plasticity of biological systems. If this is the case, it would have important implications for the field of clinical pharmacology by placing biological constraints on the magnitude of the increase in performance one could expect from drug treatments.
Since the hormetic effect is one that is highly generalizable across biological models, it suggests that this response strategy has been highly selected for, indicating that the hormetic dose–response is adaptive in nature. This is particularly seen in the context of adaptive/preconditioning responses where a prior exposure to a low dose of a toxic agent or stressful condition up-regulates adaptive mechanisms that protect against subsequent exposures to similar toxic agents or stressor conditions. The duration over which such protection occurs will vary according to the biological model and endpoint. However, it usually does not extend beyond about a 10–14 day period [5].
Since hormetic dose–responses are highly generalizable, occurring in essentially all species of plants, microbes, invertebrates and vertebrates, in all organ systems, and for a large number of endpoints, there is a generalized mechanistic strategy but no single mechanism. The 1977 paper by Szabadi [6] suggests one such general strategy which may account for numerous cases of hormetic-like biphasic dose–response relationships. In this case, a single agonist may bind to two receptor subtypes, with one activating a stimulatory pathway while the other activates an inhibitory one. The receptor subtype with the greatest agonist affinity would typically have the fewer receptors (i.e. lower capacity) and its pathway activation effects dominate at lower doses. Conversely, the second receptor subtype would have lower agonist affinity, greater capacity (i.e. more receptors) and become dominant at higher concentrations. This generalized scheme of Szabadi [6] has been supported [7–9] over the past three decades and is able to account for numerous cases of direct stimulation hormetic dose–response relationships. It may also be directly applicable to situations in environmental toxicology in which the toxin induces dose-dependent changes in the concentrations of various endogenous agonists, a situation which is known to commonly occur. In such cases one would readily expect the occurrence of an hormetic-like biphasic dose–response relationship.
Hormesis and interindividual variation
A principal concern in assessing the effects of drugs on humans is that of inter-individual variation. Numerous factors are known that contribute to such variation, including age, familial background, gender, nutritional status, the presence of pre-existing disease, amongst other factors. Using the hormesis database we subsequently identified a substantial number of experimental settings in which hormesis had been studied in individuals or closely related strains of organisms which differed in susceptibility to toxic agents. In these evaluations we compared responses where the range in susceptibility varied from less than 10 fold to well in excess of 100 fold [10]. Of particular note was that the hormetic response was generally independent of susceptibility, with hormetic responses occurring in subjects ranging from high to low susceptibility. Likewise, the quantitative features of the hormetic dose–response are independent of susceptibility. In about 20% of the cases, it appeared that the lack of an hormetic response in a susceptibile strain or subgroup was related to its increased risk. These observations have important implications in the development of treatment strategies for patients.
Drug interactions
Hormesis contributes a new dimension to the concept of chemical interactions. Hormetic dose–responses describe that portion of the dose–response that relates to performance, that is, the portion of the dose–response immediately below the threshold. This is in contrast to the portion of the dose–response above the threshold, the location where most examples are drawn for illustration purposes to demonstrate the potential magnitude of toxic interactive effects [11]. The magnitude of the hormetic dose–response interaction will also be constrained by the bounds of biological plasticity. The magnitude of the hormetic interaction will also be only 30–60% greater than the control value. The interaction is principally seen within the context of the reduction in dose that is necessary to achieve a strong interaction response. This was described in considerable detail in several papers by Flood et al. [12, 13] dealing with memory. These authors recognized that one of the important implications of these findings was that this would reduce the likelihood of adverse effects because of the very low doses of drugs needed to achieve the therapeutic effect.
Medical implications of hormesis
Anxiolytic drugs
The animal model testing of anti-anxiety drugs became extensive in the 1980s [14–18] with progressive methodological advances continuing to the present. In the screening of potential drugs the goal has been to reduce anxiety in the mouse or rat model in specific experimental settings. While there are many ways in which this problem has been studied, the basic strategy is to assess whether the drug treatment can result in the mouse and/or rat performing specific behaviours that they would normally resist or not be inclined to do. In rodent experiments it can be trying to get the animals to spend more time in lighted as compared with dark areas. The possible anxiolytic agent may be tested in a maze like apparatus in which there is choice between entering and exploring a dark or a lighted alley. If the drug increases the time spent in the lighted alley or the number of entries into the lighted alley per unit time then the drug would be judged as anxiolytic. There are more than a dozen commonly employed anxiolytic tests (e.g. elevated plus maze test, hole board test, light-dark test, social interaction test, four plates test, open-field test, staircase test, conflict test, forced swimming test, tail suspension test) for screening drugs using animal models that address a wide range of behaviours that would be indicative of anxiety, relating to dark and lightness, social interactions, social conflicts, and a variety of other aversion behaviours, all directed toward slightly different manifestations of anxiety. While the basic premise is to determine if the drug can make the animal do that which is uncomfortable, each test is unique, providing an evaluation of somewhat different behaviors.
The most common dose–response relationship seen for the broad spectrum of anxiolytic drug screening tests is the hormetic model (see [11, 211] for a review), regardless of the chemical class, strain of the animal model or gender tested [19]. Therefore, anti-anxiety drugs regardless of their class will generally not increase the so-called ‘anti-anxiety’ response (i.e. decreased anxiety) by more than about 30–60% at the optimal dose. There can be large differences in the potency of anxiolytic drugs, sometimes differing over several orders of magnitude of dose. However, the general quantitative features of the hormetic dose–response are the same regardless of the potency of the agent (Figure 2).
Figure 2.
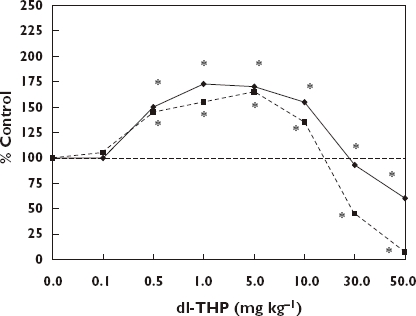
Anxiolytic effect of dl-THP, a naturally occurring alkaloid, on ICR mice of both sexes in the elevated plus-maze test. *Significantly different from controls at P ≤ 0.05 [207]. % Open arm entries, ( ); % Time in open arm (
); % Time in open arm ( )
)
Anxiolytic drugs have been shown to act through a wide range of receptors (e.g. 5-HT, dopamine, adenosine, GABA, NMDA) that mediate the response, thereby reducing anxiety via a variety of proximate mechanisms. Despite the fact that these drugs reduce anxiety by different proximate mechanisms they still show the same hormetic dose–response relationship, with the same quantitative features.
The hormetic dose response therefore is an important feature by which anxiolytic drugs act, being the basis for why the drug was selected for clinical evaluation, as well as being independent of animal model, gender, potency, and mechanism of action. This is a powerful set of parameters that converge around the hormetic dose–response relationship making it a central concept in the discovery and assessment of anxiolytic drugs.
Anti-seizure drugs
Another example where hormetic biphasic dose–response relationships have played an important role in drug discovery concerns anti-seizure medications. As in the case of anxiolytic agents, seizure drugs also undergo a screening process to eliminate the poorer performing agents and to identify those with clinical potential. One way that this is achieved is to induce seizures in animal models with standard seizure-inducing agents [e.g. pilocarpine, flurothyl(hexafluorodiethyl) ether, kainic acid, and pentylenetetrazol (PTZ)]. When this is done the researcher determines the dose required to induce a certain frequency of seizure events within a specific period of time. Drugs thought to have good potential as anti-seizure agents would be those that demonstrate the capacity to increase significantly the threshold dose of the model drug that causes seizures. That is, if the threshold dose for inducing seizures in the animal model is 100 mg kg−1, then a potential anti-seizure drug would make it more difficult for that model drug to induce the seizure response, that is, requiring even higher doses (i.e. increasing the response threshold).
The hormesis concept directly relates to how anti-seizure agents are detected in the above screening process. In the course of evaluating anti-seizure agents investigators typically assess the agent across a broad range of doses. In such evaluations anti-seizure agents at low doses increase the seizure threshold of the model seizure-inducing drug while at higher doses the anti-seizure drug typically enhances the occurrence of seizures by lowering the threshold of response. In Figure 3 morphine induced an hormetic-like biphasic dose–response on the PTZ-induced seizure threshold. Note that the threshold dose for the PTZ induced seizures increased by approximately 25%. In essence, these anti-seizure drugs follow the pattern of the hormetic biphasic dose–response relationship (see [11, 212] for a review). The extent to which the threshold increases also conforms to the quantitative features of the hormetic dose–response, usually increasing at maximum only within the 30–60% zone above the control.
Figure 3.
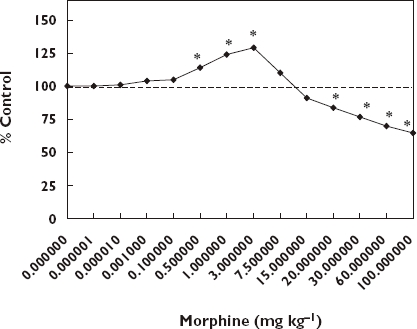
Effect of different doses of morphine on PTZ-induced seizure threshold. *Significantly different from controls at P ≤ 0.05 [208]
Learning and memory enhancement
The first major efforts to explore whether drugs could enhance learning in animal models were undertaken at the University of California at Berkeley in the Department of Psychology during the 1960s. While these efforts extended earlier preliminary investigations at the University of Chicago and elsewhere, the Berkeley group created a new research direction that lead to the development of valuable drugs in the treatment of cognitive disabilities as seen with Alzheimer's (AD) and related diseases of ageing. In fact, the initial breakthrough was undertaken by then two graduate students (James McGaugh and Lewis Petrinovitch), who hypothesized that memory was related to the concentrations of acetylcholine released by the neurons. With this guiding framework these students tried to determine why some mice were bright (i.e. smart) and others were dull (i.e. not so smart). To test this hypothesis they administered a drug over a broad dose range to the bright and dull mice that would prevent the normal breakdown (i.e. hydrolysis) of the acetylcholine. The agent used to slow down the normal breakdown of acetylcholine was physostigmine, a natural constituent of the Calabar bean, with a long history of use in medicine, especially known to cause contraction of the pupils. The treatment was expected to make the dull mice brighter and the bright mice even brighter, but only up to a point, that is, when the dose exceeded a hypothetical optimal zone, triggering a decline in performance. Both the dulls and brights exhibited the characteristic U-shaped dose–response relationship (Figure 4), thereby confirming the study hypothesis. The manuscript based on these findings was rejected with the stated reason that it was well known that chemical intervention could only decrease learning and memory, not enhance it. The two students persevered, publishing their paper several years later, opening up a new era in the psychology and pharmacology of learning and memory research.
Figure 4.
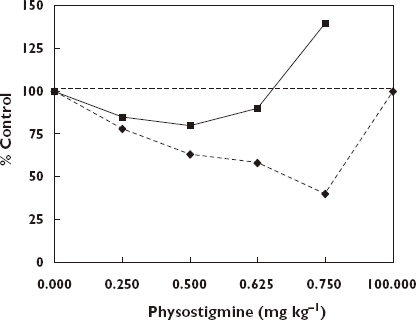
Median trials to criterion for maze bright and maze dull groups [209]. Brights, (); Dulls, ()
The work of McGaugh and Petrinovitch propelled an intellectual revolution in the neurobehavioural sciences for understanding of memory and the exploration of therapies for those with cognitive dysfunctions. The idea that one could improve learning by inhibiting the breakdown of acetylcholine was important as it established an intellectual platform for subsequent research. This was also occurring at a time when organophosphate and carbamate insecticides were being developed as agents to kill insects, with both groups of chemicals inhibiting the enzyme that hydrolyzed acetylcholine. In fact, in some of the subsequent research it was shown that administration of some of these agents appeared to increase learning and memory in the rodent models. While it has never been seriously proposed that such pesticides could be used to boost memory in people, much work was undertaken with physostigmine in clinical settings [20–50]. During this period of little more than 15 years there were about 20 AD clinical studies published involving several hundred individuals. The findings consistently noted a modest (i.e. 10–30% range, generally) improvement in various types of memory. Despite the capacity of physostigmine to enhance memory performance, it was generally accepted that the modest increases did not offer enough change to provide dramatic or often even practical improvements in patients’ lives, although there were some reports where greater degree of independent living was seen. This is especially the case given the pharmacokinetic features of this drug, with its very short half-life, requiring up to five doses per day for most subjects.
While these agents pointed the way for future research, there was the need for second and third generation agents, all with markedly improved pharmacokinetics profoundly lowering risks of side effects. Amongst those drugs extensively evaluated in this regard have been tacrine [13], heptylphosphostigmine [51], huperizine A [52, 53], arecoline [12] and gastigmine [54], all being based on an anti-ACHE concept following the premise that McGraugh and Petrinovich employed with physostigmine. The dose–response relationship for each drug was repeatedly demonstrated to be a U-shaped in multiple animal models much like that seen in Figure 4. Such findings lead to the general conclusion that the therapeutic window for such drugs in the treatment of AD patients followed the U-shaped dose–response that was causally linked to the percent of ACHE inhibition. However, more recent investigations have revealed that the neuroprotection is likely to involve multiple mechanisms, either in addition to ACHE inhibition or independent of it. Regardless of the underlying mechanistic explanation and how it may differ amongst protective agents, the quantitative features of the dose–response remain similar (see [213] for a review).
Four (i.e. Aricept, Cognex, Exelon and Razadyne) of the five drugs that have been approved for the treatment of AD by the US FDA are based on the inhibition of ACHE and demonstrate the hormetic dose–response, with the low dose stimulation being key to the increased memory improvement. The fifth approved drug, memantine, an NMDA antagonist, also acts via an hormetic-like inverted U-shaped dose response relationship [55]. Yet it is also important to recognize that the constraints of the hormetic dose–response indicates that the expected increase in performance is likely to be modest at best, observations confirmed with substantial clinical experience.
Anti-tumour drugs
A major goal of anti-tumour drug screening is to find agents that are effective in killing a broad range of tumour cell types. Those who focus on such concentration responses are typically estimating responses at the high end of the concentration–response relationship, then trying to determine the mechanism by which the killing occurred. While acknowledging the critical importance of this perspective, our interests with respect to hormesis are different, that is, focusing on the low concentration end of this relationship, that is, the zone starting immediately below the toxic threshold.
In a recent assessment Calabrese [3] provided substantial documentation that low doses of anti-tumour agents commonly enhanced the proliferation of the human tumour cells, in a manner that was fully consistent with the hormetic dose–response relationship. These hormetic dose–responses were occurring for most types of tumour cells, independent of organ. That is, Calabrese [3] reported hormetic dose–responses in 138 in vitro cell lines, and 32 primary human types induced by over 120 agents with nearly 50 being endogenous agonists while others involved the effects of various drugs, phytochemicals, and environmental contaminants.
While many of the researchers did not focus on the low dose stimulatory responses provided in their tables and figures, choosing to address only the higher concentration effects, others not only noted these findings but attempted to account for them in follow up mechanistic studies. In some instances there was a stronger interest in the clinical implications of such findings, suggesting that the low dose stimulation of human tumour cell proliferation could be potentially problematic for patients who have been treated with chemotherapeutic agents. A case in which the implications of low-dose stimulation of tumour cell growth has been extensively assessed is that of dexamethasone and neuroepithelial tumours. Dexamethasone has long been employed to prevent swelling in the brain after removal of a brain tumour [56, 57]. While this treatment is important in the management of such patients, a number of in vitro experimental studies using dexamethasone have consistently demonstrated that specific types of neuroepithelial tumours proliferate with dose–response features consistent with the 30–60% maximum stimulatory response. This low-dose enhanced response required the presence of a glucocorticoid receptor. Lacking such a receptor prevents the dexamethasone induced low-dose stimulatory response [58, 59].
Upon the publication of a paper by Kawamura et al. [60] assessing this type of biphasic response, the journal editor invited expert commentaries from other leading neuroepithelial surgical researchers on the implications of treating surgical brain tumour patients with dexamethasone [61–65]. Despite concerns raised by the experimental data and the expert commentaries, it was not clear what the tumour growth implications would be following surgery. No attempts have been published providing quantitative modeling of various possible clinical scenarios.
If tumour cells remain after treatment these data suggest the possibility that some chemotherapeutic agents could promote tumour growth once the concentration of agent entered into the low dose stimulatory zone. This would be a particular concern for agents (e.g. suramin) (Figure 5) with a very long half-life [66], while being less of a problem for agents with very short half-lives. As with other examples of hormetic dose–responses, the magnitude of the enhanced proliferation was typically in the 30–60% range at maximum. What implications this might have for tumour growth over a longer time period has not been assessed.
Figure 5.
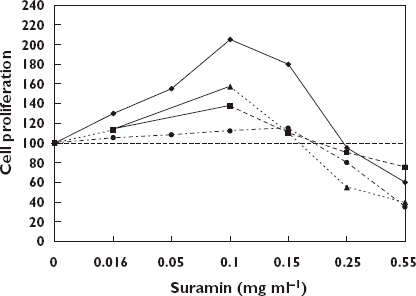
Effect of suramin on human breast cancer cells in vitro[66]. ZR/HERc Cells, (); ZR75.1 Cells, (); T-47D Cells, ( ); MDA-MB-231 Cells, (
); MDA-MB-231 Cells, ( )
)
The hormetic concentration–response observations in the human tumour cell lines in the peer-reviewed literature is mirrored in the US NCI anti-tumour agent databases. There are two databases that we have studied in some depth, the human tumour cell line database which is comprised of at least 60 different tumour cell lines representing tumour types from many areas of general clinical significance. The second database is comprised of 13 strains of yeast that have genetic defects relating to DNA repair and cell cycle control.
We have published a detailed analysis of the yeast data set that contains over 56 000 concentration–response studies [67]. This evaluation tested whether the hormetic or the threshold dose–response model best predicted the responses of the chemicals in the low dose zone. These analyses revealed that the threshold dose–response model poorly predicted responses at low concentrations. On the other hand, the hormetic dose response model performed very well, accurately predicting low concentration responses. The findings suggested that clinical oncologists need to become more cognizant of the possibility that treatments designed to kill tumour cells or suppress their proliferation in patients may have the capacity to enhance tumour growths when the drug eventually reaches a low (i.e. stimulatory) concentration in the body, in the days after the chemotherapy is administered.
Stroke and traumatic brain injuries
Given their potentially debilitating consequences strokes are a major public health concern. Considerable research has been devoted to enhance the understanding of the causes of strokes within the population in order to reduce their frequency and severity. Familial history, high blood pressure that is ineffectively treated, smoking, and excessive stress are some of the known risk factors affecting stroke.
These concerns have led numerous researchers to assess possible treatments of stroke with a wide range of animal models. Such studies, which typically involve the induction of one of a variety of specific types of stroke that occur with substantial frequency within humans, have often yielded findings of encouraging neuroprotection, only to fail once the agents were subjected to clinical trials, yielding a very low successful transition to the marketplace. There has been considerable discussion about why there has been such a disconnect between successful preclinical studies and failures of a large number of clinical trials. Amongst the many reasons for such failures in the stroke area include concerns over the validity of animal models to predict human responses in a sufficiently reliable manner, inadequately designed clinical trials, especially factors such as the entry criteria for patients may lack the necessary precision for proper comparisons, the practical problems of long delays between the onset of stroke symptoms and therapy, amongst other factors. A factor that has often gone unnoticed is the dose–response relationship of the therapeutic agent. In fact, as in many other medical conditions, the shape of the dose–response of the treatment can often take that of a U-shape, that is, showing characteristics of an hormetic dose–response. However, this type of dose–response is not usually seriously considered as far as the clinical trial is concerned. Since the biomedical and clinical domains have been so long dominated by the assumed sigmoidal nature of the dose–response, investigators have often assumed that the response for a drug may be enhanced by pushing the dose ever higher, up to a point when the response tends to flatten out [68].
Despite the fact that U-shaped dose–responses have been reported in stroke related preclinical investigations this information had never been integratively summarized, until recently [69]. U-shaped dose–responses have been reported for nearly thirty different agents in the treatment of stroke related brain damage [70, 71] and in studies directed to blunt traumatic brain injury [72, 73], both of which share similar common damage-inducing mechanisms. Different drugs that have been successfully employed in these studies are usually selected based upon a certain hypothesized mechanism by which stroke or traumatic damage is mediated. If the drug can turn off the damage mechanism switch or turn on a repair mechanism switch that could alleviate the damage then protection would be observed. Since there are many stages in the process leading to final tissue damage, intervention at separate and key points in this scheme would prevent significant damage from occurring. That is in fact what has occurred in these studies. Many experimental plans have worked well, leading to damage reduction, and essentially all displayed the same type of hormetic dose response regardless of where in the disease process the intervention occurred.
Since the U-shaped dose–response is commonly observed in these chemoprevention studies such as discussed above, it needs to be anticipated better and explored in order to enhance the success of getting new and effective therapeutic agents through the testing and evaluation process.
Benign prostate hyperplasia/cardiac glycosides
The use of digitalis in the treatment of congestive heart disease has been one of considerable historical interest with its first reporting in 1775, being credited to the British physician William Withering [74–76]. Over time it was determined that cardiac glycosides, such as digitalis, most likely acted by blocking the sodium pump (i.e. Na+/K+-ATPase) at dosages that are achieved in vitro or intravenously. Biomedical understandings of the dose–response for these agents on the sodium pump and their broader range of effects started to become considerably more complex in the mid 1970s. At this time investigators reported that cardiac glycosides affected the sodium pump biphasically, that is, stimulating activity at low concentrations while being inhibitory at higher concentrations [77]. These initial findings revealed the existence of high and low affinity binding sites, with the high affinity sites having only 3% of the capacity of the low affinity one. The binding of agents, such as ouabain, to both high and low-affinity sites has been linked to the sodium pump and/or functionally distinct Na+/K+ pumps and proposed to account for its biphasic dose–response activities, findings that have been subsequently reinforced [78, 79].
Of particular interest is that cardiac glycosides were also reported to enhance proliferation of a broad range of cell types in a biphasic fashion, starting in the mid 1970s for immune cells [80, 81]. The range of biphasically affected cell types includes smooth muscles from the canine saphegeous vein [82], smooth muscle from the human umbilical vein [83], smooth muscle from the adult prostate of subjects with benign prostate hyperplasia (BPH) [84], smooth muscle from a rat cell line (A7r5) which was selected for study due to a specific protein component of the sodium pump [83], mature human red blood cells which were selected for study due to membrane properties [85], rat renal epithelial cells [86] as well as HeLa cells which were selected for study due to their unique sensitivity to digitalis induced toxicity [87]. These diverse experimental studies have centered in large part on the development of improved understandings of the sodium pump and its capacity to function in ways that may not involve the well established ionic shift, suggesting a multiplicity of functions for the sodium pump, including diverse message signalling with broadly expanding clinical implications.
The quantitative features of the biphasic dose–response of cardiac glycosides indicate that they are fully consistent with the hormetic dose–response model. Figure 6 provides a description of the dose–response features of various cardiac glycosides in a range of biological models adjusted to the same scale for comparison purposes [83]. In each case the magnitude of the low dose stimulation is modest, with the maximum stimulation typically being in the 30–60% range above the controls. The width of the stimulatory response was also generally consistent across specific agents and biological models and usually less than 100-fold, again consistent with the hormetic dose–response.
Figure 6.
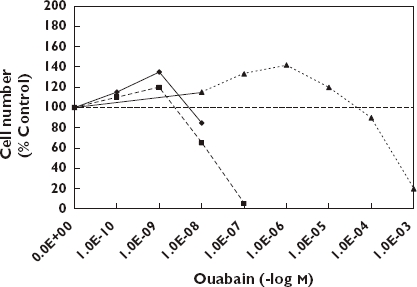
Comparison of the effect of ouabain on hormetic dose–responses in three biological models [83]. Canine (CVSMC), (); Human (HUVSMC), (); Rat (A7r5), ( )
)
Several of the above studies have obvious implications for clinical pharmacology. In 2001 Chueh et al. [84] reported that concentrations of ouabain in the therapeutic zone for cardiac glycosides enhanced cell proliferation of smooth muscle from the prostate gland of subjects with BPH. The magnitude of the increase was modest, approaching only about 20% above control values. However, these authors indicated that even such modest increases could have important clinical implications since such smooth muscle comprises about 40% of the area density of BPH tissues. Several reports have indicated that a small increase in prostate size significantly affects clinical symptoms in patients with BPH. In a similar fashion, a modest reduction in prostate volume of only about 30% has been repeatedly demonstrated to improve markedly clinical symptoms [88, 89]. Thus, the hormetic biphasic dose–response that was reported in these in vitro investigations is suggestive of possible clinical implications.
Studies with HeLa cells by Ramirez-Ortega et al. [87] revealed that four digitalis compounds biphasically affected cell proliferation, again in a manner consistent with the hormetic dose–response. In this case, the concentration ranges over which the stimulatory response range occurred were very broad and remain to be clarified. That is, the stimulatory response range was greater than 1000-fold, and could be within a concentration range approaching that of endogenous cardiac glycosides. While the biphasic dose–response was reported for four digitalis-like compounds it was not seen for digitalis when tested over similar concentrations due to its greater toxicity. Of particular mechanistic importance was the fact that the low dose stimulation occurred even in the presence of ethyacynic acid, a nonsteroidal inhibitor of Na+/K+-ATPase, thereby suggesting that the stimulation is independent of the sodium pump. Finally, even though the authors did not address the clinical implications of these findings it would appear that a broad range of digitalis compounds have the capacity to enhance cellular proliferation in numerous cell types including human tumour cell lines over a concentration range that encompasses the therapeutic zone as well as for possible normal background states.
Statins: CVD and cancer
Statins have become important drugs in the prevention of cardiovascular disease. They are also considered potentially significant in the treatment of numerous types of solid tumours and for other diseases (e.g. diabetic retinopathy and macular degeneration) that depend on capillary development. Some investigators suggest potential clinical application for statins in the treatment of AD, osteoporosis [90] and other diseases. A converging conceptual framework that is emerging on the effects of statins in this broad range of tissues and endpoints is that they often display an hormetic-like biphasic dose–response relationship, a response that was unexpected, initially overlooked, but now a factor for optimizing patient treatment strategies.
The idea that statins may act via biphasic dose–responses emerged with the publication in 2002 by Weis et al. [91]. Using primary human adult dermal microvascular endothelial cells (HMVECs) and an immortalized human dermal microvascular endothelial cell line (HMEC-1), these authors demonstrated that cerivastatin (CEV) biphasically affected endothelial cell (EC) proliferation, migration, and apoptosis (Figure 7). They also demonstrated that CEV/ATR (atorvastatin) significantly reduced lung tumour growth at high doses, a response probably related to a marked decrease in tumor capillary development. The low dose stimulatory response was consistent with their hypothesis that HMG-CoA reductase inhibition would enhance endothelial processes involved in angiogenesis and could reverse impairments associated with cardiovascular disease.
Figure 7.
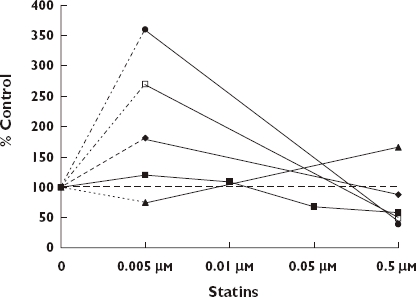
Effects of statins on human endothelial cells [91]. CEV EC Proliferation, ( ); ATV EC Migration, (
); ATV EC Migration, ( ); CEV Apoptosis, (); CEV Tube Length, (); ATV Tube Length, (
); CEV Apoptosis, (); CEV Tube Length, (); ATV Tube Length, ( )
)
The biphasic dose–response was independently confirmed about 2 months later by Urbich et al. [92] who had submitted their paper for publication consideration more than 2 months prior to the Weis et al. group. However, their paper was not accepted for publication until about 3 months after the Weis et al. paper. Using human umbilical vein endothelial cells (HUVECs) these authors also reported that ATR biphasically affected endothelial cell migration and tube formation. Similar findings were subsequently reported by Katsumoto et al. [93] with HMVECs for multiple endpoints including cell migration, chemotaxis, cell proliferation, trypan blue exclusion and others. Cooke [94], a co-author of the Weis et al. [91] paper, suggested that these findings indicate that statins may also enhance cardiovascular functioning by promoting angiogenesis in ischaemic limbs, that is, facilitating the remodeling of vessels toward a more normal and healthier condition. However, at higher doses these same agents display anti-angiogenic effects that are mediated by blocking L-mevolonate metabolism, reducing the synthesis of G-protein subunits. This process leads to an increase in apoptosis. The data of Weis et al. [91] and the other supportive findings noted above were striking, suggesting that statins could have ‘puzzling’ effects on angiogenesis – that is, proangiogenic effects at low therapeutic doses but angiostatic effects at higher doses [95].
Statin concentrations can broadly vary in the serum of patients, from a low of about 0.002 to a high of approximately 0.1 µmol l−1. The low concentration stimulation for endothelial cell parameters ranges from less than 0.001 to about 0.01 µmol l−1. Above 0.1 µmol l−1 high concentration effects start to become evident. The low concentration stimulatory effects (i.e. pro-migration, pro-angiogenic) of ATR were associated with phosphatidylinositol 3-kinase-AKT-pathway activation with phosphorylation of AKT and endothelial NO synthase (eNOS) at Ser 1177. Thus, the issue of whether statin concentrations are pro- or anti-angiogenic began to be raised and debated. However, from the available data it appears that the therapeutic concentration for lowering cholesterol concentrations spans the pro- and anti-angiogenic concentration continuum.
Proceeding along a separate intellectual track researchers interested in inhibitors of angiogenesis had not focused on the statins, which were principally seen as inhibitors of cholesterol synthesis. These researchers, following the lead of Folkman [96], identified a series of possible agents starting with interferon α (1980), plasminogen factor (1982) and some related hydrolyzed protein fragments, collagen derived endostatin and other endogenous agents, acting via a range of possible mechanisms, that inhibit angiogenesis. In the case of endothelial migration and other endpoints related to capillary development, such agents often displayed biphasic dose–response relationships. Furthermore, when tested in cancer bioassays, these anti-angiogenic agents generally displayed a U-shaped dose–response relationship [97]. In fact, even when tests failed to produce the expected U-shaped dose–response in cancer bioassays, as in the case of Eisterer et al. [98], it was most likely due to the treatment being at the high end of the U-shaped dose–response.
The mechanisms by which these U-shaped dose–responses occur has been generally viewed as ‘unclear’ [99], ‘uncertain’ [100], or ‘still to be elucidated’ [101]. Various possible mechanistic explanations have been suggested including the induction of receptor dimerization that could lead to biphasic effects or the induction of multiple signaling pathways that display an integrated U-shaped dose–response.
A complementary and/or alternative perspective on anti-angiogenic agents has been developed by Jain and colleagues [102] based on the proposal that normalization of tumour vasculature can mediate anti-angiogenic cancer therapy. This group has shown that tumour vasculature is very different from normal tissue. That is, tumorous vascular tissue can be very irregular with respect to bifurcation and the sizing of the bifurcating vessels. Vessel walls can be very tight or very leaky or intermediate. Pressure gradients within the vessels are also highly variable. The blood flow can also be very irregular with flow going in back and forth directions, or simply being stagnant. The oxygen concentrations are also usually very low, that is, hypoxic, with high acidity being common. This condition reduces the efficiency of immune cells to attack the tumour while enhancing the capacity of tumour tissue to metastasize.
Jain and colleagues [102] reduced the cancer angiogenesis capacity by using an antibody against VEGF, a promotional signal molecule abundant in most solid tumours. This treatment caused some of the aberrant tumor vessels to be ‘punned’ away. This treatment also affected a remodeling of the remaining tumour vessels toward a more normal state, becoming less leaky, dilated and tortuous. There were also higher concentrations of oxygen in the tissue and enhanced penetration of chemotherapeutic agents.
The idea of remodeling tumour blood vessels toward a more normal state seems contrary to the goal of Folkman [96, 97] to kill the tumours by preventing capillary development. The anti-angiogenic drug Avastin increased survival only in combination with standard chemotherapy, possibly because it pruned some of the aberrant blood vessels, remodeling the remaining ones, making them more susceptible to chemotherapy. Fukumura & Jain [102] believe that it is important to identify the time period when the vessels become remodeled in order to determine the optimum period of chemotherapy. The possibility exists that anti-angiogenic agent-induced pruning and remodeling may represent a biphasic dose–response, related to the findings of Weis et al. [91] and Urbich et al. [92] and later authors.
In summary, anti-angiogenic agents typically follow a U-shaped dose–response that displays the quantitative features of the hormetic dose–response. The U-shaped dose–response has become a common theme within the past 5 years of statin research, and is consistent with other effects of anti-angiogenic agents such as their effects on bone development [90]. While the mechanisms by which such hormetic effects occur for each endpoint are likely to differ, this should be seen as a basic regulatory strategy by which the biological system balances pro and anti-angiogenic signals within a framework of the constraints imposed by system-related biological plasticity.
Skin
UV light causes photo-oxidative reactions that damage the functional integrity of sub-cellular structures, cells and tissues, especially in the skin and eye. The mechanism by which UV induces skin cancer, premature ageing of skin and various opththalmological diseases such as cataracts and age-related macular degeneration involve the formation of reactive oxygen species. Within this context it is known that low level carotenoid ingestion increases the risk of cancer, aged-related macular degeneration, cataracts and CVD [103–105]. Experimental studies have also indicated that UV-induced skin damage can be reduced by carotenoid supplementation [106–109], findings generally attributed to their antioxidant activities.
Despite their antioxidant properties, carotinoids also display pro-oxidant effects [110–113] as judged by biomarkers of lipid per-oxidation. Pro-oxidant effects have been proposed to account for an enhanced risk of lung cancer observed in epidemiological studies with β-carotene. [104, 105, 114]. These studies revealed an incidence of lung cancer that was about 20% greater than control values when β-carotene was given in high doses for a prolonged period to those with an enhanced risk of this disease.
Studies with cultured human skin fibroblasts were undertaken to explore the capacity of carotenoids (i.e. lycopene, β-carotene, and lutein delivered to cells using liposomes) to show antioxidant and pro-oxidant effects following exposure to UV [115]. The cells were exposed to UV light for 20 min that increased MDA values from a background of 0.4 nmol to about 1.2 nmol. The three carotinoids decreased the UV MDA values at low concentrations while increasing MDA formation as the concentrations increased. While the magnitude of the decrease in MDA production was similar for each agent, the range of the protective responses was variable. Even though this was an in vitro study, the carotinoid concentrations in human tissues are in the range used in this study (Figure 8).
Figure 8.
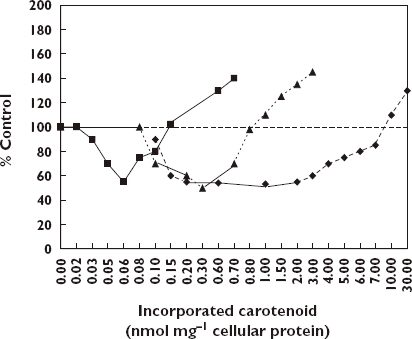
Effects of lycopene, beta-carotene, and lutein on UVB-induced TBARS formation in human skin fibroblasts [115]. Lycopene, (); Beta-carotene, (); Lutein, ()
While each carotinoid displayed the J-shaped dose–response there are notable differences with respect to the concentrations of carotinoids where the protection is initiated, ceases and where toxicity begins. These authors suggested that there are optimum levels of protection in vivo, which would like vary by individual.
Minoxidil and human keratinocytes
While attempts to grow hair have had a long history, a major breakthrough occurred in 1980 when Zappacosta [116] reported that a systemic anti-hypertensive agent, minoxidil, enhanced the growth of hair in patients. Over the next three decades minoxidil emerged as the most widely used drug for the treatment of androgenetic alopecia (AGA). While the mechanism(s) by which minoxidil increases hair growth remain to be elucidated more fully, the general emerging perspective suggests that it involves the restoration of normal keratinocyte proliferation [117, 118] via the regulation of calcium channels by its metabolite minoxidil sulfate. Particularly comprehensive in this regard have been the studies of Boyera et al. [117] involving a systematic investigation of the effects of minoxidil on human keratinocytes from different donors using different biological sources (i.e. interfollicular keratinocytes, follicular keratinocytes from microdissected hairs or plucked hairs), a range of experimental conditions (e.g. high- and low-calcium medium, with or without serum, high or low epidermal growth factor concentration) using multiple but complementary endpoints to assess cellular proliferation (i.e. mitochondrial dehydrogenase activity, BrdU incorporation, lysosome content, protein content, lactate dehydrogenase released, and involucrin expression). Regardless of the specific experimental conditions assessed, minoxidil induced biphasic concentration responses with stimulatory effects at low (i.e. micro-molar concentration) and anti-proliferative, pro-differentiative and partial cytotoxic effects at higher concentrations (i.e. millimolar concentrations). Figure 9 illustrates this general relationship in a comparison of mitochrondrial dehydrogenase activities (i.e. biomarker for the number of viable cells) relative to controls across a range of experimental systems. Similar qualitative and quantitative concentration relationships also were reported for BrdU incorporation (i.e. marker of proliferative cells in S phase), neutral red (i.e. marker for the number of viable cells) and BioRad protein (i.e. biomarker for the total number of cells). The high degree of consistency of the concentration–response relationships for complementary endpoints strongly supported the overall reliability of the findings. The concentration–response relationships generally indicated that the maximum stimulatory responses regardless of endpoint were modest, being in the 15–30% range greater than the controls. The stimulatory concentration range is also consistent across endpoints and experimental systems, being approximately 10- to 100-fold.
Figure 9.
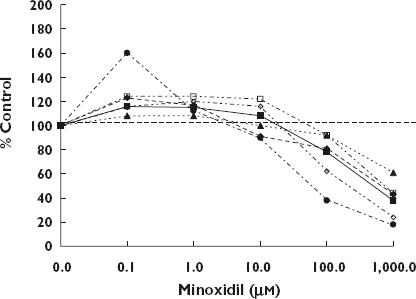
Effect of Minoxidil on normal human keratinocytes [117]. 1st Passage Epidemal NHK – high, (); 2nd Passage Epidemal NHK – low, (); 1st Passage Follicular NHK – high microdissected, (); 1st Passage Follicular NHK – High plucked, (); Follicular NHK – Low, (); NHK Proliferation, ( )
)
The quantitative findings of the in vitro investigations of Boyera et al. [117] are remarkably consistent across the broad range of experimental protocols and suggest the possibility of providing mechanistic insight of clinical findings. In fact, the maximum plasma concentrations of minoxidil with a hair growth promoting dose is approximately 0.775 µm, a concentration seen to consistently induce stimulatory cell proliferation responses in the Boyera et al. [117] study. Therefore, the minoxidil treatment at low concentrations would be expected to maintain keratinocyte proliferation in conditions such as AGA while conconmitantly preventing premature commitment of cells to differentiative pathways. However, Boyera et al. [117] raised the possibility that minoxidil may accumulate in some follicle compartments, such as the hair shaft which has particularly high concentrations of keratins and melanins. Such millimolar concentrations of the drug could occur adjacent to the keratogenic zone, thereby supporting keratinocyte differentiation along with hair shaft thickening.
Bone
Bisphosphonates
Osteoporosis is a major public health issue for ageing women. In the US alone nearly 10 million women over 50 years of age have osteoporosis with nearly double that many being at risk because they have low bone mass [119]. Bisphosphonates can prevent bone resorption and therefore have been employed in the treatment of osteoporosis. Bisphosphonates are synthetic analogues of pyrophosphate where a carbon atom replaces the oxygen at the center of the pyrophosphate. This chemical substitution results in the bisphosphonate becoming resistant to hydrolysis. It permits two additional side chains (R1, R2) of potential variable structure. The R1 side chain contains an hydroxyl moiety leading to high affinity for calcium crystals and bone mineral. The chemical differences at R2 have been exploited for the development of bone anti-resorptive potency [120, 121]. Despite their successful chemical applications, mechanistic understanding of bisphosphonates has emphasized their direct inhibitory effects on mature osteoclasts [122, 123]. However, in addition to the inhibitory effects on osteoclastic bone resorption, Giuliani et al. [124] explored whether the chemical effectiveness of these agents may result from a direct effect on osteoblasts, thereby representing an alternative target for bisphosphonate-induced beneficial effects on the bone formation process. In their investigation Giuliani et al. [124] evaluated potential effects of etidronate and alendronate on the formation of early and late osteoblastic cell precursors by measuring the number of colony-forming units for fibroblasts (CFU-F) and colony-forming units for osteoblasts (CFU-OB) in murine and human bone marrow cultures. Using aspirates from the femurs of three-month old Swiss-Webster female mice, 10 to 12 treatment concentrations over nine orders of magnitude were employed along with controls. In the mouse marrow cultures, etidronate (10−5–10−9 mol l−1) enhanced the formation of CFU-F by 50–100% more than the controls. The alendronate treatment displayed a biphasic effect with stimulation occurring below 10−7m with inhibitory responses occurring at higher concentrations. The maximum stimulatory response was 78% greater than controls. In the case of CFU-OB (i.e. mineralized nodule formation) both compounds displayed biphasic concentration responses with stimulation occurring at low, inhibition at higher concentrations. A comparable concentration–response relationship was also reported for CFU-OB in human bone marrow cells following alendronate treatment. The two lowest concentrations of alendronate which were effective in the formation of the early osteoblast precursors in this in vitro study were generally equivalent to those doses (5–10 mg day−1) employed to treat patients with osteoporosis [125, 126].
In addition to the in vitro testing the authors also assessed the effects of both agents on CFU-F formation in young (3 months) and old (18 months) mice. There was good agreement between the in vitro and in vivo studies with low doses being stimulatory for both agents and each age group [124].
The mechanisms by these agents enhance the formation of early and late osteoblast precursors suggest an involvement with basic fibroblast growth factor (bFGF-α) [124]. The production of bFGF-α, a powerful mitogen for mesenchymal cells, increased by 50% with alendronate treatment over the same concentration range (10−8–10−12 mol l−1) capable of stimulating osteoblastogenesis in vitro (Figure 10) [127]. bFGF-α stimulates the proliferation of CFU-F and the formation of mineralization nodules in both animal and human cultures [128–132]. These findings suggest that bisphosphonates are most likely affecting an increase in the number of mesenchymal bone marrow cells committed to the osteoblast phenotype. Thus, the overall effectiveness of these compounds may represent a beneficial influence of osteoblast precursors to complement the previously recognized high dose inhibitory effect on osteoblastic bone resorption.
Figure 10.
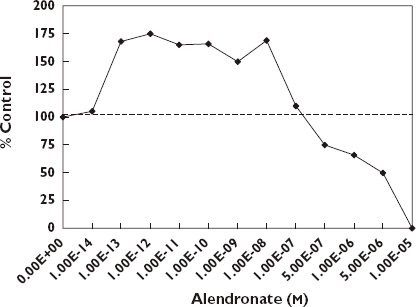
Effects of alendronate on CFU-F formation in murine bone marrow cells [124]
Since the ground breaking study of Giuliani et al. [124], a series of independent investigations have confirmed their basic conclusions, while generalizing the bisphosphonate induced biphasic dose–response on osteoblasts to a broader array of compounds, experimental models and endpoints. The quantitative features of the dose–response were remarkably consistent, including not only the magnitude and width of the stimulatory response but also absolute tissue sensitivity. That is, the response range for the CFU-F and CFU-OB for the female Swiss-Webster mouse bone marrow cells was generally consistent with MG-63 human osteoblast [133] and human osteoprotegerin production [134]. While the consistency in the quantitative features of the hormetic dose–response was not unexpected, the striking similarities in the absolute sensitivity across tissues to a range of bisphosphonates was unexpected and has not been previously discussed. Furthermore, the stimulatory range over which the osteoblast-proliferation occurs is extraordinary, being well over a 10 million-fold, and likely even wider. No mechanistic foundation to account for the broad stimulatory range has been offered (Figure 10).
Of relevance to the above assessment of bisphosphonate-induced biphasic dose–responses is that the molecular mechanism of the biphosphonates differ between their two structure subgroups, that is, those with single substrates (e.g. H, OH, Cl, CH5) and lacking nitrogenation, and those with common substituents being a hydroxyl group together with a nitrogen-containing aliphate side chain or heterocyclic rings. Non-nitrogen compounds such as etidronate appear to form cytotoxic ATP-analogues following interactions. The nitrogen-based compounds inhibit farneyl diphosphosphate synthase, an enzyme of the mevalonate pathway, thereby reducing the prenylation of small GTP-binding proteins needed for normal cell function and survival. However, despite such marked differences in mechanism both groups biphasically induced osteoblast proliferation, suggesting other mechanisms that are shared [120].
A further possible mechanism by which nitrogen containing bisphosphonates may prevent bone resorption is via an anti-angiogenesis process as reported by Wood et al. [135] with the drug zolerdronic acid (ZOL) using multiple endpoints. These authors noted the osteoclastic bone resorption depends upon efficient vascularization of the haemangiogenic endothelials cells. The Wood et al. [135] study also reported a biphasic effect of biphosphonates [e.g. ZOL, pamidronate (PAM)] on endothelial cell adhesion consistent with the quantitative features of the hormetic dose–response, thereby suggesting a more complex and integrative biological response.
Statins
In 1999 Mundy et al. [136] were the first to report that a number of statins, in particular simvastatin and lovastatin, stimulated bone formation following a subcutaneous injection over the murine calvaria. This same treatment enhanced the expression of BMP-2 mRNA in osteoblasts. Since these findings were confirmed and extended to bone diseases, such as osteoporosis [137, 138], Yazawa et al. [139] explored whether statins could affect the regeneration of periodontal cells that affect hard tissue regeneration. Using human periodontal pigment (PDL) cells these investigators observed the capacity of simvastatin to biphasically enhance the proliferation of PDL cells (24 h after treatment) in vitro as well as alkaline phosphatase (ALP) activity (7 days after treatment). Since the effects of simvastatin on ALP activity were prevented by mevalonate, it suggested that these effects were caused by the inhibition of the mevalonate pathway. Other published findings supported the earlier observations of the capacity of simvastatin to affect bone formation. That is, osteoblast differentiation and mineralization in MC3T3-E1 osteoblasts and bone marrow cells (in vitro) were induced by simvastatin [140–142].
From a clinical perspective simvastatin is taken orally in 5, 10, 20, 40 or 80 mg tablets, with 20–100 mg tablets yielding maximum plasma concentrations in the range of 4.4 × 10−8−3.0 × 10−7m[143]. According to Yazawa et al. [139] the optimal in vitro concentration appeared to correlate with the 20 mg tablet dose, although it is difficult to relate in vitro concentrations with dose to target tissue.
It is noteworthy that bisphosphonates stimulate the formation of osteoblasts and possibly do so via the inhibition of the mevalonate pathway, thus mechanistically linking bone stimulation in osteoblasts and in human PDL cells.
Ocular diseases and cell proliferation
The successful treatment of intraocular proliferative diseases, such as proliferative vitreopathy (PVR), progressive traumatic traction retinal detachment (PTTRD) and intraocular neovascularization remain problematic despite notable advances in intraocular microsurgery in the direct handling of vitreoretinal tissues. This is due to the excessive accumulation of fibrous vascular tissue within the eye. Surgical treatment alone has also had limitations in the treatment of a range of other disorders characterized by progressive conjunctival or extraocular cicatrization including ocular pemphigoid (i.e. blisters), restrictive motility syndromes, and aphakic (lack of a natural lens) or neovascular glaucoma filtration surgery. According to Blumenkranz et al. [144], the central underlying feature that is common to these apparently unrelated clinical disorders is the rapid and uncontrolled proliferation of non-neoplastic cells within or about the eye. In such ocular-related proliferative disorders, the cells may be of diverse origin, including the retinal pigment epithelium, astroglia, macrophages, vascular endothelium, myoblasts, myofibroblasts, or fibroblasts. Nonetheless, the induced damage is generally accepted as being due to fibrocellular proliferation, active contraction of cellular membranes, and the formation and cross-linkage of newly formed collagen by fibroblasts and myofibroblasts. Such observations suggested that non-toxic pharmacological agents that inhibit the growth of rapidly proliferating cells may have value in the treatment of such diseases. Such reasoning led to a substantial series of cell culture investigations to find appropriate drugs which would be able to inhibit the proliferative response while not being toxic to ocular tissue. Other notable considerations included the toxicity of carrier solvents as well as the pharmacokinetics of the agent that might affect the periodicity of re-treatment.
In his 1988 reflective essay, Machemer [145] noted that he first thought to supplement surgical advances in the treatment of proliferative vitreopathy with pharmacological agents in the early 1980s with a consideration of the gout medication, colchicine, because of its capacity to be an inhibitor of mitosis. However, the selection of this agent for clinical application was problematic since clinical doses used for gout were far too low to affect cellular proliferation whereas higher doses were thought to markedly increase the risk of inducing toxicity in the neural tissue of the eye. This led to consideration of cancer chemotherapeutic agents, such as 5FU and daunomycin, since they were strongly anti-proliferative. While Machemer had concerns about their use due to toxicity, others proceeded to establish their therapeutic potential. Nonetheless, Machemer became interested in the possible use of corticosteroids since they had been reported to inhibit mitosis [146, 147] while having a very low likelihood of causing ocular toxicity. These collective perspectives lead to follow up investigations with a wide range of steroids, non-steroidal anti-inflammatory agents, as well as anti-metabolites and potent biopeptides.
An evaluation was undertaken of such agents in which a broad concentration range was assessed, including concentrations below the proliferation inhibition threshold. This evaluation revealed that hormetic-like biphasic dose–responses commonly occur and that these responses were independent of the chemical agent and the biological system employed. The maximum stimulatory responses were modest, generally being only 30–60% greater than the control value. However, there was considerable variation in the width of the stimulatory concentration range. For example, in the case of DEX, Blumenkranz et al. [144] reported a low concentration stimulatory response across greater than five orders of magnitude whereas the fluoridated pyrimidines displayed a stimulatory response of approximately only 10-fold [148]. The causes of such inherent variability in the width of low dose stimulatory response have not been addressed. The occurrence of inter-individual variation in low dose proliferative stimulatory responses of cultured retinal pigment epithelium proliferation was reported in by Yang & Cousins [149]. They noted that concentrations that were stimulatory for one individual might be inhibitory for another.
The implications of the inter-chemical and inter-individual variation in low dose stimulatory responses may have important implications. The ideal pharmacological agent would inhibit cell proliferation without being toxic and without being stimulatory in the low concentration zone. Investigators have yet to address explicitly the biomedical implications of a 30–60% stimulatory response over a certain number of days. Agents such as DEX could have a highly variable stimulatory zone depending on the individual and cell type. Even in the case of agents which have not shown the type of low dose stimulatory response as reported for DEX or trimicolon, these have not been typically well studied in the low dose zone. However, there is considerable evidence that these antimetabolites can be very effective in enhancing cell proliferation in the low dose zone [150]. The potential of pharmacological therapeutic agents such as DEX (Figure 11) to stimulate cell proliferation in ocular tissue at low doses may be a risk factor that needs to be considered in the treatment of surgical patients.
Figure 11.
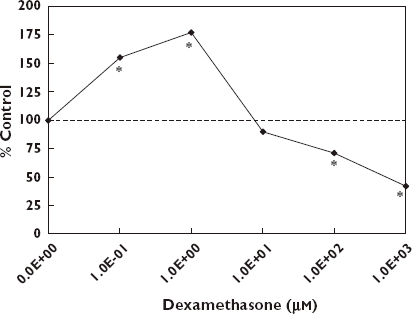
Effects of dexamethasone on cell growth and viability of cultured human RPE. *Significantly different from controls at P ≤ 0.05 [210]
Male sexual behaviour
Considerable pharmaceutical interest has been directed toward improving aspects of male sexual behaviour, with particular emphasis on erectile dysfunction (ED). Despite the rapid commercialization of products over the past decade, research on endogenous and exogenous agents that could enhance male sexual performance extends back to the 1960s. Of particular interest is that essentially all chemical groups that have been shown to enhance male sexual performance (e.g. penile erection, ejaculatory functions) have generally been shown to display hormetic-like biphasic dose–response relationships [151–156] (Figures 12 and 13). There are a sizeable number of such agents affecting various receptor systems, with particular research interest having been directed to dopamine, α-adrenergic agents, serotonin, opioids, nitric oxide, cholinergic agents, histamine, prostaglandin and oxytocin, amongst others.
Figure 12.
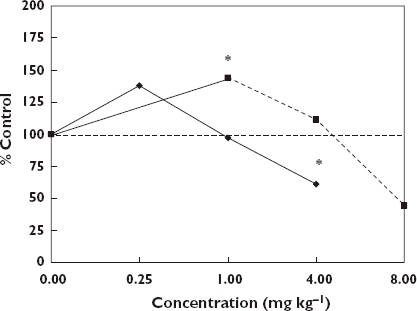
Effects of yohimbine and idazoxan on erections exhibited by rats in the reflex test. *Significantly different from controls at P ≤ 0.05 [153]. Yohimbine, (); Idazoxan, ()
Figure 13.
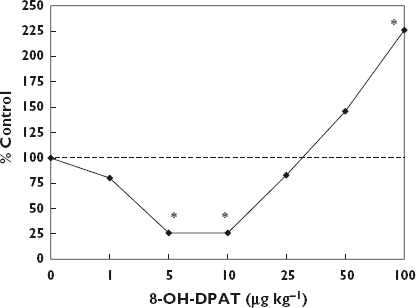
Effects of yohimbine on ejaculation latency of male rhesus monkeys. *Significantly different from controls at p ≤ 0.05 [151]
An evaluation of the pharmacological foundations of each of these general areas of research concerning male sexual behaviour have typically utilized male rats of multiple strains with particular emphasis on the use of Wistar and Sprague-Dawley rats, with dogs being employed as the second most common animal model. The investigators have tended to use penile erection as the most commonly used general endpoint in studies with rats. Penile erection has generally been studied using the spontaneous erection model in which drugs are tested for their capacity to increase the incidence of erections as compared to non-stimulated male, that is, without the influence of a female. A relatively low proportion of studies dealing with the penile erection have used a penile reflex test to estimate the frequency of erections. Copulatory investigations have also been common with multiple endpoints being assessed including mounting behavior, intromissions and ejaculatory parameters. Particular applications in the research have been directed to castrated males or other models of dysfunction. In the case of the dog model the principal endpoint assessed has been ejaculatory performance. Limited studies with large numbers of doses have been published using non-human primates.
The receptor systems that have been assessed most extensively with respect to detailed dose–response evaluation and male sexual behaviour have been dopamine [157–167], α2-adrenoceptor antagonists [152–154, 168–171], and serotonin [151, 172–179]. Each has a unique data base and scientific foundation that reflects investigator goals and research strategies. For example, in the case dopamine there has been extensive investigation of the dopamine agonist apomorphine, its metabolites, and other agonists. Considerable attention has been directed to clarification of the receptor subtype(s) that mediated the enhanced male sexual performance. Thus, extensive research has involved agonists and antagonists for receptor subtypes D1–D5 [164]. Similarly, there has been considerable research dealing with receptor interactions and the implications that specific agonists often display differential affinities for multiple receptors, each potentially affecting the sexual behaviour endpoint of interest. The case of dopamine has been generally similar for other receptor systems.
Since this general area of research was initiated in the 1960s there has been a progressive and, at times, rapid re-assessment of receptor pathways involved in the stimulation of male sexual behaviour. For example, each major receptor area has displayed a notable increase in the number of receptor subtypes, sometimes requiring the reclassification of agonist affinity to a newly discovered subtype with a consequent re-assessment of the stimulatory pathway. Likewise, numerous stimulatory agonists have been found to have affinities within multiple receptor systems after the initial findings that classified the agent as stimulating male sexual behaviour via a specific pathway. Despite this progressive growth in complexity of pathway interactions and signal convergences, the dose–response features have continued to reflect that of the hormetic-like biphasic dose–response.
Even though essentially all papers with evidence of a biphasic dose–response relationship for a male sexual behaviour have acknowledged their occurrence, very few such papers have provided even speculative hypotheses that might account for such common and reproducible observations. Several have been provided in areas such as with dopamine [159] and α2-adrenoceptor antagonists [152] but even these were put forth as speculative. Most of the detailed focus was directed to trying to clarify pathway involvement. In addition to the above major research areas, there have been reports published using botanically derived agents [180] which also have commonly displayed the hormetic-like biphasic dose–response relationships.
Of particular current interest have been the phosphodiesterase 5 inhibitors, which include the commercial products of Viagra, Cialis and Levitra. However, perhaps because of the history of their discoveries which are principally within the pharmaceutical industrial setting there has not been the same type of extensive dose–response studies to evaluate in the open literature. However, in the case of sildenafil recent studies have indicated that it displays hormetic-like biphasic dose–response relationships for human sperm motility [181] and in animal model studies concerning memory performance [182].
The dose–response features have generally conformed to that of the hormetic dose–response model with respect to the maximum stimulatory response and the width of the stimulatory range. However, in the case of spontaneous penile erection it is not uncommon to observe a four-fold increase in erections. While this is nearly double what might be expected, it may be an artifact of the use of a spontaneous rat model in which the control response rate is very low. There were also interspecies differences in the width of the stimulatory responses. For example, research assessing the effects of yohimbine in the rat model indicated that the width of the stimulatory response was very narrow, implying that this was due to its non-selective α2-adrenoceptor agonist binding [170]. However, research with the beagle dog model for ejaculatory endpoints revealed a considerably greater width of the stimulatory response which exceeded 30-fold [154].
Prion diseases, protein folding and hormetic dose–responses
Prion diseases have been shown to occur via both inherited and infectious processes, with infectious human prion diseases comprising only about 1% despite their notoriety [183]. Considerable evidence now exists which indicates that prions are comprised of a misfolded prion protein (PrP) isoform (PrPSc) of a glycolipid-anchored host protein (PrPC). While prions lack nucleic acid, their diversity is related to the conformation of PrPSc[184].
Prion diseases are progressive neurological disorders found in animals and humans. While rare, they are invariably fatal. Prion diseases have been of particular societal concern since there is the potential for cross species transmission of the scrapie condition (named from the symptoms of the condition due to compulsive scraping off of the fleece) from sheep to cattle where it has produced bovine spongiform encephalopathy (BSE) [185]. Hundreds of thousands of BSE-affected cattle have been slaughtered in order to prevent this epidemic in cattle and to protect the human food supply. As a result of public health concerns with this and other prion disease conditions, substantial efforts have been initiated to discover drugs effective in preventing the onset and/or progression of the disease process.
The normal cellular prion protein PrPc is relatively susceptible to proteolysis compared with an abnormal isoform PfPSc. PrPc may be converted to PrPSc and this process is considered a biochemical correlate of agent replication in cell culture and animals with the prion disease. According to Rudyk et al. [186, 187], one therapeutic strategy in the treatment of prion diseases is the discovery and use of agents that slow the replication of the infectious agent and delay the occurrence of the clinical disease when administered directly into the brain or peritoneum at or near the time of infection in the animal model. Using the non-neuronal scrapie infected mouse cell line model (SMB) originally cloned from an infected mouse brain [188], these authors assessed the capacity of Congo red and a number of its structural analogues to prevent the formation of PrP-res, a biomarker (protease resistant prion protein) of the infectious agent in this cell line. Congo red was selected because of its well recognized affinity for amyloid protein, its capacity to inhibit the replication of prion agents and thereby prevent the accumulation of PrPSc[186], and its ability to prolong the survival time of hamsters infected intraperitoneally with two different strains of scrapie [189]. However, Congo red is limited by its poor capacity to pass the blood brain barrier and that it is metabolized to benzidine, a human carcinogen. These factors lead to a broader assessment of structural analogues with improved penetration of the blood brain barrier and whose metabolites would be non-carcinogenic. In an initial follow up dose–response assessment 18 agents were tested including Congo red using only two concentrations (100 and 1 µm). While the 100 µm concentration markedly prevented the formation of PrPSc there was the general observation of an enhancement of PrPSc formation at the 1 µm concentration by numerous sulphonic acid derivatives of Congo red but not with the two cases of carboxylate derivatives. The increase of PrP-res formation ranged from approximately 140% to 225% (relative to a control value of 100%) depending on the specific analogue tested. In follow-up experiments selected agents were tested over six concentrations with log concentration spacing. Not only was the low concentration stimulation response confirmed in representative sulphonic acid derivative compounds, but even Congo red (Figure 14), which was negative in the preliminary studies with only the two concentrations (1 and 100 µm), was positive once the bioassay included even lower concentrations [186].
Figure 14.
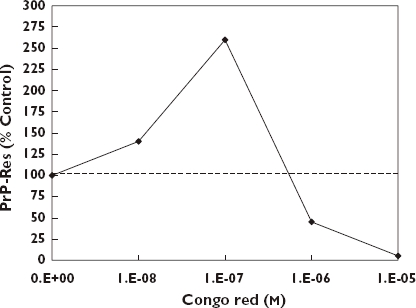
Effects of Congo red in the SMB cell assay [186]
These dose–response features of Congo red and its sulphonic acid derivatives are consistent with the quantitative characteristics of the hormetic dose–response model. These findings are of medical concern because they raise the possibility that such potential therapeutic agents may have the possibility of enhancing the occurrence of the prion disease within the low concentration range.
The mechanism by which such low concentration related increases in PrPSc may occur have been addressed to a limited extent. The low concentration stimulatory response was hypothesized by Rudyk et al. [186] to be related to an observation by Caspi et al. [190] that Congo red inhibited new PrPSc synthesis and PrPSc degradation in scrapie-infected neuroblastoma cells. Rudyk et al. [186] also speculated that these compounds, as monomers at low concentrations, may stimulate PrPSc formation due to binding to PrPC at just one site, while at higher concentrations in physiological salt, they may form a supramolecular ligand, a liquid crystal, which binds PrPSc and/or PrPC, preventing their interaction [191, 192].
Discussion
The paper asserts that the hormetic dose–response is more common and fundamental than other dose–response models, including the long revered threshold model, far out-competing them in fair head-to-head evaluations. It is argued further that we are in the midst of a major dose–response revolution that has highly significant implications for essentially all branches of science that are concerned with dose–response relationships and adaptive responses.
The last three decades have witnessed growing interdisciplinary evidence of hormetic-biphasic dose–responses that are characterized by remarkably similar quantitative features of the dose–response and similar underlying mechanistic explanatory strategies. It is the emergence and integration of these findings from diverse biomedical fields that has lead to the consolidation of the hormesis dose–response concept and motivation to re-discover the historical foundations of the dose–response [193–197].
The concept of hormesis can also be considered within a preventive context. Considerable research has associated beneficial responses from low to moderate levels of exercise with hormetic mechanisms [198–201]. Likewise, the benefits of caloric restriction [202, 203] or certain fasting regimes [204, 205] have also been proposed as manifestations of hormetic effects. These developments are believed to have the potential to enhance the quality of life within ageing populations [206].
This paper presented a spectrum of examples in which hormesis is having or could have an important role in clinical pharmacology. The examples selected are illustrative of the potential of the hormesis to affect the biomedical sciences and to improve its capacity not only to enhance human health and performance but also to avoid harm in patient treatments. A key point is that the concept of hormetic-biphasic dose–responses is already an important feature within clinical pharmacology, especially within the areas of drug discovery for anxiolytic drugs, anti-seizure drugs, memory, neuroprotection, drug addiction, pain, and others. In fact, hormetic-biphasic dose–responses are common to these fields, affect decisions on drug development but almost never is the term hormesis used to describe these versions of the dose–response relationship. The hormesis concept is also important in areas of clinical pharmacology in which high doses are important for killing harmful organisms or tumour cells. In these cases, there may be health concerns over the potential of hormetic-biphasic dose responses, such as tumour cell proliferation or prostate enlargement, at hormetically acting low concentrations. In these cases as well, the low dose stimulatory response has not been referred to as a hormetic dose–response. Yet it is argued that such cases are manifestations of the hormetic dose–response, reflecting its centrality within the biomedical sciences, its generalizability across biological models, gender and age groups, endpoints and chemical classes and the important constraints it imposes on the quantitative features of the dose–response as an indicator of biological plasticity. Recently a large number of biomedical scientists have proposed the use of a common terminology that more effectively places biphasic dose–responses and stress responses within a broad hormetic framework [214]. Finally, it is time for the hormetic dose–response to be integrated within the education and training of current and future biomedical scientists and to improve the design and conduct of studies affecting drug discovery and safety evaluation as well as providing a sound dose–response framework in the refining or fine-tuning of drug doses for patients in clinical settings.
Acknowledgments
Effort sponsored by the Air Force Office of Scientific Research, Air Force Material Command, USAF, under grant number FA9550-07-1-0248. The US Government is authorized to reproduce and distribute for governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsement, either expressed or implied, of the Air Force Office of Scientific Research or the US Government.
REFERENCES
- 1.Calabrese EJ. Historical blunders: how toxicology got the dose-response relationship half right. Cell Mol Biol. 2005;51:643–54. [PubMed] [Google Scholar]
- 2.Calabrese EJ, Blain R. The occurrence of hormetic dose responses in the toxicological literature, the hormesis database: an overview. Toxicol Appl Pharmacol. 2005;202:289–301. doi: 10.1016/j.taap.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 3.Calabrese EJ. Cancer biology and hormesis: human tumor cell lines commonly display hormetic (biphasic) dose responses. Crit Rev Toxicol. 2005;35:463–582. doi: 10.1080/10408440591034502. [DOI] [PubMed] [Google Scholar]
- 4.Davies JMS, Lowry CV, Davies KJA. Transcient adaptation to oxidative stressing yeast. Arch Biochem Biophys. 1995;317:1–6. doi: 10.1006/abbi.1995.1128. [DOI] [PubMed] [Google Scholar]
- 5.Samson L, Cairns J. A new pathway for DNA repair in Escherichia coli. Nature. 1977;267:281–3. doi: 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- 6.Szabadi E. Model of two functionally antagonistic receptor populations activated by same agonist. J Theor Biol. 1977;69:101–12. doi: 10.1016/0022-5193(77)90390-3. [DOI] [PubMed] [Google Scholar]
- 7.Leff P. Theoretical treatment of one-agonist-2-receptor systems. Trends Pharmacol Sci. 1994;15:320–1. doi: 10.1016/0165-6147(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 8.Rovati GE, Nicosia S. An alternative model for bell-shaped concentrations-response curve – reply. Trends Pharmacol Sci. 1994;15:321–2. doi: 10.1016/0165-6147(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 9.Jarv J. A model of nonexclusive binding of agonist and antagonist on G-protein coupled receptors. J Theoret Biol. 1995;175:577–82. doi: 10.1006/jtbi.1995.0166. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese EJ, Baldwin LA. Hormesis and high-risk groups. Regul Toxicol Pharmacol. 2002;35:414–28. doi: 10.1006/rtph.2001.1529. [DOI] [PubMed] [Google Scholar]
- 11.Calabrese EJ. Converging concepts: adaptive response, preconditioning, and the Yerkes-Dodson Law are manifestations of hormesis. Ageing Res Rev. 2008;7:8–20. doi: 10.1016/j.arr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Flood JF, Landry DW, Jarvik ME. Cholinergic receptor interactions and their effects on long-term memory processing. Brain Res. 1981;215:177–85. doi: 10.1016/0006-8993(81)90500-x. [DOI] [PubMed] [Google Scholar]
- 13.Flood JF, Smith GE, Cherkin A. Memory enhancement: supra-additive effect of subcutaneous cholinergic drug combinations in mice. Psychopharmacology. 1985;86:61–7. doi: 10.1007/BF00431685. [DOI] [PubMed] [Google Scholar]
- 14.Crawley JN. Neuropharmacologic specificity of a simple animal model for the behavioral actions of benzodiazepine. Pharmacol Biochem Behav. 1981;15:695–9. doi: 10.1016/0091-3057(81)90007-1. [DOI] [PubMed] [Google Scholar]
- 15.Crawley JN, Goodwin FK. Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol Biochem Behav. 1980;13:167–70. doi: 10.1016/0091-3057(80)90067-2. [DOI] [PubMed] [Google Scholar]
- 16.Pellow S, File SE. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze – a novel test of anxiety in the rat. Pharmacol Biochem Behav. 1986;24:525–9. doi: 10.1016/0091-3057(86)90552-6. [DOI] [PubMed] [Google Scholar]
- 17.File SE. The use of social interaction as a method for detecting anxiolytic activity of chlordiazepoxide-like drugs. J Neurosci Methods. 1980;2:219–38. doi: 10.1016/0165-0270(80)90012-6. [DOI] [PubMed] [Google Scholar]
- 18.File SE, Hyde JRG. Can social interaction be used to measure anxiety? Br J Pharmacol. 1978;62:19–24. doi: 10.1111/j.1476-5381.1978.tb07001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calabrese EJ. Hormesis: changing view of the dose–response, a personal account of the history and current status. Mut Res. 2002;511:181–9. doi: 10.1016/s1383-5742(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 20.Slanska J, Vojtechovsky M, Votava Z. The influence of physostigmine on individual phases of learning in man. Act Nerv Super (Praha) 1972;14:110–1. [PubMed] [Google Scholar]
- 21.Drachman DA, Leavitt J. Human memory and the cholinergic system. A relationship to aging? Arch Neurol. 1974;30:113–21. doi: 10.1001/archneur.1974.00490320001001. [DOI] [PubMed] [Google Scholar]
- 22.Davis KL, Hollister LE, Overall J, Johnson A, Train K. Physostigmine: effects on cognition and affect in normal subjects. Psychopharmacology. 1976;51:23–7. doi: 10.1007/BF00426316. [DOI] [PubMed] [Google Scholar]
- 23.Peters BH, Levin HS. Memory enhancement after physostigmine treatment in the amnesic syndrome. Arch Neurol. 1977;34:215–9. doi: 10.1001/archneur.1977.00500160029004. [DOI] [PubMed] [Google Scholar]
- 24.Peters BH, Levin HS. Effects of physostigmine and lecithin on memory in Alzheimer disease. Ann Neurol. 1979;6:219–21. doi: 10.1002/ana.410060307. [DOI] [PubMed] [Google Scholar]
- 25.Davis KL, Mohs RC, Tinklenberg JR, Pfefferbaum A, Hollister LE, Kopell BS. Physostigmine: improvement of long-term memory processes in normal humans. Science. 1978;201:272–4. doi: 10.1126/science.351807. [DOI] [PubMed] [Google Scholar]
- 26.Davis KL, Berger PA. Pharmacological investigations of the cholinergic imbalance hypotheses of movement disorders and psychosis. Biol Psychiatry. 1978;13:23–49. [PubMed] [Google Scholar]
- 27.Muramoto O, Suishita M, Sugita H, Toyokura Y. Effect of physostigmine on constructional and memory tasks in Alzheimer's disease. Arch Neurol. 1979;36:501–3. doi: 10.1001/archneur.1979.00500440071014. [DOI] [PubMed] [Google Scholar]
- 28.Davis KL, Mohs RC. Enhancement of memory by physostigmine. N Engl J Med. 1979;301:946. doi: 10.1056/NEJM197910253011722. [DOI] [PubMed] [Google Scholar]
- 29.Delwaide PJ, Devoitille JM, Ylieff M. Acute effect of drugs upon memory of patients with senile dementia. Acta Psychiatr Belg. 1980;80:748–54. [PubMed] [Google Scholar]
- 30.Drachman DA, Sahakian BJ. Memory and cognitive function in the elderly. A preliminary trial of physostigmine. Arch Neurol. 1980;37:674–5. doi: 10.1001/archneur.1980.00500590098022. [DOI] [PubMed] [Google Scholar]
- 31.Ashford JW, Soldinger S, Schaeffer J, Cochran L, Jarvik LF. Physostigmine and its effect on six patients with dementia. Am J Psychiatry. 1981;138:829–30. doi: 10.1176/ajp.138.6.829. [DOI] [PubMed] [Google Scholar]
- 32.Christie JE, Shering A, Ferguson J, Glen AIM. Physostigmine and arecholine: effects of intravenous infusions in Alzheimer presenile dementia. Br J Psychiatry. 1981;138:46–50. doi: 10.1192/bjp.138.1.46. [DOI] [PubMed] [Google Scholar]
- 33.Laurent B, Hibert-Kuntzler O, Chazot G, Michel D, Schott B. Effects de la physostigmine sur les syndromes amnesiques. Rev Neurol (Paris) 1981;137:649–60. [PubMed] [Google Scholar]
- 34.Caltagirone C, Gainotti G, Masullo C. Oral administration of chronic physostigmine does not improve cognitive or amnesic performances in Alzheimer's presenile dementia. Int J Neurosci. 1982;16:247–9. doi: 10.3109/00207458209147153. [DOI] [PubMed] [Google Scholar]
- 35.Davis KL, Mohs RC. Enhancement of memory process in Alzheimer's disease with multiple-dose intravenous physostigmine. Am J Psychiatry. 1982;139:1421–4. doi: 10.1176/ajp.139.11.1421. [DOI] [PubMed] [Google Scholar]
- 36.Wettstein A. No effect from double-blind trial of physostigmine and lecithin in Alzheimer's disease. Ann Neurol. 1983;13:210–2. doi: 10.1002/ana.410130220. [DOI] [PubMed] [Google Scholar]
- 37.Jotkowitz S. Lack of clinical efficacy of chronic oral physostigmine in Alzheimer's disease. Ann Neurol. 1983;14:690–1. doi: 10.1002/ana.410140616. [DOI] [PubMed] [Google Scholar]
- 38.Thal LJ, Fuld PA, Masur DM, Sharpless NS. Oral physostigmine and lecithin improve memory in Alzheimer's disease. Ann Neurol. 1983;13:491–6. doi: 10.1002/ana.410130504. [DOI] [PubMed] [Google Scholar]
- 39.Thal LJ, Masur DM, Blau AD, Fuld PA, Klauber MR. Chronic oral physostigmine without lecithin improves memory in Alzheimer's disease. J Am Geriatr Soc. 1989;37:42–8. doi: 10.1111/j.1532-5415.1989.tb01567.x. [DOI] [PubMed] [Google Scholar]
- 40.Johns CA, Haroutunian V, Greenwald BS, Mohs RC, Davis BM, Kanof P, Horvath TB, Davis KL. Development of cholinergic drugs for the treatment of Alzheimer's disease. Drug Dev Res. 1985;5:77–96. [Google Scholar]
- 41.Mohs RC, Davis BM, Greenwald BS, Mathe AA, Johns CA, Horvath TB, Davis KL. Clinical studies of the cholinergic deficit in Alzheimer's disease. II. Psychopharmacologic studies. J Am Geriatr Soc. 1985;33:749. doi: 10.1111/j.1532-5415.1985.tb04185.x. [DOI] [PubMed] [Google Scholar]
- 42.Mohs RC, Davis BM, Johns CA, Mathe AA, Greenwald BS, Horvath TB, Davis KL. Oral physostigmine treatment of patients with Alzheimer's disease. Am J Psychiatry. 1985;142:28–33. doi: 10.1176/ajp.142.1.28. [DOI] [PubMed] [Google Scholar]
- 43.Mohs RC, Davis BM, Mathe AA, Rosen WG, Johns CA, Greenwald BS, Horvath TB, Davis KL. Intravenous and oral physostigmine in Alzheimer's disease. Interdiscipl Topics Geront. 1985;20:140–52. [Google Scholar]
- 44.Beller SA, Overall JE, Swann AC. Efficacy of oral physostigmine in primary degenerative dementia. A double-blind study of response to different dose level. Psychopharmacology. 1985;87:147–51. doi: 10.1007/BF00431798. [DOI] [PubMed] [Google Scholar]
- 45.Blackwood DHR, Christie JE. The effects of physostigmine on memory and auditory P300 in Alzheimer-type dementia. Biol Psychiatry. 1986;21:557–60. doi: 10.1016/0006-3223(86)90201-5. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz AS, Kohlstaedt EV. Physostigmine effects in Alzheimer's disease: relationships to dementia severity. Life Sci. 1986;38:1021–8. doi: 10.1016/0024-3205(86)90236-5. [DOI] [PubMed] [Google Scholar]
- 47.Gustafson L, Edvinsson L, Dahlgren N, Hagberg B, Risberg J, Rosen I, Ferno H. Intravenous physostigmine treatment of Alzheimer's disease evaluated by psychometric testing, regional cerebral blood flow (rCBF) measurement, and EEG. Psychopharmacology. 1987;93:31–5. doi: 10.1007/BF02439583. [DOI] [PubMed] [Google Scholar]
- 48.Levin Y, Elizure A, Korczyn AD. Physostigmine improves ECT-induced memory disturbances. Neurology. 1987;37:871–5. doi: 10.1212/wnl.37.5.871. [DOI] [PubMed] [Google Scholar]
- 49.Stern Y, Sano M, Mayeux R. Effects of oral physostigimine in Alzheimer's disease. Ann Neurol. 1987;22:306–10. doi: 10.1002/ana.410220305. [DOI] [PubMed] [Google Scholar]
- 50.Stern Y, Sano M, Mayeux R. Long-term administration of oral physostigmine in Alzheimer's disease. Neurology. 1988;38:1837–41. doi: 10.1212/wnl.38.12.1837. [DOI] [PubMed] [Google Scholar]
- 51.Braida D, Paladini E, Griffini P, Lamperti M, Maggi A, Sala M. An inverted U-shaped curve for heptylphysostigmine on radial maze performance in rats: comparison with other cholinesterase inhibitors. Eur J Pharmacol. 1996;302:13–20. doi: 10.1016/0014-2999(96)00072-6. [DOI] [PubMed] [Google Scholar]
- 52.Liu J, Zhang H-Y, Tang X-C, Wang B, He X-C, Bai D-L. Effects of synthetic (−)-huperzine A on cholinesterase activities and mouse water maze performance. Acta Pharmacol Sin. 1998;19:413–6. [PubMed] [Google Scholar]
- 53.Wang T, Tang XC. Reversal of scopolamine-induced deficits in radial maze performance by (−)-hyperzine A: comparison with E2020 and tacrine. Eur J Pharmacol. 1998;349:137–42. doi: 10.1016/s0014-2999(98)00199-x. [DOI] [PubMed] [Google Scholar]
- 54.Windisch M, Hutter-Paier B, Jerkovic L, Imbimbo B, Villetti G. The protective effect of ganstigmine against amyloid beta25-35 neurotoxicity on chicken cortical neurons is independent from the cholinesterase inhibition. Neurosci Lett. 2003;341:181–4. doi: 10.1016/s0304-3940(03)00125-3. [DOI] [PubMed] [Google Scholar]
- 55.Wise LE, Lichtman AH. The uncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist memantine prolongs spatial memory in a rat delayed radial-arm maze memory task. Eur J Pharmacol. 2007;575:98–102. doi: 10.1016/j.ejphar.2007.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guner M, Freshney RI, Morgan D, Freshney MG, Thomas DGT, Graham DI. Effects of dexamethasone and betamethasone on in vitro cultures from human astrocytoma. Eur J Cancer. 1977;35:439–47. doi: 10.1038/bjc.1977.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freshney RI, Sherry A, Hassanzadan M, Freshney M, Crilly P, Morgan D. Control of cell proliferation in human glioma by glucocorticoids. Br J Cancer. 1980;41:857–66. doi: 10.1038/bjc.1980.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paoletti P, Butti G, Zibera C, Scerrati M, Gibelli N, Roselli R, Magrassi L, Sica G, Rossi G, Cuna GR. Characteristics and biological role of steroid hormone receptors in neuroepithelial tumors. J Neurosurg. 1990;73:736–42. doi: 10.3171/jns.1990.73.5.0736. [DOI] [PubMed] [Google Scholar]
- 59.Gibelli N, Zibera C, Butti G, Assietti R, Sica G, Scerrati M, Iacopino F, Roselli R, Paoletti P, Cuna GR, Rossi GF. Hormonal modulation of brain tumour growth: a cell culture study. Acta Neurochir (Wien) 1989;101:129–33. doi: 10.1007/BF01410528. [DOI] [PubMed] [Google Scholar]
- 60.Kawamura A, Tamaki N, Kokunai T. Effect of dexamethasone on cell proliferation of neuroepithelial tumor cell lines. Neurol Med Chir (Tokyo) 1998;38:633–40. doi: 10.2176/nmc.38.633. [DOI] [PubMed] [Google Scholar]
- 61.Kuratsu J. Commentary. Neurol Med Chir (Tokyo) 1998;38:639. [Google Scholar]
- 62.Laws ER. Commentary. Neurol Med Chir (Tokyo) 1998;38:639. [Google Scholar]
- 63.Rutka J. Commentary. Neurol Med Chir (Tokyo) 1998;38:639. [Google Scholar]
- 64.Tabuchi K. Commentary. Neurol Med Chir (Tokyo) 1998;38:639–40. [Google Scholar]
- 65.Yoshida J. Commentary. Neurol Med Chir (Tokyo) 1998;38:633–40. [Google Scholar]
- 66.Foekens JA, Sieuwerts AM, Stuurman-Smeets EMJ, Dorssers LCJ, Berns EMJJ, Klijn JGM. Pleiotropic actions of suramin on the proliferation of human breast cancer cells in vitro. Int J Cancer. 1992;51:439–44. doi: 10.1002/ijc.2910510317. [DOI] [PubMed] [Google Scholar]
- 67.Calabrese EJ, Staudenmayer JW, Stanek EJ, Hoffmann GR. Hormesis outperforms threshold model in NCI anti-tumor drug screening data. Toxicol Sci. 2006;94:368–78. doi: 10.1093/toxsci/kfl098. [DOI] [PubMed] [Google Scholar]
- 68.Calabrese EJ. A dose of common sense. Good Clin Pract J. 2007:12–6. [Google Scholar]
- 69.Calabrese EJ. Drug therapies for stroke and traumatic brain injury often display U-shaped dose responses: Occurrence, mechanisms, and clinical implications. Crit Rev Toxicol. 2008;38:557–77. doi: 10.1080/10408440802014287. [DOI] [PubMed] [Google Scholar]
- 70.Sakakibara Y, Mitha AP, Ogilvy CS, Maynard KI. Post-treatment with nicotinamide (vitamin B3) reduces the infarct volume following permanent focal cerebral ischemia in female Sprague-Dawley and Wistar rats. Neurosci Lett. 2000;281:111–4. doi: 10.1016/s0304-3940(00)00854-5. [DOI] [PubMed] [Google Scholar]
- 71.Sakakibara Y, Mitha AP, Ayoub IA, Ogilvy CS, Maynard KI. Delayed treatment with nicotinamide (vitamin B3) reduces the infarct volume following focal cerebral ischemia in spontaneously hypertensive rats, diabetic and non-diabetic Fischer 344 rats. Brain Res. 2002;931:68–73. doi: 10.1016/s0006-8993(02)02263-1. [DOI] [PubMed] [Google Scholar]
- 72.Mauler F, Mittendorg J, Horvath E, De Vry J. Characterization of the diarylether sulfonylester (−)-(R)-3-(2-hydroxymethylindanyl-4-oxy)phenyl-4,4,4-trigluoro-1-sulfonate (BAY 38-7271) as a potent cannabinoid receptor agonist with neuroprotective properties. J Pharmacol Exp Ther. 2002;302:259–68. doi: 10.1124/jpet.302.1.359. [DOI] [PubMed] [Google Scholar]
- 73.Mauler F, Hinz V, Horvath E, Schuhmacher J, Hofmann HA, Wirtz S, Hahn MG, Urbahns K. Selective intermediate-/ small-conductance calcium-activated potassium channel (KCNN4) blockers are potent and effective therapeutics in experimental brain oedema and traumatic brain injury caused by acute subdural haematoma. Eur J Neurosci. 2004;20:1761–8. doi: 10.1111/j.1460-9568.2004.03615.x. [DOI] [PubMed] [Google Scholar]
- 74.Roddis LH. William Withering and the introduction of digitalis into medical practice. Ann Med Hist. 1936;8:93. [PMC free article] [PubMed] [Google Scholar]
- 75.Nylin G. William Withering – A bicentenary tribute. Am Heart J. 1943;25:285. [Google Scholar]
- 76.Rahimtoola SH. Digitalis and William Withering, the clinical investigator. Circulation. 1975;52:969–71. doi: 10.1161/01.cir.52.6.969. [DOI] [PubMed] [Google Scholar]
- 77.Godfraind T, Ghysel-Burton J. Binding sites related to ouabain-induced stimulation or inhibition of the sodium pump. Nature. 1977;365:165–6. doi: 10.1038/265165a0. [DOI] [PubMed] [Google Scholar]
- 78.Noble D. Mechanism of action of therapeutic levels of cardiac glycosides. Cardiovasc Res. 1980;14:495–514. doi: 10.1093/cvr/14.9.495. [DOI] [PubMed] [Google Scholar]
- 79.Gao J, Wymore RS, Wang Y, Gaudette GR, Krukenkamp IB, Cohen IS, Mathias RT. Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J Gen Physiol. 2002;119:297–312. doi: 10.1085/jgp.20028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christen Y, Sasportes M, Mawas C, Dausewt J, Kaplan JG. The mixed lymphocyte reaction selective activation and inactivation of the stimulating cells. Cell Immunol. 1975;19:137–42. doi: 10.1016/0008-8749(75)90298-1. [DOI] [PubMed] [Google Scholar]
- 81.Dornand J, Kaplan JG. Persistent effects of ouabain treatment on human lymphocytes: synthesis of DNA, RNA and protein in stimulated and unstimulated cells. Can J Biochem. 1976;54:280–6. doi: 10.1139/o76-041. [DOI] [PubMed] [Google Scholar]
- 82.Aydemir-Koksoy A, Abramowitz J, Allen JC. Ouabain-induced signaling and vascular smooth muscle cell proliferation. Biol Chem. 2001;276:46605–11. doi: 10.1074/jbc.M106178200. [DOI] [PubMed] [Google Scholar]
- 83.Abramowitz J, Dai C, Hirschi KK, Dmitrieva RI, Doris PA, Liu L, Allen JC. Ouabain- and marinobufagenin-induced proliferation of human umbilical vein smooth muscle cells and a rat vascular smooth muscle cell lines, A7r5. Circulation. 2003;108:3048–53. doi: 10.1161/01.CIR.0000101919.00548.86. [DOI] [PubMed] [Google Scholar]
- 84.Chueh S-C, Guh J-H, Chen J, Lai M-K, Teng C-M. Dual effects of ouabain on the regulation of proliferation and apoptosis in human prostatic smooth muscle cells. J Urol. 2001;166:347–53. [PubMed] [Google Scholar]
- 85.Balzan S, D’Urso G, Nicolini G, Forini F, Pellegrino M, Montali U. Erythrocyte sodium pump stimulation by ouabain and an endogenous ouabain-like factor. Cell Biochem Funct. 2007;25:297–303. doi: 10.1002/cbf.1387. [DOI] [PubMed] [Google Scholar]
- 86.Dmitrieva R, Doris PA. Ouabain is a potent promoter of growth and activator of ERK1/2 in ouabain-resistant rat renal epithelial cells. Biol Chem. 2003;278:28160–8. doi: 10.1074/jbc.M303768200. [DOI] [PubMed] [Google Scholar]
- 87.Ramirez-Ortega M, Maldonado-Lagunas V, Melendez-Zajgla J, Carrillo-Hernandez JF, Pastelin-Hernandez G, Picazo-Picazo O, Ceballos-Reyes G. Proliferation and apoptosis of HeLa cells induces by in vitro stimulation with digitalis. Eur J Pharmacol. 2006;534:71–86. doi: 10.1016/j.ejphar.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 88.McConnell JD, Bruskewitz R, Walsh P, Andriole G, Lieber M, Holtgrewe HL, Albertsen P, Roehrborn CG, Nickel JC, Wang DZ, Taylor AM, Waldstreicher J. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. Finasteride Long-Term Efficacy and Safety Study Group. N Engl J Med. 1998;338:557. doi: 10.1056/NEJM199802263380901. [DOI] [PubMed] [Google Scholar]
- 89.Jonler M, Riehmann M, Bruskewitz RC. Benign prostatic hyperplasia. Current pharmacological treatment. Drugs. 1994;47:66. doi: 10.2165/00003495-199447010-00005. [DOI] [PubMed] [Google Scholar]
- 90.Gutierrez GE, Lalka D, Garrett IR, Rossini G, Mundy GR. Transdermal application of lovastatin to rats causes profound increases in bone formation and plasma concentrations. Osteoporos Int. 2006;17:1033–42. doi: 10.1007/s00198-006-0079-0. [DOI] [PubMed] [Google Scholar]
- 91.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–45. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 92.Urbich C, Dernbach E, Zeiher AM, Dimmeler S. Double-edged role of statins in angiogenesis signaling. Circ Res. 2002;90:737–44. doi: 10.1161/01.res.0000014081.30867.f8. [DOI] [PubMed] [Google Scholar]
- 93.Katsumoto M, Shingu T, Kuwashima R, Nakata A, Nomura S, Chayama K. Biphasic effect of HMG-CoA reductase inhibitor, pitavastatin, on vascular endothelial cells and angiogenesis. Circ J. 2005;69:1547–55. doi: 10.1253/circj.69.1547. [DOI] [PubMed] [Google Scholar]
- 94.Cooke JP. NO and angiogenesis. Atherosclerosis. 2003;4(Suppl):53–60. doi: 10.1016/s1567-5688(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 95.Sata M. Biphasic effects of statins on angiogenesis. Letter to the Editor. Circulation. 2002;105:739–45. doi: 10.1161/01.cir.0000030081.54465.2d. [DOI] [PubMed] [Google Scholar]
- 96.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 97.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–85. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 98.Eisterer W, Jiang XY, Bachelot T, Pawliuk R, Abramovich C, Leboulch P, Hogge D, Eaves C. Unfulfilled promise of endostatin in a gene therapy-xenotransplant model of human acute lymphocytic leukemia. Mol Ther. 2002;5:352–9. doi: 10.1006/mthe.2002.0573. [DOI] [PubMed] [Google Scholar]
- 99.Slaton JW, Perrotte P, Inoue K, Dinney CPN, Fidler IJ. Interferon-α-mediated down-regulation of angiogenesis-related genes and therapy of bladder cancer are dependent on optimization of biological dose and schedule. Clin Cancer Res. 1999;5:2726–34. [PubMed] [Google Scholar]
- 100.Celik I, Surucu O, Dietz C, Heymach JV, Force J, Hoschele I, Becker CM, Folkman J, Kisker O. Therapeutic efficacy of endostatin exhibits a biphasic dose-response curve. Cancer Res. 2005;65:11044–50. doi: 10.1158/0008-5472.CAN-05-2617. [DOI] [PubMed] [Google Scholar]
- 101.Jockovich M-E, Murray TG, Escalona-Benz E, Hernandez E, Feuer W. Anecortave acetate as single and adjuvant therapy in the treatment of retinal tumors of LHβTag mice. Invest Ophthalmol Vis Sci. 2006;47:1264–8. doi: 10.1167/iovs.05-1194. [DOI] [PubMed] [Google Scholar]
- 102.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Landrum JT, Bone RA. Lutein, zeaxanthin, and the macular pigment. Arch Biochem Biophys. 2001;385:28–40. doi: 10.1006/abbi.2000.2171. [DOI] [PubMed] [Google Scholar]
- 104.Mayne ST. Beta-carotene, carotenoids, and disease prevention in humans. FASEB J. 1996;10:690–701. [PubMed] [Google Scholar]
- 105.Pryor WA, Stahl W, Rock CL. Beta-carotene: from biochemistry to clinical trials. Nutr Rev. 2000;58:39–53. doi: 10.1111/j.1753-4887.2000.tb07810.x. [DOI] [PubMed] [Google Scholar]
- 106.Stahl W, Heinrich U, Jungmann H, Sies H, Tronnier H. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am J Clin Nutr. 2000;71:795–8. doi: 10.1093/ajcn/71.3.795. [DOI] [PubMed] [Google Scholar]
- 107.Stahl W, Heinrich U, Wiseman S, Eichler O, Sies H, Tronnier H. Dietary tomato paste protects against UV-induced erythema in humans. J Nutr. 2001;131:1449–51. doi: 10.1093/jn/131.5.1449. [DOI] [PubMed] [Google Scholar]
- 108.Lee J, Jiang S, Levine N, Watson RR. Carotenoid supplementation reduces erythema in human skin after simulated solar radiation exposure. Proc Soc Exp Biol Med. 2000;223:170–4. doi: 10.1046/j.1525-1373.2000.22323.x. [DOI] [PubMed] [Google Scholar]
- 109.Gollnick HPM, Hopfenmuller W, Hemmes C, Chun SC, Schmid C, Sundermeier K, Biesalski HK. Systemic beta-carotene plus topical UV-sunscreen are an optimal protection against harmful effects of natural UV-sunlight: results of the Berlin-Eilath study. Eur J Dermatol. 1996;6:200–5. [Google Scholar]
- 110.Young AJ, Lowe GM. Antioxidant and pro-oxidant properties of carotenoids. Arch Biochem Biophys. 2001;385:20–7. doi: 10.1006/abbi.2000.2149. [DOI] [PubMed] [Google Scholar]
- 111.Lowe GM, Booth LA, Young AJ, Bilton RF. Lycopene and beta-carotene protect against oxidative damage in HT29 cells at low concentrations but rapidly lose this capacity at higher doses. Free Radic Res. 1999;30:141–51. doi: 10.1080/10715769900300151. [DOI] [PubMed] [Google Scholar]
- 112.Palozza P, Luberto C, Calviello G, Ricci P, Bartoli GM. Antioxidant and pro-oxidant role of beta-carotene in murine normal and tumor thymocytes: effects of oxygen partial pressure. Free Radic Biol Med. 1997;22:1065–73. doi: 10.1016/s0891-5849(96)00498-4. [DOI] [PubMed] [Google Scholar]
- 113.Biesalski HK, Obermueller-Jevic UC. UV light, beta-carotene and huma skin – beneficial and potentially harmful effects. Arch Biochem Biophys. 2001;389:1–6. doi: 10.1006/abbi.2001.2313. [DOI] [PubMed] [Google Scholar]
- 114.Omaye ST, Krinsky NI, Kagan VE, Mayne ST, Liebler DC, Bidlack WR. Beta-carotene: friend or foe? Fundam Appl Toxicol. 1997;40:163–74. doi: 10.1006/faat.1997.2387. [DOI] [PubMed] [Google Scholar]
- 115.Eichler O, Sies H, Stahl W. Divergent optimum levels of lycopene, β-carotene and lutein protecting against UVB irradiation in human fibroblasts. Photochem Photobiol. 2002;75:503–6. doi: 10.1562/0031-8655(2002)075<0503:dololc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 116.Zappacosta AR. Reversal of baldness in a patient receiving minoxidil for hypertension. N Engl J Med. 1980;303:1480–1. doi: 10.1056/nejm198012183032516. [DOI] [PubMed] [Google Scholar]
- 117.Boyera N, Galey I, Bernard BA. Biphasic effects of minoxidil on the proliferation and differentiation of normal human keratinocytes. Skin Pharmacol. 1997;10:206–20. doi: 10.1159/000211506. [DOI] [PubMed] [Google Scholar]
- 118.Yeo H, Beck LH, McDonald JM, Zayzafoon M. Cyclosporin A elicits dose-dependent biphasic effects on osteoblast differentiation and bone formation. Bone. 2007;40:1502–16. doi: 10.1016/j.bone.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Foreman J. A cure for osteoporosis may be near. Boston Globe. pp. pC1–pC4.
- 120.Santini D, Fratto ME, Vincenzi B, La Cesa A, Dianzani C, Tonini G. Bisphosphonate effects in cancer and inflammatory diseases. BioDrugs. 2004;18:269–78. doi: 10.2165/00063030-200418040-00004. [DOI] [PubMed] [Google Scholar]
- 121.Verdijk R, Franke HR, Wolbers F, Vermes I. Differential effects of bisphosphonates on breast cancer cell lines. Cancer Lett. 2007;246:308–12. doi: 10.1016/j.canlet.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 122.Sato M, Grasser W, Endo N, Akins R, Simmons H, Thompson DD, Golub E, Rodan GA. Bisphosphonate action. Alendronate localization in rat bone and effects on osteoclast ultrastructure. J Clin Invest. 1991;88:2095–105. doi: 10.1172/JCI115539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carano A, Teitelbaum SL, Konsek JD, Schlesinger PH, Blair HC. Bisphosphonates directly inhibit the bone resorption activity of isolated avian osteoclasts in vitro. J Clin Invest. 1990;85:456–61. doi: 10.1172/JCI114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Giuliani N, Pedrazzoni M, Negri G, Passeri G, Impicciatore M, Girasole G. Bisphosphonates stimulate formation of osteoblast precursors and mineralized nodules in murine and human bone marrow cultures in vitro and promote early osteoblastogenesis in young and aged mice in vivo. Bone. 1998;22:455–61. doi: 10.1016/s8756-3282(98)00033-7. [DOI] [PubMed] [Google Scholar]
- 125.Liberman UA, Weiss SR, Broll J, Minne HW, Quan H, Bell NH, Rodriguez-Portales J, Downs RW, Jr, Dequeker J, Favus M, Seeman E, Recker RR, Capizzi T, Santora AC, II, Lombardi A, Shah RV, Hirsch LJ, Karpf DB. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. N Engl J Med. 1995;333:1437–43. doi: 10.1056/NEJM199511303332201. [DOI] [PubMed] [Google Scholar]
- 126.Rossini M, Gatti D, Zamberlan N, Braga V, Dorizzi R, Adami S. Long-term effects of a treatment course with oral alendronate of postmenopausal osteoporosis. J Bone Miner Res. 1994;9:1833–7. doi: 10.1002/jbmr.5650091121. [DOI] [PubMed] [Google Scholar]
- 127.Giuliani N, Girasole G, Pedrazzoni M, Passeri G, Gatti C, Passeri M. Alendronate stimulate b-FGF production and mineralized nodule formation in human osteoblastic cells and osteoblastogenesis in human bone marrow cultures. J Bone Miner Res. 1995;10:S171. [Google Scholar]
- 128.Canalis E, Centrella M, McCarthy T. Effects of basic fibroblast growth factor on bone formation in vitro. J Clin Invest. 1988;81:1572–7. doi: 10.1172/JCI113490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Long MW, Robinson JA, Ashcraft EA, Mann KG. Regulation of human bone marrow-derived osteoprogenitor cells by osteogenic growth factors. J Clin Invest. 1995;95:881–7. doi: 10.1172/JCI117738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Noff D, Pitaru S, Savion N. Basic fibroblast growth factor enhances the capacity of bone marrow cells to form bone-like nodules in vitro. FEBS Lett. 1989;250:619–21. doi: 10.1016/0014-5793(89)80808-7. [DOI] [PubMed] [Google Scholar]
- 131.Savion N, Pri-Chen S, Pitaru S. bFGF enhances the growth and the expressions of the osteogenic phenotype of dexamethasone treated human bone marrow derived bone-like cells in culture. J Bone Miner Res. 1996;11:S176. doi: 10.1016/s8756-3282(98)00087-8. [DOI] [PubMed] [Google Scholar]
- 132.Wang QR, Yan ZJ, Wolf NS. Dissecting the hematopoietic microenvironment. VI. The effects of several growth factors on the in vitro growth of murine bone marrow CFU-F. Exp Hematol. 1990;18:341–7. [PubMed] [Google Scholar]
- 133.Im G-I, Qureshi SA, Kenney J, Rubash HE, Shanbhag AS. Osteoblast proliferation and maturation by bisphosphonates. Biomaterials. 2004;25:4105–15. doi: 10.1016/j.biomaterials.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 134.Viereck V, Emons G, Lauck V, Frosch K-H, Blaschke S, Grundker C, Hofbauer LC. Bisphosphonates pamidronate and zoledronic acid stimulate osteoprotegerin production by primary human osteoblasts. Biochem Biophys Res Commun. 2002;291:680–6. doi: 10.1006/bbrc.2002.6510. [DOI] [PubMed] [Google Scholar]
- 135.Wood J, Bonjean K, Ruetz S, Bellahcene A, Devy L, Foidart JM, Castronovo V, Green JR. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055–61. doi: 10.1124/jpet.102.035295. [DOI] [PubMed] [Google Scholar]
- 136.Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, Boyce B, Zhao M, Gutierrez G. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–9. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 137.Meier CR, Schlienger RG, Kraenzlin ME, Schlegel B, Jick H. HMG-CoA reductase inhibitors and the risk of fractures. JAMA. 2000;283:3205–10. doi: 10.1001/jama.283.24.3205. [DOI] [PubMed] [Google Scholar]
- 138.Maeda T, Kawane T, Horiuchi N. Statins augment vascular endothelial growth factor expression in osteoblastic cells via inhibition of protein prenylation. Endocrinology. 2003;144:681–92. doi: 10.1210/en.2002-220682. [DOI] [PubMed] [Google Scholar]
- 139.Yazawa H, Zimmermann B, Asarni Y, Bernimoulin J-P. Simvastatin promotes cell metabolism, proliferation, and osteoblastic differentiation in human periodontal ligament cells. J Periodontol. 2005;76:295–302. doi: 10.1902/jop.2005.76.2.295. [DOI] [PubMed] [Google Scholar]
- 140.Song CL, Guo ZQ, Ma QJ, Chen ZQ, Liu ZJ, Jia HT, Dang GT. Simvastatin induces osteoblastic differentiation and inhibits adipocytic differentiation in mouse bone marrow stromal cells. Biochem Biophys Res Commun. 2003;308:458–62. doi: 10.1016/s0006-291x(03)01408-6. [DOI] [PubMed] [Google Scholar]
- 141.Thylin MR, McConnell JC, Schmid MJ, Reckling RR, Ojha J, Bhattacharyya I, Marx DB, Reinhardt RA. Effects of simvastatin gels on murine calvarial bone. J Periodontol. 2002;73:1141–8. doi: 10.1902/jop.2002.73.10.1141. [DOI] [PubMed] [Google Scholar]
- 142.Sugiyama M, Kodama T, Konishi K, Abe K, Asami S, Oikawa S. Compactin and simvastatin, but not pravastatin, induce bone morphogenetic protein-2 in human osteosarcoma cells. Biochem Biophys Res Commun. 2000;271:688–92. doi: 10.1006/bbrc.2000.2697. [DOI] [PubMed] [Google Scholar]
- 143.Desager JP, Horsmans Y. Clinical pharmacokinetics of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. Clin. Pharmacokinet. 1996;31:348–71. doi: 10.2165/00003088-199631050-00003. [DOI] [PubMed] [Google Scholar]
- 144.Blumenkranz MS, Claflin A, Hajek AS. Selection of therapeutic agents for intraocular proliferative disease. Arch Ophthalmol. 1984;102:598–604. doi: 10.1001/archopht.1984.01040030470029. [DOI] [PubMed] [Google Scholar]
- 145.Machemer R. Proliferative vitreoretinopathy (PVR): a personal account of its pathogenesis and treatment. Invest Ophthalmol Vis Sci. 1988;29:1771–83. [PubMed] [Google Scholar]
- 146.Grossfield H, Ragan C. Action of hydrocortisone on cells in tissue culture. Proc Soc Exp Biol Med. 1954;86:63. doi: 10.3181/00379727-86-21011. [DOI] [PubMed] [Google Scholar]
- 147.Ruhmann AG, Berliner DL. Effect of steroids on growth of mouse fibroblasts in vitro. Endocrinology. 1965;76:916. doi: 10.1210/endo-76-5-916. [DOI] [PubMed] [Google Scholar]
- 148.Lee DA, Shapourifar-Tehrani S, Stephenson TR, Kitada S. The effects of the fluorinated pyrimidines FUR, FUdR, FUMP, and FdUMP on human tendon's fibroblasts. Invest Ophthalmol Vis Sci. 1991;32:2599–609. [PubMed] [Google Scholar]
- 149.Yang CM, Cousins SW. Quantitative assessment of growth stimulating activity of the vitreous during PVR. Invest Ophthalmol Vis Sci. 1992;33:2436–42. [PubMed] [Google Scholar]
- 150.Vichi P, Tritton TR. Stimulation of growth in human and murine cells by adriamycin. Cancer Res. 1989;49:2679–82. [PubMed] [Google Scholar]
- 151.Pomerantz SM, Hepner BC, Wertz JM. Serotonergic influences on male sexual behavior of rhesus monkeys: effects of serotonin agonists. Psycopharmacology. 1993;111:47–54. doi: 10.1007/BF02257406. [DOI] [PubMed] [Google Scholar]
- 152.Smith ER, Lee RL, Schnur SL, Davidson JM. Alpha2-adrenoceptor antagonists and male sexual behavior: I. Mating Behavior. Physiol Behav. 1987;41:7–14. doi: 10.1016/0031-9384(87)90123-5. [DOI] [PubMed] [Google Scholar]
- 153.Smith ER, Lee RL, Schnur SL, Davidson JM. Alpha2-adrenoceptor antagonists and male sexual behavior: II. Erectile and Ejaculatory Reflexes. Physiol Behav. 1987;41:15–9. doi: 10.1016/0031-9384(87)90124-7. [DOI] [PubMed] [Google Scholar]
- 154.Yonezawa A, Kawamura S, Ando R, Tadano T, Nobunaga T, Kimura Y. Biphasic effects of yohimbine on the ejaculatory response in the dog. Life Sci. 1991;48:PL103, 9. doi: 10.1016/0024-3205(91)90233-2. [DOI] [PubMed] [Google Scholar]
- 155.Yonezawa A, Ando R, Watanabe C, Furuta S, Kutsuwa M, Sakurada S, Kimura Y. α2-Adrenoceptor antagonists: effects on ejaculation, penile erection and pelvic thrusting behavior in dogs. Pharmacol Biochem Behav. 2001;70:141–7. doi: 10.1016/s0091-3057(01)00584-6. [DOI] [PubMed] [Google Scholar]
- 156.Zarrindast M-R, Nojoomi K, Sharifzadeh M, Mokri A. Nitric oxide agents and apomorphine-induced rat behaviors. Pharmacology. 2004;71:169–73. doi: 10.1159/000078082. [DOI] [PubMed] [Google Scholar]
- 157.Baraldi M, Benassi-Benelli A, Bernabei MT, Cameroni R, Rferrari F, Ferrari P. Apocodeine-induced stereotypes and penile erection in rats. Pharmacology. 1979;18:165–9. doi: 10.1016/0028-3908(79)90057-1. [DOI] [PubMed] [Google Scholar]
- 158.Ferrari F, Baggio G. Potentiation of the aphrodisiac effect of N-n-propyl-norapomorphine by naloxone. Eur J Pharmacol. 1982;81:321–6. doi: 10.1016/0014-2999(82)90451-4. [DOI] [PubMed] [Google Scholar]
- 159.Pehek EA, Thompson JT, Eaton RC, Bazzett TJ, Hull EM. Apomorphine and haloperidol, but not domperidone, affect penile reflexes in rats. Pharmacol Biochem Behav. 1988;31:201–8. doi: 10.1016/0091-3057(88)90334-6. [DOI] [PubMed] [Google Scholar]
- 160.Matsuoka N, Maeda N, Yamazaki M, Yamaguchi I. Brain somatostatin depletion by cysteamine attenuates the penile erection induced by serotonergic and dopaminergic, but not by cholinergic, activation in rats. Brain Res. 1996;729:132–6. [PubMed] [Google Scholar]
- 161.Ferrari F, Pelloni F, Giuliani D. Behavioural evidence that different neurochemical mechanisms underly stretching-yawning and penile erection induced in male rats by SND 919, a new selective D2 dopamine receptor agonist. Psychopharmacology. 1993;113:172–6. doi: 10.1007/BF02245694. [DOI] [PubMed] [Google Scholar]
- 162.Ferrari F, Baggio G, Mangiafico V. The dopamine autoreceptor agonist B-HT 920 markedly stimulates sexual behavior in male rats. Experientia. 1985;41:636–8. doi: 10.1007/BF02007696. [DOI] [PubMed] [Google Scholar]
- 163.Benassi-Benelli A, Ferrari F, Quarantotti BP. Penile erection induced by apomorphine and N-n-propyl-norapomorphine in rats. Arch Int Pharmacodyn Ther. 1979;242:241–7. [PubMed] [Google Scholar]
- 164.Brioni JD, Moreland RB, Cowart M, Hsieh GC, Stewrt AO, Hedlund P, Donnelly-Roberts DL, Nakane M, Lynch JJ, III, Kolasa T, Polakowski JS, Osinski MA, Marsh K, Andersson K-E, Sullivan JP. Activation of dopamine D4 receptors by ABT-724 induced penile erection in rats. Proc Natl Acad Sci USA. 2004;101:6758–63. doi: 10.1073/pnas.0308292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sharifzadeh M, Dehpout AR, Samini M, Hassan-Mazandarani H, Samadian T, Asghari GhR. Alterations of bromocriptine-induced penile erection by chronic lithium in rats. J Psychopharmacol. 1996;10:157–61. doi: 10.1177/026988119601000212. [DOI] [PubMed] [Google Scholar]
- 166.Dehpour AR, Samini M, Sharifzadeh M, Hasan-Mazandarani H. Effects of chronic lithium pretreatment on apomorphine-induced penile erections. Gen Pharmacol. 1995;26:1015–20. doi: 10.1016/0306-3623(94)00276-s. [DOI] [PubMed] [Google Scholar]
- 167.Hsieh GC, Hollingsworth PR, Martino B, Chang R, Terranova MA, O’Neill AB, Lynch JJ, Moreland RB, Donnelly-Roberts DL, Kolasa T, Mikusa JP, McVey JM, Marsh KC, Sullivan JP, Brioni JD. Central mechanisms regulating penile erection in conscious rats: the dopaminergic systems related to the proerectile effect of apomorphine. J Pharmacol Exp Ther. 2004;308:330–8. doi: 10.1124/jpet.103.057455. [DOI] [PubMed] [Google Scholar]
- 168.Sala M, Braida D, Leone MP, Calcaterra P, Monti S, Gori E. Central effect of yohimbine on sexual behavior in the rat. Physiol Behav. 1990;47:165–73. doi: 10.1016/0031-9384(90)90057-b. [DOI] [PubMed] [Google Scholar]
- 169.Yonezawa A, Ando R, Imai M, Watanabe C, Furuta S, Kutsuwa M, Kimura Y, Sakurada S. Differential effects of yohimbine, naloxone and 8-OH-DPAT on ejaculatory response in male dogs. Methods Find Exp Clin Pharmacol. 2004;26:47–51. doi: 10.1358/mf.2004.26.1.793472. [DOI] [PubMed] [Google Scholar]
- 170.Tallentire D, McRae G, Spedding M, Clark R, Vickery B. Modulation of sexual behaviour in the rat by a potent and selective α2-adrenoceptor antagonist, delequamine (RS-15385-197) Br J Pharmacol. 1996;118:63–72. doi: 10.1111/j.1476-5381.1996.tb15367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yonezawa A, Yoshizumi M, Ebiko M, Amano T, Kimura Y, Sakurada S. Long-lasting effects of yohimbine on the ejaculatory function in male dogs. Biomed Res. 2005;26:201–6. doi: 10.2220/biomedres.26.201. [DOI] [PubMed] [Google Scholar]
- 172.Berendsen HHG, Broekkamp CLE. Drug-induced penile erections in rats: indications of serotonin1B receptor mediation. Eur J Pharmacol. 1987;135:279–87. doi: 10.1016/0014-2999(87)90676-5. [DOI] [PubMed] [Google Scholar]
- 173.Berendsen HHG, Jenck F, Broekkamp CLE. Involvement of 5-HT1C-receptors in drug-induced penile erections in rats. Psychopharmacology. 1990;101:57–61. doi: 10.1007/BF02253718. [DOI] [PubMed] [Google Scholar]
- 174.Bagdy G, Kalogeras KT, Szemeredi K. Effect of 5-HT1C and 5-HT2 receptor stimulation on excessive grooming, penile erection and plasma oxytocin concentrations. Eur J Pharmacol. 1992;229:9–14. doi: 10.1016/0014-2999(92)90279-d. [DOI] [PubMed] [Google Scholar]
- 175.Yonezawa A, Yoshizumi M, Ebiko M, Ise S-N, Watanabe C, Mizoguchi H, Kimura Y, Sakurada S. Ejaculatory response induced by a 5-HT2 receptor agonist m-CPP in rats: differential roles of 5-HT2 receptor subtypes. Pharmacol Biochem Behav. 2008;88:367–73. doi: 10.1016/j.pbb.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 176.Millan MJ, Peglion J-L, Lavielle G, Perrin-Monneyron S. 5-HT2C receptors mediate penile erections in rats: actions of novel and selective agonists and antagonists. Eur J Pharmacol. 1997;325:9–12. doi: 10.1016/s0014-2999(97)89962-1. [DOI] [PubMed] [Google Scholar]
- 177.Stancampiano R, Melis MR, Argiolas A. Penile erection and yawning induced by 5-HT1C receptor agonists in male rats: relationship with dopaminergic and oxytocinergic transmission. Eur J Pharmacol. 1994;261:149–55. doi: 10.1016/0014-2999(94)90313-1. [DOI] [PubMed] [Google Scholar]
- 178.Kimura Y, Hatanaka K-I, Naitou Y, Maeno K, Shimada I, Koakutsu A, Wanibuchi F, Yamaguchi T. Pharmacological profile of YM348, a novel, potent and orally active 5-HT2C receptor agonist. Eur J Pharmacol. 2004;483:37–43. doi: 10.1016/j.ejphar.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 179.Kimura Y, Naitou Y, Wanibuchi F, Yamaguchi T. Characterization of intracavernous pressure increase induced by Ym348, a novel 5-HT2C receptor agonist, in anesthetized rats. J Urol. 2006;175:1953–7. doi: 10.1016/S0022-5347(05)00920-1. [DOI] [PubMed] [Google Scholar]
- 180.Tharakan B, Manyam BV. Botanical therapies in sexual dysfunction. Phytother Res. 2005;19:457–63. doi: 10.1002/ptr.1634. [DOI] [PubMed] [Google Scholar]
- 181.Mostafa T. In vitro sildenafil citrate use as a sperm motility stimulant. Fertil Steril. 2007;88:994–6. doi: 10.1016/j.fertnstert.2006.11.182. [DOI] [PubMed] [Google Scholar]
- 182.Edwards TM, Lindley N. Phosphodiesterase type 5 inhibition coupled to strong reinforcement results in two periods of transient retention loss in the young chick. Behav Brain Res. 2007;183:231–5. doi: 10.1016/j.bbr.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 183.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98:9836–41. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Frid P, Anisimov SV, Popovic N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev. 2007;53:135–60. doi: 10.1016/j.brainresrev.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 185.Wells GAH, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, Dawson M, Bradley R. A novel progressive spongiform encephalopathy in cattle. Vet Rec. 1987;121:419–20. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 186.Rudyk H, Vasiljevic S, Hennion RM, Birkett CR, Hope J, Gilbert IH. Screening Congo red and its analogues for their ability to prevent the formation of PrP-res in scrapie-infected cells. J Gen Virol. 2000;81:1155–64. doi: 10.1099/0022-1317-81-4-1155. [DOI] [PubMed] [Google Scholar]
- 187.Rudyk H, Knaggs MH, Vasiljevic S, Hope J, Birkett C, Gilbert IH. Synthesis and evaluation of analogues of Congo Red as potential compounds against transmissible spongiform encephalopathies. Eur J Med Chem. 2003;38:567–79. doi: 10.1016/s0223-5234(03)00081-3. [DOI] [PubMed] [Google Scholar]
- 188.Clarke M. Infection of cell cultures with the scrapie agent. In: Prusiner S, Hadlow W, editors. Slow transmissible diseases of the nervous system. New York: Academic Press; 1979. pp. 225–33. [Google Scholar]
- 189.Ingrosso L, Ladogana A, Pocchiari M. Congo red prolongs the incubation period in scrapie-infected hamsters. J Virol. 1995;69:506–8. doi: 10.1128/jvi.69.1.506-508.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Caspi S, Halimi M, Yanai A, Ben Sasson S, Taraboulos A, Gabizon R. The anti-prior activity of Congo red. Putative mechanism. J Biol Chem. 1998;273:3484–9. doi: 10.1074/jbc.273.6.3484. [DOI] [PubMed] [Google Scholar]
- 191.Skowronek M, Stopa B, Konieczny L, Rybarska J, Piekarska B, Szneler E, Bakalarski G, Roterman I. Self-assembly of Congo red – a theoretical and experimental approach to identify its supramolecular organization in water and salt solutions. Biopolymers. 1998;46:267–81. [Google Scholar]
- 192.Stopa B, Gorny M, Konieczny L, Piekarska B, Rybarska J, Skowronek M, Roterman I. Supramolecular ligands: monomer structure and protein ligation capability. Biochimie. 1998;80:963–8. doi: 10.1016/s0300-9084(99)80001-7. [DOI] [PubMed] [Google Scholar]
- 193.Calabrese EJ, Baldwin LA. Chemical hormesis: its historical foundations as a biological hypothesis. Hum Exp Toxicol. 2000;19:2–31. doi: 10.1191/096032700678815585. [DOI] [PubMed] [Google Scholar]
- 194.Calabrese EJ, Baldwin LA. The marginalization of hormesis. Hum Exp Toxicol. 2000;19:32–40. doi: 10.1191/096032700678815594. [DOI] [PubMed] [Google Scholar]
- 195.Calabrese EJ, Baldwin LA. Radiation hormesis: its historical foundations as a biological hypothesis. Hum Exp Toxicol. 2000;19:41–75. doi: 10.1191/096032700678815602. [DOI] [PubMed] [Google Scholar]
- 196.Calabrese EJ, Baldwin LA. Radiation hormesis: the demise of a legitimate hypothesis. Hum Exp Toxicol. 2000;19:76–84. doi: 10.1191/096032700678815611. [DOI] [PubMed] [Google Scholar]
- 197.Calabrese EJ, Baldwin LA. Tales of two similar hypotheses: the rise and fall of chemical and radiation hormesis. Hum Exp Toxicol. 2000;19:85–97. doi: 10.1191/096032700678815620. [DOI] [PubMed] [Google Scholar]
- 198.Ji LL. Physical activity: a strong stimulant for hormesis during aging. In: Le Bourg E, Rattan SIS, editors. Mild Stress and Healthy Aging. Berlin: Springer; 2008. pp. 97–114. [Google Scholar]
- 199.Radak Z, Chung HY, Goto S. Exercise and hormesis: oxidative stress-related adaptation for successful aging. Biogerontology. 2005;6:71–5. doi: 10.1007/s10522-004-7386-7. [DOI] [PubMed] [Google Scholar]
- 200.Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing Res Rev. 2008;7:34–42. doi: 10.1016/j.arr.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 201.Gomez-Pinilla F. The influences of diet and exercise on mental health through hormesis. Ageing Res Rev. 2008;7:49–62. doi: 10.1016/j.arr.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Turturro A, Hass B, Hart RW. Hormesis – implications for risk assessment caloric intake (body weight) as an exemplar. Hum Exp Toxicol. 1998;17:454–9. doi: 10.1177/096032719801700810. [DOI] [PubMed] [Google Scholar]
- 203.Turturro A, Hass BS, Hart RW. Does caloric restriction induce hormesis? Hum Exp Toxicol. 2000;19:320–9. doi: 10.1191/096032700678815981. [DOI] [PubMed] [Google Scholar]
- 204.Mattson MP. Energy intake, meal frequency, and health: a neurobiological perspective. Annu Rev Nutr. 2005;25:237–60. doi: 10.1146/annurev.nutr.25.050304.092526. [DOI] [PubMed] [Google Scholar]
- 205.Mattson MP, Wan R. Beneficial effects of intermittent fasting and caloric restriction on the cardiovascular and cerebrovascular systems. J Nutr Biochem. 2005;16:129–37. doi: 10.1016/j.jnutbio.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 206.Abete P, Calabrese E, Ji LL, Kristensen T, Le Bourg E, Loeschcke V, Morris B, Rengo F, Rattan SIS, Safwat A, Sarup P, Sorensen J, Vaiserman A. Conclusion. Mild stress and healthy aging: perspectives for human beings. In: Le Bourg E, Rattan SIS, editors. Mild Stress and Healthy Aging. Berlin: Springer; 2008. pp. 171–83. [Google Scholar]
- 207.Leung WC, Zheng H, Huen M, Law SL, Xue H. Anxiolytic-like action of orally administered dl-tetrahydropalmatine in elevated plus-maze. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:775–9. doi: 10.1016/s0278-5846(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 208.Honar H, Riazi K, Homayoun H, Sadeghipour H, Rashidi N, Ebrahimkhani MR, Mirazi N, Dehpour AR. Ultra-low dose naltrexone potentiates the anticonvulsant effect of low dose morphine on clonic seizures. Neuroscience. 2004;129:733–42. doi: 10.1016/j.neuroscience.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 209.Stratton LO, Petrinovich L. Post-trial injections of an anti-cholinesterase drug and maze-learning in 2 strains of rats. Psychopharmacologia. 1963;5:47–54. doi: 10.1007/BF00405574. [DOI] [PubMed] [Google Scholar]
- 210.Wu W-C, Kao Y-H, Hu D-N. A comparative study of effects of antiproliferative drugs on human retinal pigment epithelial cells in vitro. J Ocul Pharmacol Ther. 2002;18:251–64. doi: 10.1089/108076802760116179. [DOI] [PubMed] [Google Scholar]
- 211.Calabrese EJ. An assessment of anxiolytic drug screening tests: Hormetic dose responses predominate. Crit Rev Toxicol. 2008;38:489–542. doi: 10.1080/10408440802014238. [DOI] [PubMed] [Google Scholar]
- 212.Calabrese EJ. Modulation of the epileptic seizure threshold: Implications of biphasic dose responses. Crit Rev Toxicol. 2008;38:543–56. doi: 10.1080/10408440802014261. [DOI] [PubMed] [Google Scholar]
- 213.Calabrese EJ. Alzheimer's disease drugs: An application of the hormetic dose-response model. Crit Rev Toxicol. 2008;38:419–51. doi: 10.1080/10408440802003991. [DOI] [PubMed] [Google Scholar]
- 214.Calabrese EJ, Bachmann KA, Bailer AJ, Bolger PM, Borak J, Cai L, Cedergreen N, Cherian MG, Chiueh CC, Clarkson TW, Cook RR, Diamond DM, Doolittle DJ, Dorato MA, Duke SO, Feinendegen L, Gardner DE, Hart RW, Hasting KL, Hayes AW, Hoffmann GR, Ives JA, Jaworowski Z, Johnson TE, Jonas WB, Kaminski NE, Keller JG, Klaunig JE, Knudsen TB, Kozumbo WJ, Lettieri T, Liu SZ, Maisseu A, Maynard KI, Masoro EJ, McClellan RO, Mehendale HM, Mothersill C, Newlin DB, Nigg HN, Oehme FW, Phalen RF, Philbert MA, Rattan SI, Riviere JE, Rodricks J, Sapolsky RM, Scott BR, Seymour C, Sinclair DA, Smith-Sonneborn J, Snow ET, Spear L, Stevenson DE, Thomas Y, Tubiana M, Williams GM, Mattson MP. Biological stress terminology: integrating the concepts of adapative response and preconditioning stress within a hormetic dose-response framework. Toxicol Appl Pharmacol. 2007;222:122–8. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]