

Abstract
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT.
Most β-lactams are excreted by filtration and to a greater extent by tubular secretion, which is a capacity-limited saturable pathway.
Pharmacokinetic interactions between co-administered β-lactams have been frequently reported; however, their mechanism and possible clinical benefits are not well defined.
We are not aware of the interaction between piperacillin and flucloxacillin being reported in the literature.
WHAT THIS STUDY ADDS.
Piperacillin inhibits the renal and nonrenal elimination of flucloxacillin to a clinically significant extent, but not vice versa.
Modelling suggests that the mechanism for the decrease of renal clearance of flucloxacillin is probably competitive inhibition of renal tubular secretion by piperacillin.
Piperacillin has a 15-times higher affinity for the renal transporter than flucloxacillin based on the molar ratio.
AIMS
To explore the extent, time course, site(s), mechanism and possible clinical relevance of the pharmacokinetic (PK) interaction between piperacillin and flucloxacillin.
METHODS
A single-dose, randomized, six-way crossover study in 10 healthy volunteers where all subjects received all of the following as 5-min intravenous infusions: (i) 1.5 g piperacillin, (ii) 0.5 g flucloxacillin, (iii) 1.5 g piperacillin + 0.5 g flucloxacillin, (iv) 3 g piperacillin, (v) 1 g flucloxacillin, and (vi) 3 g piperacillin + 1 g flucloxacillin. Drug concentrations in plasma and urine were determined by high-performance liquid chromatography. WinNonlin® was used for PK modelling and statistics.
RESULTS
Piperacillin significantly decreased the renal clearance of flucloxacillin from 5.44 to 2.29 l h−1 (medians, P < 0.01) and the nonrenal clearance of flucloxacillin from 2.67 to 1.80 l h−1 (P < 0.01). The renal clearance of flucloxacillin was reduced to 45% (point estimate, 90% confidence interval 40 to 50%) and the nonrenal clearance to 66% (59, 73). The extent of interaction was larger at the higher doses. Competitive inhibition of tubular secretion by piperacillin was identified as the most likely mechanism for the decreased renal clearance of flucloxacillin. Piperacillin had a 15-times higher affinity for the renal transporter than flucloxacillin based on the molar ratio. Piperacillin PK was only slightly affected by flucloxacillin.
CONCLUSIONS
Piperacillin inhibits renal and nonrenal elimination of flucloxacillin. This interaction seems clinically significant, as total clearance was reduced by a factor of 1.5 for the lower and 2.1 for the higher doses. PK interactions, especially with piperacillin, are likely to occur also with other β-lactam combinations and might be useful to improve the effectiveness of antibacterial treatment.
Keywords: flucloxacillin, pharmacokinetic interaction, piperacillin
Introduction
β-Lactams have remained a main therapeutic option in Gram-positive and Gram-negative infections. Piperacillin, an acylureido penicillin, in combination with the β-lactamase inhibitor tazobactam, is a frequently used intravenous antibiotic, especially in the empirical treatment of hospital-acquired infections. Other combination therapy with piperacillin involves β-lactams such as flucloxacillin [1–5] or aminoglycosides [6–8]. The combination of piperacillin and flucloxacillin together with an aminoglycoside has been used successfully for the treatment of infections in children with cancer [2, 3]. These examples show that combination therapy is very important in using β-lactams efficiently, but also that the pharmacokinetic (PK) interactions need special attention, since they can be of therapeutic importance by altering the pharmacodynamic profile.
Like most β-lactams, piperacillin and flucloxacillin are predominantly eliminated renally [9, 10] by glomerular filtration and tubular secretion. Tubular secretion is an active saturable process. If two drugs that are primarily eliminated by tubular secretion are given concomitantly, clinically significant decreases in clearance may result [11]. This has been frequently shown with β-lactams and probenecid, a well-known inhibitor of tubular secretion [9, 12, 13]. Renal interactions have also been reported when two β-lactams were given together [14–17]. Therefore, the possibility of PK drug–drug interactions should be considered when two or more β-lactams are given in combination.
Interactions at transporters have been frequently studied in vitro. With animal studies it needs to be considered that activities of transporters may exhibit species differences, as has been shown for the organic anion transporter 3 (OAT 3) [18]. Most recently, Nozaki et al. [19] reported the use of human kidney slices to study the uptake of organic anions, including benzylpenicillin, by OAT 1 and OAT 3. They could show that human kidney slices maintain transport activities and are useful in characterization of renal drug transport. However, they also concluded that one should be cautious when extrapolating their data to the normal human response. Therefore, PK studies in humans are necessary. Several PK interaction studies have been published [15, 20–25]. However, little has been reported about the exact mechanism, which is important to know in order to predict the extent and time-course of interactions for different agents and dosage regimens. Noncompartmental analysis, which has been used in the vast majority of published studies, does not allow these predictions.
The aim of our study was to describe the extent of interaction between flucloxacillin and piperacillin at different dose levels in healthy volunteers. Our secondary objective was to explore the time course, site(s) and possible mechanisms of the interaction between the two β-lactams by compartmental modelling techniques.
Methods
Study participants
The study included 10 healthy White volunteers (five male and five female). All subjects had to undergo laboratory tests including urinalysis and screening for drugs of abuse, a physical examination, and electrocardiography before the start of the study. The volunteers were asked to complete a questionnaire on their health status on each study day. Subjects were asked to report any adverse reactions or discomfort, and were closely observed by physicians during the study periods. All subjects gave their written informed consent prior to entry into the study. The study was approved by the investigational review board of the Medical Faculty and University Hospital of the University of Essen, Germany.
Study design and drug administration
A randomized, controlled, six-way crossover study was conducted. Each subject received all of the following treatments: (i) 1.5 g piperacillin, (ii) 0.5 g flucloxacillin, (iii) 1.5 g piperacillin + 0.5 g flucloxacillin, (iv) 3 g piperacillin, (v) 1 g flucloxacillin, and (vi) 3 g piperacillin + 1 g flucloxacillin. The doses were administered as 5-min intravenous infusions. Food and fluid intake were strictly standardized on each study day. Treatment periods were separated by a wash-out period of at least 4 days. Subjects were requested to abstain from alcohol and caffeine-containing foods and beverages and other medications during the study periods.
Sampling schedule
Blood samples were drawn via an intravenous catheter from a forearm vein contralateral to the one used for drug administration. Blood samples were drawn immediately before start of infusion, at the end of infusion as well as at 5, 10, 15, 20, 25, 45, 60, 75, 90 min and 2, 2.5, 3, 3.5, 4, 5, 6, 8 and 24 h after the end of the infusion. The samples were cooled in an ice-water bath for 10–15 min before centrifugation. After centrifugation plasma samples were immediately frozen and stored at −70°C until analysis. Urine samples were collected immediately before start of the infusion, from start of the infusion until 1 h after end of infusion and in the following time intervals: 1–2, 2–3, 3–4, 4–5, 5–6, 6–8, 8–12 and 12–24 h after the end of the infusion. The urine samples were stored at 4°C during the collection period. The amount and pH of the urine were measured. Samples were immediately frozen and stored at −70°C until analysis.
Determination of plasma and urine concentrations
Piperacillin and flucloxacillin concentrations in plasma and urine were determined by high-performance liquid chromatography (HPLC). For drug determination in plasma, 100 µl of the sample was deproteinized with 200 µl acetonitrile containing the internal standard (mezlocillin). After mixing and centrifugation at 21 885 g, 40 µl was injected onto the HPLC system. For determination of piperacillin and flucloxacillin in urine, 20 µl of the sample was diluted with 180 µl water. After mixing, 40 µl was injected onto the HPLC system. Drug concentrations were determined using a reversed phase column (C18, 5 µm, 250 × 4.6 mm i.d.), 0.01 M potassium dihydrogen phosphate (pH 6.2)/acetonitrile mobile phase (4 : 1; v : v) with a flow of 2 ml min−1. Piperacillin, flucloxacillin and the internal standard were detected at 220 nm.
The plasma and urine samples were measured against a plasma or urine calibration row. For control of interassay variation, spiked quality controls in plasma and urine were prepared. No interference was observed in plasma and urine for piperacillin, flucloxacillin and the internal standard. Calibration was performed by linear regression. The linearity of piperacillin calibration curves in plasma and urine was shown from 0.200 to 150 mg l−1 and 1.00 to 1000 mg l−1 for piperacillin, and from 0.500 to 250 mg l−1 and 5.00 to 400 mg l−1 for flucloxacillin. The quantification limits were identical to the lowest calibration levels. The interday precision and the analytical recovery of the spiked quality control standards in human plasma ranged from 3.5 to 9.2% and 95.0 to 106.9% for piperacillin and from 4.1 to 7.7% and 84.9 to 106.0% for flucloxacillin. The interday precision and the analytical recovery of the spiked quality control standards in human urine ranged from 3.0 to 5.5% and 92.0 to 97.9% for piperacillin, and from 3.3 to 5.1% and 100.0 to 103.0% for flucloxacillin.
Noncompartmental analysis
The maximum plasma concentrations for each subject were read directly from the plasma concentration–time curves. The area under the plasma concentration–time curve (AUC) for each subject was calculated using the linear up/log down method in WinNonlin™ Professional (version 4.0.1; Pharsight Corp., Mountain View, CA, USA).
Compartmental modelling
We modelled all plasma and urine profiles from the six different treatments simultaneously by the standard two-stage approach in WinNonlin™. Model discrimination was based on the following four criteria: (i) visual inspection of the observed and predicted plasma and urine concentration–time curves, (ii) visual comparison of the patterns of systematic and random residuals, (iii) intrasubject comparison of Akaike criteria (AIC) between competing models, and (iv) the frequency of subjects for each model who had the best AIC for the respective model.
For models that gave similar performance when assessed against the above criteria, we simulated the plasma and urine concentration–time profiles for 5000 subjects for each competing model in nonmem version V release 1.1 (NONMEM Project Group, University of California, San Francisco, CA, USA). From these data we calculated the median and the nonparametric 80% prediction interval (10–90% percentile) for the predicted plasma concentrations and amounts in urine. These prediction interval lines were then overlayed the original raw data. If the model described the data adequately, then 20% of the observed data points should fall outside the 80% prediction interval at each time point. We compared the median predicted concentrations and the 80% prediction interval with the raw data. For the visual predictive check we tested whether the median and the 80% prediction interval mirrored the central tendency and the variability of the raw data for the respective model.
Models
We tested one-, two- and three-compartment disposition models for both piperacillin and flucloxacillin. The drug input was modelled as a zero order process with 5 min duration. We could separate between renal and nonrenal elimination because we determined the amounts of piperacillin and flucloxacillin excreted in urine. We described renal clearance as parallel first-order and mixed-order elimination process and used the following equation for the renal clearances of piperacillin and flucloxacillin:
CLR,linear is the first-order renal clearance, VmaxR is the maximum rate of the mixed-order renal elimination, KmR is the piperacillin or flucloxacillin concentration associated with a half maximal rate (VmaxR/2) for the mixed-order renal elimination, and [C] is the plasma concentration of piperacillin or flucloxacillin. As all subjects had normal renal function and the nonprotein-bound fraction in plasma (fU) is reported as 6% [26, 27] for flucloxacillin, the renal filtration clearance of flucloxacillin (fU ·glomerular filtration rate) is about 0.432 l h−1, which accounts for only approximately 5% of total body clearance. Therefore, we fixed CLR,linear to 0.432 l h−1 for flucloxacillin and did not estimate CLR,linear for flucloxacillin.
For models without a mechanistic interaction for nonrenal clearance of flucloxacillin, we described the nonrenal elimination as a first-order process with a nonrenal clearance (CLNR,FLU). For models with a mechanistic interaction for nonrenal elimination we described the nonrenal clearance of flucloxacillin as a mixed-order process:
VmaxNR,FLU is the maximum rate of the nonrenal elimination, KmNR,FLU is the flucloxacillin concentration associated with a half maximal rate (VmaxNR,FLU/2) for the mixed-order nonrenal elimination of flucloxacillin, and [F] is the plasma concentration of flucloxacillin.
Interaction models
We assumed that the first-order renal elimination (glomerular filtration) of flucloxacillin was not influenced by piperacillin. The following four mechanistic interactions for the mixed-order renal elimination process were modelled: competitive, uncompetitive, mixed and noncompetitive inhibition (Table 1). Besides those mechanistic interactions we also studied static interactions. The static interactions were expressed as two different nonrenal clearances for flucloxacillin with (CLNR,FLU with P) and without (CLNR,FLU without P) piperacillin. The studied models with different combinations of interactions at the renal and nonrenal sites are shown in Table 2. For competitive interactions we calculated the relative affinity (=ratio of Km,FLU/Kic) of piperacillin and flucloxacillin to the transporter for each subject (Table 1), while accounting for differences in molecular weight between piperacillin and flucloxacillin. The molecular weight is 517 for piperacillin base and 453 for flucloxacillin base.
Table 1.
Drug–drug interaction models
| Type of inhibition | Inhibition constant | Apparent Km | Apparent Vmax |
|---|---|---|---|
| Competitive | Kic | Vmax | |
| Uncompetitive | Kiu | 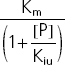 |
 |
| Mixed | Kic, Kiu | 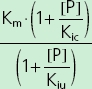 |
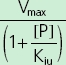 |
| Non-competitive | Ki | Km | 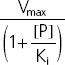 |
Vmax (mg h−1), maximum rate of the active transport process; Km (mg l−1), Michaelis–Menten constant, flucloxacillin concentration at half Vmax; [P] (mg l−1), piperacillin concentration; Kic (mg l−1), competitive inhibition constant, describes the affinity of piperacillin to the drug transporter without the bound substrate (i.e. without flucloxacillin); Kiu (mg l−1), uncompetitive inhibition constant, describes the affinity of piperacillin to the transporter–flucloxacillin complex; Ki (mg l−1), noncompetitive inhibition constant, represents the special case that piperacillin binds to both the transporter without the bound substrate and the transporter–flucloxacillin complex with the same affinity (i.e. Ki = Kic = Kiu).
Table 2.
Interaction models studied
| Model | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Interaction in renal elimination | C | UC | M | NC | C |
| Interaction in nonrenal elimination | S | S | S | S | C |
Statistical analysis
The noncompartmental and compartmental parameter estimates were tested for differences between treatments. anova statistics on log scale and an α-level of significance of 0.05 were used.
Results
All 10 subjects completed the study. The average ± SD weight was 75.6 ± 7.3 kg for men and 63.6 ± 8.3 kg for women, height 183.2 ± 5.0 cm for men and 171.8 ± 6.1 cm for women, and age 26.8 ± 4.2 years for men and 24.6 ± 0.55 years for women. Plasma concentrations of flucloxacillin after infusion of 0.5 g and 1 g were considerably higher with piperacillin compared with flucloxacillin given alone (Figure 1). Flucloxacillin amounts excreted in urine during the first 2 h after administration were much lower with the interaction treatments compared with baseline (Figure 1).
Figure 1.
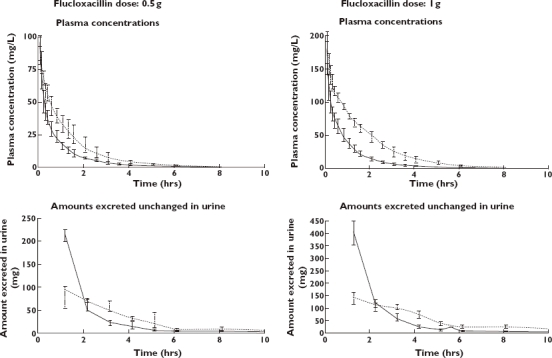
Median [P25%–P75%] profiles of flucloxacillin in healthy volunteers after a 5-min infusion of 0.5 g and 1 g flucloxacillin with or without piperacillin. Solid lines: flucloxacillin alone (—); Dashed lines: flucloxacillin 0.5 g with piperacillin 1.5 g or flucloxacillin 1 g with piperacillin 3 g (––)
Noncompartmental analysis
The results of the noncompartmental analysis (NCA) are shown in Table 3. Addition of the three times (on a mg basis) higher dose of piperacillin reduced the renal clearance of flucloxacillin by 46% (from 5.44 to 2.90 l h−1) for the low dose and by 63% (from 5.47 to 2.06 l h−1) for the high dose (P < 0.01). Nonrenal clearance was reduced by 33% (from 2.66 to 1.77 l h−1) for the low dose and by 35% (from 2.79 to 1.80 l h−1) for the high dose (P < 0.01). Figure 2 shows box plots for renal and nonrenal clearance of flucloxacillin with and without piperacillin. The time (and concentration) dependence of the observed interaction is shown in Figure 3. Median renal clearance of flucloxacillin in the presence of piperacillin increased from 1.2 l h−1 at 1 h after the infusion to 5.2 l h−1 at 8 h. The inhibition was more pronounced for the high-dose than for the low-dose regimen. Towards the end of the observation period, renal clearances were similar for the interaction treatments and flucloxacillin given alone.
Table 3.
Pharmacokinetic parameters from noncompartmental analysis for 0.5 g and 1 g flucloxacillin (FLU) with or without piperacillin (PIP)
| Median (P25%–P75%) | Point estimate (90% CI) | P-value* | Median (P25%–P75%) | Point estimate (90% CI) | P-value* | |||
|---|---|---|---|---|---|---|---|---|
| Dose FLU | 0.5 g | 0.5 g | With PIP/without PIP | 1 g | 1 g | With PIP/without PIP | ||
| Dose PIP | 1.5 g | – | 3 g | – | ||||
| CL (l h−1) | 5.04 (3.80–6.07) | 7.82 (7.37–9.36) | 59% (54, 65) | <0.01 | 3.79 (3.53–4.21) | 7.96 (6.81–9.57) | 47% (44, 50) | <0.01 |
| CLR (l h−1) | 2.90 (2.20–3.46) | 5.44 (4.63–6.27) | 54% (46, 64) | <0.01 | 2.06 (1.88–2.35) | 5.47 (4.52–6.07) | 37% (34, 41) | <0.01 |
| CLNR (l h−1) | 1.77 (1.53–2.60) | 2.66 (2.18–3.44) | 67% (56, 80) | <0.01 | 1.80 (1.64–1.99) | 2.79 (2.17–3.48) | 65% (55, 76) | <0.01 |
| fR (%) | 56.7 (53.9–72.0) | 68.6 (61.2–70.6) | 92% (82, 102) | 0.18 | 51.8 (50.8–58.5) | 64.0 (59.7–74.6) | 80% (73, 87) | <0.01 |
| Vss (l) | 7.14 (6.23–7.77) | 9.52 (8.38–11.1) | 72% (64, 80) | <0.01 | 6.56 (5.99–6.93) | 10.4 (8.89–11.6) | 64% (59, 70) | <0.01 |
| Cmax (mg l−1) | 95.8 (82.0–101) | 87.7 (79.1–93.0) | 106% (94, 120) | 0.40 | 183 (164–209) | 173 (144–192) | 109% (99, 120) | 0.13 |
| t1/2 (h) | 1.35 (1.18–1.45) | 1.44 (1.31–1.75) | 95% (74, 121) | 0.70 | 1.24 (1.13–1.39) | 1.59 (1.32–1.84) | 77% (70, 85) | <0.01 |
| MRT (h) | 1.44 (1.33–1.56) | 1.17 (1.06–1.41) | 122% (115, 130) | <0.01 | 1.64 (1.47–1.79) | 1.18 [1.13–1.34) | 136% (129, 144) | <0.01 |
P-value from anova. CI, confidence interval; CL, total body clearance; CLR, renal clearance; CLNR, nonrenal clearance; Cmax, maximum plasma concentration; fR, fraction excreted unchanged in urine; MRT, mean residence time; t1/2, terminal half-life in plasma; Vss, volume of distribution at steady state.
Figure 2.
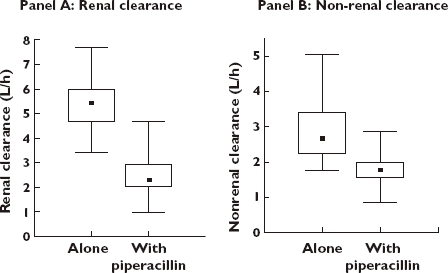
Renal and nonrenal clearance of flucloxacillin without and with piperacillin (results from noncompartmental analysis). Median (▪); 25%–75% ( ); Min–Max (
); Min–Max ( )
)
Figure 3.
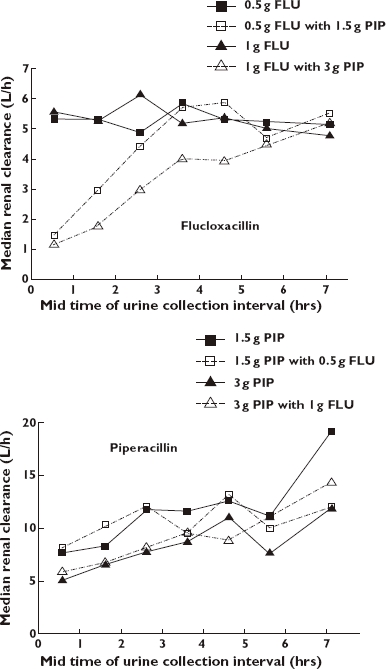
Median renal clearances of piperacillin (PIP) and flucloxacillin (FLU) from noncompartmental analysis
According to NCA, volume of distribution at steady state and mean residence time was significantly changed for both interaction treatments compared with baseline. We simulated the plasma concentration–time profiles for flucloxacillin with and without piperacillin based on the final parameter estimates from model 1. In this simulation, all flucloxacillin PK parameters except nonrenal clearance were identical between the treatments. The volume of distribution at steady state determined by NCA of these curves was 25% (median) too low compared with the true volume for the interaction treatment, but only 4.6% too low for the treatment without interaction. Therefore, the decrease in volume of distribution was probably an artefact from NCA.
NCA of the observed data (Table 3) showed that terminal half-life in plasma and fraction excreted unchanged in urine was only significantly affected with the high-dose interaction treatment. A higher extent of the interaction was seen for all PK parameters at the higher dose level compared with the lower dose. Maximum concentrations in plasma were not affected significantly by the interaction.
PK parameters of piperacillin were very similar after the treatments with and without flucloxacillin. No significant differences were seen, except for a decrease in nonrenal clearance from 5.2 to 4.3 l h−1 (P < 0.01) and an increase from 0.53 to 0.60 (P = 0.02) in the fraction excreted unchanged in urine, when 3 g piperacillin was given together with 1 g flucloxacillin.
Compartmental modelling
For the plasma and urine concentration–time data of both piperacillin and flucloxacillin a three-compartment model was chosen based on the Akaike criterion (AIC) and visual inspection of the observed and predicted plasma concentration–time curves and profiles of amounts in urine.
The results from the AIC (data available on request) and the visual predictive checks showed that models 1 and 4 (Table 2) both had excellent predictive performance. The visual predictive checks gave virtually identical results for those two models. Model 3 achieved AIC values similar to models 1 and 4, but was probably over-parameterized, and models 2 and 5 were ranked less likely (median AIC at least 30 points higher). The visual predictive checks of model 1 are shown in Figure 4 for the interaction treatment. The visual predictive checks for the profiles of piperacillin and flucloxacillin alone showed also a highly sufficient predictive performance for model 1. Table 4 lists the PK parameter estimates of flucloxacillin for model 1. Overall, piperacillin reduced the nonrenal clearance of flucloxacillin significantly (P < 0.01) by 33% (23–42%), point estimate (90% confidence interval from anova statistics).
Figure 4.
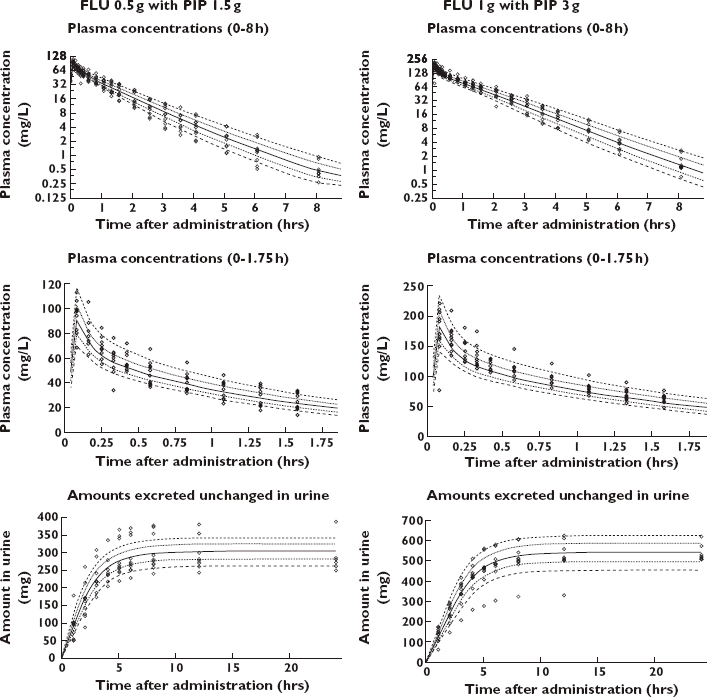
Visual predictive check for plasma concentrations and amounts excreted unchanged in urine of flucloxacillin for model 1. The plots show the raw data, the 80% prediction interval [10–90% percentile] and the interquartile range [25–75% percentile]. Ideally, 50% of the raw data points should fall inside the interquartile range at each time point and 80% of the raw data should fall inside the 80% prediction interval. 10% percentile ( ); 25% percentile (
); 25% percentile ( ); 50% percentile (
); 50% percentile ( ); 75% percentile (
); 75% percentile ( ); 90% percentile (
); 90% percentile ( ); Raw data (◊)
); Raw data (◊)
Table 4.
Pharmacokinetic parameter estimates for flucloxacillin for model 1 (see Table 2)
| Parameter | Unit | Median (range) |
|---|---|---|
| CLR* | l h−1 | 0.432 |
| KmR,FLU | mg l−1 | 334 (46.1–1406) |
| VmaxR,FLU | mg h−1 | 1786 (337–7194) |
| Kic | mg l−1 | 21.7 (8.95–28.3) |
| CLNR, FLU without PIP | l h−1 | 2.86 (1.74–4.66) |
| CLNR, FLU with PIP | l h−1 | 1.76 (1.36–2.52) |
| V1FLU | l | 4.45 (3.61–7.41) |
| V2FLU | l | 1.99 (0.86–3.51) |
| V3FLU | l | 2.30 (1.60–2.92) |
| CLicshallow,FLU | l h−1 | 16.3 (2.65–22.5) |
| CLicdeep,FLU | l h−1 | 1.49 (0.876–2.91) |
Fixed, not estimated. CLR, linear renal clearance; KmR,FLU, Michaelis–Menten constant of the mixed order renal elimination; VmaxR,FLU, apparent maximum rate of the mixed order renal elimination; Kic, competitive inhibition constant; CLNR,FLU, linear nonrenal clearance; V1FLU, volume of distribution of central compartment; V2FLU, volume of distribution of shallow peripheral compartment; V3FLU, volume of distribution of deep peripheral compartment; CLicshallow,FLU, intercompartmental clearance between the central and the shallow peripheral compartment; CLicdeep,FLU, intercompartmental clearance between the central and the deep peripheral compartment.
The affinity of piperacillin for the renal transporter was 13.3 times (median) higher than the affinity of flucloxacillin, based on the competitive interaction model and the drug concentrations in mg l−1. After accounting for differences in molecular weight, the affinity of piperacillin was 15.1 times higher compared with flucloxacillin. As piperacillin had a higher affinity than flucloxacillin and similar (about 10% higher) average plasma concentrations, piperacillin inhibited the renal elimination of flucloxacillin, whereas the effect of flucloxacillin on piperacillin was small.
Discussion
In serious hospital-related infections, multiple pathogens may be involved and empirical treatment needs to be implemented as soon as possible. In order to widen the spectrum of pathogenic organisms covered, combination antibiotic treatment is frequently used. β-Lactams are often given together with a β-lactamase inhibitor [28, 29], an aminoglycoside [6–8] or another β-lactam [30–33]. Combinations of β-lactams are frequently used, especially when there is concern about nephrotoxicity with aminoglycosides [1, 34, 35]. The combination of piperacillin and moxalactam was as effective as moxalactam together with amikacin in the treatment of febrile granulocytopenic cancer patients, and was associated with significantly less frequent nephrotoxicity [36]. Indeed a synergistic effect of β-lactam combinations against some Pseudomonas aeruginosa isolates has been shown recently [37].
Concomitant administration of multiple drugs carries the potential for PK drug–drug interactions. This is especially true when these drugs share the same primary elimination pathway [38]. β-Lactams are ionized at physiological pH and are excreted by the kidneys by glomerular filtration and active tubular secretion [39, 40]. Tubular secretion is a capacity limited process that can be inhibited by other substances. As β-lactams are similar in their chemical structure and physicochemical properties, they may compete for the same transporter for the renal tubular secretion when given concomitantly. Reported PK interactions between β-lactams often involve acylureidopenicillins like piperacillin. Both azlocillin [14] and mezlocillin [15] reduce the renal and nonrenal clearance of cefotaxime, and mezlocillin also reduces the clearance of oxacillin [17]. Piperacillin inhibits the renal clearance of cefazolin and the renal and nonrenal clearance of cefoperazone in rabbits [16], as well as the total clearance of moxalactam [17]. Concomitant administration of piperacillin increased plasma concentrations of imipenem in rabbits [41]. In addition to the interactions with other β-lactams, azlocillin reduces the renal and nonrenal clearance of ciprofloxacin [24].
These findings show that acylureidopenicillins have a high potential to inhibit the elimination of other β-lactams given concomitantly. In vitro and animal studies are valuable tools to explore transport of drugs by various transporters. However, caution should be used when extrapolating from these studies to the situation in humans [42]. Both data from well-controlled clinical studies and adequate data analysis are necessary to gain insight into the extent, site and mechanism of a PK interaction in humans. NCA is an adequate method to explore the extent of an interaction for the dose levels used in this study. However, NCA cannot predict the extent of interaction with other doses, because it does not directly account for the time (and especially concentration) dependence of an interaction. Therefore, compartmental modelling is a much more powerful method to study the sites and mechanisms of interactions, as compartmental modelling explicitly accounts for the time course of interaction. We used compartmental analysis to study the time course of the PK interaction between piperacillin and flucloxacillin as model drugs that are co-administered in certain clinical situations. The time course of the PK interaction was studied at two therapeutic dose levels and various interaction models were tested to explore possible mechanisms and site(s) of the interaction.
We performed a well-controlled, six-way crossover study and collected frequent plasma and urine samples in healthy volunteers. For practical reasons it is difficult to perform such a trial in patients who might also have varying degrees of renal dysfunction. However, flucloxacillin clearance has been reported to be decreased in haemodialysis patients [43] and elderly patients [44]. A trial to investigate the extent of the interaction in elderly and renally impaired patients would be helpful. Our model can be used to predict the extent of interaction based on the renal and nonrenal clearance of piperacillin and flucloxacillin in these special populations. In clinical practice, piperacillin is frequently given together with tazobactam, also in the combination with flucloxacillin, to prevent degradation of piperacillin by β-lactamases. It has been reported that the PK of piperacillin is not affected by concomitant administration of tazobactam at dose ratios of 4 : 1 or 8 : 1 [45]. Therefore, any effect of tazobactam on piperacillin PK is likely to be small.
Although NCA indicated a significant decrease of volume of distribution at steady state for flucloxacillin under the influence of piperacillin, we have shown by subsequent simulations that this is probably an artefact from NCA. We confirmed this by simulations using the compartmental PK model and estimation of PK parameters by NCA (data not shown). Jungbluth and Jusko have shown that a decreased volume of distribution with increasing doses of mezlocillin in rats was an artefact from NCA due to saturable elimination of mezlocillin, as NCA assumes linear disposition [46].
Piperacillin PK parameters were not affected significantly by the low-dose interaction treatment. For the high-dose interaction treatment nonrenal clearance of piperacillin was reduced by 23% and the fraction excreted unchanged in urine increased by 15% compared with 3 g piperacillin alone. As nonrenal clearance accounted for only about 40% of piperacillin elimination, the small decrease in nonrenal clearance did not result in a significant decrease in total body clearance. The increase in piperacillin renal clearance with decreasing plasma concentrations (Figure 3) was independent of flucloxacillin and may be attributed to saturable elimination of piperacillin [12, 47, 48]. Therefore, we used a parallel nonsaturable (first-order) and saturable (mixed-order) elimination model for piperacillin and did not include an influence of flucloxacillin on piperacillin PK.
We used compartmental modelling and tested several mechanistic models (Table 2). Models 1, 3 and 4 showed similar AIC values, whereas models 2 and 5 were ranked less likely. Model 5 included a competitive interaction at the nonrenal site and therefore had two more parameters to describe the nonrenal interaction compared with models with static nonrenal interaction. Probably because of the relatively small extent of the nonrenal interaction, our data did not support this more complicated model with a mechanistic interaction at both the renal and nonrenal site. Thus, a specific mechanism for the nonrenal interaction could not be identified. Model 3 (mixed interaction) was probably over-parameterized. For eight of 10 subjects, model 3 (mixed inhibition) indicated that the contribution of the noncompetitive interaction was not significant in addition to the competitive interaction component.
Therefore, we further considered the competitive (model 1) and the noncompetitive model (model 4) and evaluated their predictive performance. Models 1 and 4 both had excellent predictive performance and virtually identical visual predictive checks. In noncompetitive inhibition the inhibitor binds to a site different from the active site of the transporter or enzyme, and this site is not changed by substrate binding. The noncompetitive inhibitor is not transported by the transporter [49]. As many β-lactams are transported by OATs in the renal tubules, and both piperacillin and cloxacillin were identified as competitive inhibitors of OAT 1 [50], a competitive interaction at an OAT seems a likely reason for the interaction between piperacillin and flucloxacillin. As piperacillin was shown to be both an inhibitor and a substrate of OAT, it seems more likely to be a competitive interaction. Also, in models with mixed inhibition, which include both a competitive and a noncompetitive interaction, the noncompetitive part was estimated to contribute notably in only two of 10 subjects. Therefore we chose model 1 with competitive renal and static nonrenal interaction as our final model.
Model 1 indicated that the affinity of piperacillin for the renal transporter was 15.1 times (median) higher than the affinity of flucloxacillin after accounting for the molecular ratio. The average plasma concentration (in mg l−1) of piperacillin from 0 to 8 h was about 10% higher than for flucloxacillin when both were co-administered. Because of the higher affinity to the renal transporter protein, there was a marked effect of piperacillin on flucloxacillin, but not vice versa. The extent of interaction can be quantified by the ratio of flucloxacillin clearance under the influence of piperacillin divided by the flucloxacillin clearance when given alone. A competitive interaction model predicts that this ratio is lower when both substrates are given at higher doses, i.e. 1 g flucloxacillin and 3 g piperacillin, compared with lower doses, i.e. 0.5 g flucloxacillin and 1.5 g piperacillin, although the dose ratio stays constant at 1 : 3. These predictions agree with the results from NCA. Overall, the estimates from the compartmental model were in good agreement with the results from NCA.
We observed that concomitant administration of piperacillin decreased the renal clearance and, to a lesser extent, the nonrenal clearance of flucloxacillin. This is in agreement with literature data on acylureidopenicillins as discussed above [14, 15, 17, 24]. Both piperacillin and flucloxacillin are excreted by renal tubular secretion [9, 10]. Piperacillin increases the AUC of methotrexate at concomitant administration [51], mainly due to decreasing renal clearance [52]. Probenecid decreased the renal clearance of piperacillin [12] and flucloxacillin AUC [53]. From studies in rats and rabbits it was concluded that piperacillin inhibits renal clearance of imipenem by inhibition of its OAT-mediated transport [41]. The interaction involves inhibition of transporters, but it is not possible to identify these transporter(s) by a PK study in humans. Knowledge about transport proteins and their substrate specificity has increased considerably during the last decade(s) [38, 54, 55]. β-Lactams have been reported to be secreted by and also to inhibit various members of the family of OATs in the proximal renal tubules [13, 50, 56, 57].
More specifically, Jariyawat et al. [50] tested 25 different antibiotics, among them 17 β-lactams including piperacillin and cloxacillin. All of the tested β-lactams showed competitive inhibition of p-aminohippuric acid transport by OAT 1 [50]. Cloxacillin is structurally very similar to flucloxacillin, with one additional fluorine as the only difference. These findings suggest that the site of the renal interaction of piperacillin and flucloxacillin could be an OAT, possibly OAT 1, in the proximal renal tubules. As OATs have been found in various organs of the body, including the liver [58–61], a competitive interaction process could also be the reason for the decreased nonrenal clearance of flucloxacillin when given with piperacillin. Further studies are needed to identify exactly which transporter(s) are involved in this interaction. Recently, a potentially significant genetic contribution of OAT to the variability in renal clearance of amoxicillin and cefaclor in Chinese subjects has been proposed [62].
We calculated the nonprotein bound time above minimum inhibitory concentration (MIC) (fT > MIC) for the flucloxacillin treatments without and with piperacillin to explore possible pharmacodynamic benefits of this PK interaction. fT > MIC of flucloxacillin increased twofold under interaction (1 g flucloxacillin: 1.90 ± 0.31 h vs. 1 g flucloxacillin with 3 g piperacillin: 3.72 ± 0.53 h) for a MIC of 1 mg l−1. Therefore this interaction might be used to improve the effectiveness of flucloxacillin. The dosing interval for flucloxacillin might potentially be prolonged when flucloxacillin and piperacillin are given together. In addition, potential synergistic effects for bacterial killing of piperacillin and flucloxacillin should be explored.
In conclusion, compartmental modelling identified competitive or noncompetitive inhibition of flucloxacillin tubular secretion by piperacillin as the most likely mechanism of the renal interaction. Based on physiological considerations and interaction studies from literature reports on other β-lactams, and the similarity in chemical structure of piperacillin and flucloxacillin, a competitive interaction was chosen as the final model. Further in vitro studies on the interaction of these two antibiotics are required to elucidate further details on the mechanism of inhibition. Piperacillin had a 15.1 times (median) higher affinity to the renal transporter than flucloxacillin when concentrations of both drugs were expressed in molar units. The average plasma concentration of piperacillin from 0 to 8 h was about 10% higher than for flucloxacillin when both were co-administered. Therefore piperacillin considerably inhibited the active tubular secretion of flucloxacillin. Piperacillin significantly decreased the renal clearance of flucloxacillin from 5.44 to 2.29 l h−1 (P < 0.01) and the nonrenal clearance of flucloxacillin from 2.67 to 1.80 l h−1 (P < 0.01). The extent of the interaction was larger for the higher doses. The interaction is potentially clinically significant, since the total clearance decreased from 7.96 to 4.00 l h−1 (P < 0.01) for flucloxacillin. Piperacillin PK was not affected by the concomitant administration of flucloxacillin, except for a decrease in the nonrenal clearance from 5.2 to 4.3 l h−1 (P < 0.01) with the high-dose interaction treatment compared with piperacillin alone. We used piperacillin and flucloxacillin as model drugs to explore interactions between β-lactams. The inhibition of flucloxacillin elimination led to a prolonged time of nonprotein-bound concentrations above the MIC, which may be used to improve the effectiveness of flucloxacillin. As piperacillin showed a higher affinity to the renal transporter than flucloxacillin, it is the more likely drug to be used as inhibitor in the further studies that are required to show the clinical relevance of PK interactions between β-lactams.
Competing interests
None to declare.
Acknowledgments
Part of this work has been presented as a poster at the AAPS Annual Meeting, San Antonio, TX, USA; 29 October to 2 November 2006.
REFERENCES
- 1.Joint Formulary Committee. British National Formulary. 49th edn. London: British Medical Association and Royal Pharmaceutical Society of Great Britain; 2005. [Google Scholar]
- 2.Jones PD, Henry RL, Stuart J, Francis L. Suspected infection in children with cancer. J Qual Clin Pract. 1998;18:275–84. doi: 10.1046/j.1440-1762.1998.00284.x. [DOI] [PubMed] [Google Scholar]
- 3.Mahmood S, Revesz T, Mpofu C. Febrile episodes in children with cancer in the United Arab Emirates. Pediatr Hematol Oncol. 1996;13:135–42. doi: 10.3109/08880019609030803. [DOI] [PubMed] [Google Scholar]
- 4.Smith CL, Milliken S, Powles R, Da Costa F, Gore M, Benjamin S, Talbot D, Ellis L, Large J, Jameson B. Teicoplanin compared to flucloxacillin for antibiotic treatment of neutropenic patients. Br J Haematol. 1990;76(Suppl. 2):6–9. doi: 10.1111/j.1365-2141.1990.tb07927.x. [DOI] [PubMed] [Google Scholar]
- 5.Perkkio M, Hovi L, Rajantie J, Lanning M, Salmi T, Williams K, Makipernaa A, Ruuskanen O, Renkonen OV, Herva E, Toivanen P, Peltola H, Siimes M. A randomised comparison of ceftazidime and piperacillin, both in combination with flucloxacillin for treatment of febrile episodes in neutropenic children. Finnish Three-Centre Study. Scand J Infect Dis. 1990;22:209–18. doi: 10.3109/00365549009037904. [DOI] [PubMed] [Google Scholar]
- 6.Joshi M, Metzler M, McCarthy M, Olvey S, Kassira W, Cooper A. Comparison of piperacillin/tazobactam and imipenem/cilastatin, both in combination with tobramycin, administered every 6 h for treatment of nosocomial pneumonia. Respir Med. 2006;100:1554–65. doi: 10.1016/j.rmed.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Flidel-Rimon O, Friedman S, Leibovitz E, Shinwell ES. The use of piperacillin/tazobactam (in association with amikacin) in neonatal sepsis: efficacy and safety data. Scand J Infect Dis. 2006;38:36–42. doi: 10.1080/00365540500372879. [DOI] [PubMed] [Google Scholar]
- 8.Sanz MA, Bermudez A, Rovira M, Besalduch J, Pascual MJ, Nocea G, Sanz-Rodriguez C. Imipenem/cilastatin versus piperacillin/tazobactam plus amikacin for empirical therapy in febrile neutropenic patients: results of the COSTINE study. Curr Med Res Opin. 2005;21:645–55. doi: 10.1185/030079905X43631. [DOI] [PubMed] [Google Scholar]
- 9.GlaxoSmithKline. Uxbridge, Middlesex, UK: 2005. Flucloxacillin Product Information (Floxapen) [Google Scholar]
- 10.Wyeth . Maidenhead, Berks, UK: 2005. Piperacillin/Tazobactam Product Information (Tazocin) [Google Scholar]
- 11.Endres CJ, Hsiao P, Chung FS, Unadkat JD. The role of transporters in drug interactions. Eur J Pharm Sci. 2006;27:501–17. doi: 10.1016/j.ejps.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Tjandramaga TB, Mullie A, Verbesselt R, De Schepper PJ, Verbist L. Piperacillin: human pharmacokinetics after intravenous and intramuscular administration. Antimicrob Agents Chemother. 1978;14:829–37. doi: 10.1128/aac.14.6.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee W, Kim RB. Transporters and renal drug elimination. Annu Rev Pharmacol Toxicol. 2004;44:137–66. doi: 10.1146/annurev.pharmtox.44.101802.121856. [DOI] [PubMed] [Google Scholar]
- 14.Kampf D, Borner K, Moller M, Kessel M. Kinetic interactions between azlocillin, cefotaxime, and cefotaxime metabolites in normal and impaired renal function. Clin Pharmacol Ther. 1984;35:214–20. doi: 10.1038/clpt.1984.29. [DOI] [PubMed] [Google Scholar]
- 15.Rodondi LC, Flaherty JF, Schoenfeld P, Barriere SL, Gambertoglio JG. Influence of coadministration on the pharmacokinetics of mezlocillin and cefotaxime in healthy volunteers and in patients with renal failure. Clin Pharmacol Ther. 1989;45:527–34. doi: 10.1038/clpt.1989.68. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi T, Watanabe Y, Kumano K, Kitayama R, Yasuda T, Saikawa I, Totsuka K, Kumada T, Shimizu K. Pharmacokinetic studies on the concomitant administration of piperacillin and cefazolin, and piperacillin and cefoperazone in rabbits. J Antibiot (Tokyo) 1986;39:699–712. doi: 10.7164/antibiotics.39.699. [DOI] [PubMed] [Google Scholar]
- 17.Barriere SL. Therapeutic considerations in using combinations of newer beta-lactam antibiotics. Clin Pharm. 1986;5:24–33. [PubMed] [Google Scholar]
- 18.Tahara H, Shono M, Kusuhara H, Kinoshita H, Fuse E, Takadate A, Otagiri M, Sugiyama Y. Molecular cloning and functional analyses of OAT1 and OAT3 from cynomolgus monkey kidney. Pharm Res. 2005;22:647–60. doi: 10.1007/s11095-005-2503-0. [DOI] [PubMed] [Google Scholar]
- 19.Nozaki Y, Kusuhara H, Kondo T, Hasegawa M, Shiroyanagi Y, Nakazawa H, Okano T, Sugiyama Y. Characterization of the uptake of organic anion transporter (OAT) 1 and OAT3 substrates by human kidney slices. J Pharmacol Exp Ther. 2007;321:362–9. doi: 10.1124/jpet.106.113076. [DOI] [PubMed] [Google Scholar]
- 20.Adamis G, Papaioannou MG, Giamarellos-Bourboulis EJ, Gargalianos P, Kosmidis J, Giamarellou H. Pharmacokinetic interactions of ceftazidime, imipenem and aztreonam with amikacin in healthy volunteers. Int J Antimicrob Agents. 2004;23:144–9. doi: 10.1016/j.ijantimicag.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Adam D, Koeppe P, Heilmann HD. Pharmacokinetics of amoxicillin and flucloxacillin following the simultaneous intravenous administration of 4 g and 1 g, respectively. Infection. 1983;11:150–4. doi: 10.1007/BF01641294. [DOI] [PubMed] [Google Scholar]
- 22.Bernabeu-Wittel M, Pichardo C, Garcia-Curiel A, Pachon-Ibanez ME, Ibanez-Martinez J, Jimenez-Mejias ME, Pachon J. Pharmacokinetic/pharmacodynamic assessment of the in-vivo efficacy of imipenem alone or in combination with amikacin for the treatment of experimental multiresistant Acinetobacter baumannii pneumonia. Clin Microbiol Infect. 2005;11:319–25. doi: 10.1111/j.1469-0691.2005.01095.x. [DOI] [PubMed] [Google Scholar]
- 23.Uber WE, Brundage RC, White RL, Brundage DM, Bromley HR. In vivo inactivation of tobramycin by piperacillin. DICP. 1991;25:357–9. doi: 10.1177/106002809102500405. [DOI] [PubMed] [Google Scholar]
- 24.Barriere SL, Catlin DH, Orlando PL, Noe A, Frost RW. Alteration in the pharmacokinetic disposition of ciprofloxacin by simultaneous administration of azlocillin. Antimicrob Agents Chemother. 1990;34:823–6. doi: 10.1128/aac.34.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overbosch D, Van Gulpen C, Hermans J, Mattie H. The effect of probenecid on the renal tubular excretion of benzylpenicillin. Br J Clin Pharmacol. 1988;25:51–8. doi: 10.1111/j.1365-2125.1988.tb03281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergan T, Engeset A, Olszewski W, Ostby N, Solberg R. Extravascular penetration of highly protein-bound flucloxacillin. Antimicrob Agents Chemother. 1986;30:729–32. doi: 10.1128/aac.30.5.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergan T. Penicillins. Antibiot Chemother. 1978;25:1–122. doi: 10.1159/000401058. [DOI] [PubMed] [Google Scholar]
- 28.Lee N, Yuen KY, Kumana CR. Clinical role of beta-lactam/beta-lactamase inhibitor combinations. Drugs. 2003;63:1511–24. doi: 10.2165/00003495-200363140-00006. [DOI] [PubMed] [Google Scholar]
- 29.Klein JO. Amoxicillin/clavulanate for infections in infants and children: past, present and future. Pediatr Infect Dis J. 2003;22:S139–48. doi: 10.1097/00006454-200308001-00005. [DOI] [PubMed] [Google Scholar]
- 30.Joshi JH, Newman KA, Brown BW, Finley RS, Ruxer RL, Moody MA, Schimpff SC. Double beta-lactam regimen compared to an aminoglycoside/beta-lactam regimen as empiric antibiotic therapy for febrile granulocytopenic cancer patients. Support Care Cancer. 1993;1:186–94. doi: 10.1007/BF00366445. [DOI] [PubMed] [Google Scholar]
- 31.Menichetti F, Del Favero A, Bucaneve G, Minotti V, Patoia L, Pauluzzi S. Ceftriaxone versus aztreonam plus cefazolin for infections in cancer patients with adequate neutrophil counts. Infection. 1990;18:166–9. doi: 10.1007/BF01642106. [DOI] [PubMed] [Google Scholar]
- 32.Rotstein C, Cimino M, Winkey K, Cesari C, Fenner J. Cefoperazone plus piperacillin versus mezlocillin plus tobramycin as empiric therapy for febrile episodes in neutropenic patients. Am J Med. 1988;85:36–43. doi: 10.1016/0002-9343(88)90173-8. [DOI] [PubMed] [Google Scholar]
- 33.Rolston KV, Jones PG, Fainstein V, Elting L, Bodey GP. Ceftizoxime plus ticarcillin: double beta-lactam therapy for infections in cancer patients. J Antimicrob Chemother. 1987;19:367–71. doi: 10.1093/jac/19.3.367. [DOI] [PubMed] [Google Scholar]
- 34.Gambertoglio JG, Barriere SL, Lin ET, Conte JE., Jr Amdinocillin pharmacokinetics. Simultaneous administration with cephalothin and cerebrospinal fluid penetration. Am J Med. 1983;75:54–9. doi: 10.1016/0002-9343(83)90094-3. [DOI] [PubMed] [Google Scholar]
- 35.Winston DJ, Ho WG, Champlin RE, Gale RP, Busuttil RW. Ureidopenicillins, aztreonam, and thienamycin: efficacy as single-drug therapy of severe infections and potential as components of combined therapy. J Antimicrob Chemother. 1986;17(Suppl. A):55–66. doi: 10.1093/jac/17.suppl_a.55. [DOI] [PubMed] [Google Scholar]
- 36.De Jongh CA, Joshi JH, Thompson BW, Newman KA, Finley RS, Moody MR, Salvatore PC, Tenney JH, Drusano GL, Schimpff SC. A double beta-lactam combination versus an aminoglycoside-containing regimen as empiric antibiotic therapy for febrile granulocytopenic cancer patients. Am J Med. 1986;80:101–11. [PubMed] [Google Scholar]
- 37.Chen YH, Peng CF, Lu PL, Tsai JJ, Chen TP. In vitro activities of antibiotic combinations against clinical isolates of Pseudomonas aeruginosa. Kaohsiung J Med Sci. 2004;20:261–7. doi: 10.1016/s1607-551x(09)70116-0. [DOI] [PubMed] [Google Scholar]
- 38.Masereeuw R, Russel FG. Mechanisms and clinical implications of renal drug excretion. Drug Metab Rev. 2001;33:299–351. doi: 10.1081/dmr-120000654. [DOI] [PubMed] [Google Scholar]
- 39.Barza M, Weinstein L. Pharmacokinetics of the penicillins in man. Clin Pharmacokinet. 1976;1:297–308. doi: 10.2165/00003088-197601040-00004. [DOI] [PubMed] [Google Scholar]
- 40.Bergan T. Overview of acylureidopenicillin pharmacokinetics. Scand J Infect Dis. 1981;29:33–48. [PubMed] [Google Scholar]
- 41.Saitoh H, Oda M, Gyotoku T, Kobayashi M, Fujisaki H, Sekikawa H. A beneficial interaction between imipenem and piperacillin possibly through their renal excretory process. Biol Pharm Bull. 2006;29:2519–22. doi: 10.1248/bpb.29.2519. [DOI] [PubMed] [Google Scholar]
- 42.Nozaki Y, Kusuhara H, Kondo T, Hasegawa M, Shiroyanagi Y, Nakazawa H, Okano T, Sugiyama Y. Characterization of the uptake of OAT1 and OAT3 substrates by human kidney slices. J Pharmacol Exp Ther. 2007;321:362–9. doi: 10.1124/jpet.106.113076. [DOI] [PubMed] [Google Scholar]
- 43.Nauta EH, Mattie H. Pharmacokinetics of flucloxacillin and cloxacillin in healthy subjects and patients on chronic intermittent haemodialysis. Br J Clin Pharmacol. 1975;2:111–21. doi: 10.1111/j.1365-2125.1975.tb01566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gath J, Charles B, Sampson J, Smithurst B. Pharmacokinetics and bioavailability of flucloxacillin in elderly hospitalized patients. J Clin Pharmacol. 1995;35:31–6. doi: 10.1002/j.1552-4604.1995.tb04742.x. [DOI] [PubMed] [Google Scholar]
- 45.Sorgel F, Kinzig M. The chemistry, pharmacokinetics and tissue distribution of piperacillin/tazobactam. J Antimicrob Chemother. 1993;31(Suppl. A):39–60. doi: 10.1093/jac/31.suppl_a.39. [DOI] [PubMed] [Google Scholar]
- 46.Jungbluth GL, Jusko WJ. Dose-dependent pharmacokinetics of mezlocillin in rats. Antimicrob Agents Chemother. 1989;33:839–43. doi: 10.1128/aac.33.6.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergan T, Williams JD. Dose dependence of piperacillin pharmacokinetics. Chemotherapy. 1982;28:153–9. doi: 10.1159/000238070. [DOI] [PubMed] [Google Scholar]
- 48.Batra VK, Morrison JA, Lasseter KC, Joy VA. Piperacillin kinetics. Clin Pharmacol Ther. 1979;26:41–53. doi: 10.1002/cpt197926141. [DOI] [PubMed] [Google Scholar]
- 49.Segel IH. Enzyme Kinetics. New York, NY: John Wiley & Sons, Inc.; 1993. [Google Scholar]
- 50.Jariyawat S, Sekine T, Takeda M, Apiwattanakul N, Kanai Y, Sophasan S, Endou H. The interaction and transport of beta-lactam antibiotics with the cloned rat renal organic anion transporter 1. J Pharmacol Exp Ther. 1999;290:672–7. [PubMed] [Google Scholar]
- 51.Najjar TA, Abou-Auda HS, Ghilzai NM. Influence of piperacillin on the pharmacokinetics of methotrexate and 7-hydroxymethotrexate. Cancer Chemother Pharmacol. 1998;42:423–8. doi: 10.1007/s002800050840. [DOI] [PubMed] [Google Scholar]
- 52.Iven H, Brasch H. Influence of the antibiotics piperacillin, doxycycline, and tobramycin on the pharmacokinetics of methotrexate in rabbits. Cancer Chemother Pharmacol. 1986;17:218–22. doi: 10.1007/BF00256687. [DOI] [PubMed] [Google Scholar]
- 53.Hedstrom SA, Kahlmeter G. Dicloxacillin and flucloxacillin twice daily with probenecid in staphylococcal infections. A clinical and pharmacokinetic evaluation. Scand J Infect Dis. 1980;12:221–5. doi: 10.3109/inf.1980.12.issue-3.10. [DOI] [PubMed] [Google Scholar]
- 54.Inui KI, Masuda S, Saito H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000;58:944–58. doi: 10.1046/j.1523-1755.2000.00251.x. [DOI] [PubMed] [Google Scholar]
- 55.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–46. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Burckhardt BC, Burckhardt G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev Physiol Biochem Pharmacol. 2003;146:95–158. doi: 10.1007/s10254-002-0003-8. [DOI] [PubMed] [Google Scholar]
- 57.Hasegawa M, Kusuhara H, Endou H, Sugiyama Y. Contribution of organic anion transporters to the renal uptake of anionic compounds and nucleoside derivatives in rat. J Pharmacol Exp Ther. 2003;305:1087–97. doi: 10.1124/jpet.102.046847. [DOI] [PubMed] [Google Scholar]
- 58.Shitara Y, Sato H, Sugiyama Y. Evaluation of drug–drug interaction in the hepatobiliary and renal transport of drugs. Annu Rev Pharmacol Toxicol. 2005;45:689–723. doi: 10.1146/annurev.pharmtox.44.101802.121444. [DOI] [PubMed] [Google Scholar]
- 59.Meier PJ, Eckhardt U, Schroeder A, Hagenbuch B, Stieger B. Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology. 1997;26:1667–77. doi: 10.1002/hep.510260641. [DOI] [PubMed] [Google Scholar]
- 60.Faber KN, Muller M, Jansen PL. Drug transport proteins in the liver. Adv Drug Deliv Rev. 2003;55:107–24. doi: 10.1016/s0169-409x(02)00173-4. [DOI] [PubMed] [Google Scholar]
- 61.Wright SH, Dantzler WH. Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev. 2004;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- 62.Yin OQ, Tomlinson B, Chow MS. Variability in renal clearance of substrates for renal transporters in Chinese subjects. J Clin Pharmacol. 2006;46:157–63. doi: 10.1177/0091270005283838. [DOI] [PubMed] [Google Scholar]