

Abstract
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The only existing study of CYP2D6*10-associated alterations in flecainide pharmacokinetics was retrospective.
Paroxetine has been known as a strong inhibitor of CYP2D6.
WHAT THIS STUDY ADDS
This study reports that the extent of drug interaction between flecainide and paroxetine is influenced by the CYP2D6*10 allele in healthy subjects, which is frequent in Asians.
AIMS
The objectives were to evaluate the effect of CYP2D6 genetic polymorphism on the pharmacokinetics of flecainide, and also on the extent of drug interaction with paroxetine as a CYP2D6 inhibitor after a single oral administration in healthy subjects.
METHODS
An open-label, two-period, single-sequence, cross-over study was performed in 21 healthy Korean male volunteers (seven for CYP2D6*1/*1 or *1/*2, group 1; seven for CYP2D6*1/*10, group 2; seven for CYP2D6*10/*10 or *10/*36, group 3). Subjects were administered 200 mg of flecainide on day 1. After a 7-day wash-out period, subjects were administered 20 mg of paroxetine from day 8 to 14, and 200 mg of flecainide on day 15. Blood sampling was performed up to 72 h after flecainide administration.
RESULTS
Terminal elimination half-life and mean residence time (MRT) were significantly different among three genotype groups after a single oral administration of flecainide (P = 0.021, 0.011, respectively). Area under the concentration–time curve, terminal elimination half-life and MRT increased significantly after paroxetine co-administration only in groups 1 and 2.
CONCLUSIONS
This study reports that the extent of drug interaction between flecainide and paroxetine is influenced by the CYP2D6*10 allele in healthy subjects, which is frequent in Asians.
Keywords: CYP2D6, drug interaction, flecainide, paroxetine, pharmacokinetics
Introduction
There is a considerable variation between individuals regarding cytochrome P450 (CYP) 2D6 activity [1]. CYP2D6 protein and enzymatic activity is completely absent in subjects with two null alleles (e.g. *4 and *5), with a frequency of up to 10% in Caucasians and <1% in Asians [2]. Compared with these ‘poor metabolizers’ (PMs), a subgroup of individuals is termed ‘intermediate metabolizers’ (IMs) of CYP2D6, to reflect reduced protein activity. However, the most prevalent IM allele is different among different ethnic groups such as Caucasians, Africans and East Asians [3]. Yoon et al. reported that the frequency of the CYP2D6*10 allele, one of the most common IM alleles in East Asians, is approximately 50% in Koreans [4]. Moreover, the magnitude of the effect of the CYP2D6*10 allele varies between substrates [3]. Recently, Lim et al. reported that CYP2D6*10/*10 was associated with lower steady-state plasma concentrations of active tamoxifen metabolites and influenced the clinical outcome of tamoxifen in Korean breast cancer patients [5].
Flecainide acetate (Tambocor™; 3M Pharmaceuticals, St Paul, MN, USA) is a class Ic antiarrythmic agent indicated for paroxysmal supraventricular tachycardia and paroxysmal atrial flutter/fibrillation. It is metabolized by CYP2D6 [6], and the target therapeutic range is relatively narrow, i.e. 200–1000 ng ml−1[7]. Increased adverse effects including proarrythmic potential are reported to be related to high plasma flecainide concentrations [8]. In a previous study, exposure to flecainide was increased after amiodarone co-administration to Caucasian healthy subjects, but the extent of the increase was not significantly different between CYP2D6 extensive metabolizers (EMs) and PMs [9]. In other studies, quinidine decreased total and nonrenal clearance of flecainide by 24 and 28%, respectively, in six nongenotyped or phenotyped healthy subjects [10] and decreased total clearance of flecainide by 15% in five EM patients [11]. Doki et al. recently reported that the pharmacokinetics (PK) of flecainide differs between subjects with the CYP2D6 wild-type (*1 or *2) allele and the *10 allele in Japanese patients with supraventricular tachyarrhythmia using routine therapeutic drug monitoring data [12], which might be clinically relevant in the East Asian population. However, paroxetine, known as a strong inhibitor of CYP2D6 [13], has not yet been reported to inhibit metabolism of flecainide.
The objectives of this study were to evaluate the effect of CYP2D6 genetic polymorphism on the PK of flecainide, and also on the extent of drug interaction with paroxetine as a CYP2D6 inhibitor, after a single oral administration in healthy Korean subjects.
Methods
Subjects and study design
Twenty-one healthy nonsmoking Korean male volunteers were enrolled in the study. None showed any abnormalities on physical examination, vital signs, routine laboratory tests, or 12-lead electrocardiograms, and none had any relevant medical disorders. All of the subjects denied any medication use within the period of 4 weeks before the study, and this was confirmed by urine drug screening using REMEDi HS® (Bio-Rad Laboratories, Hercules, CA, USA). Written informed consent was obtained from all subjects after the study procedures had been fully explained. The study was approved by the Institutional Review Board of Seoul National University Hospital.
This was an open-label, two-period, single-sequence, cross-over study. Subjects were administered 200 mg of rac-flecainide acetate (four 50-mg tablets) on day 1. After a 7-day wash-out period, subjects were administered 20 mg of paroxetine once a day from day 8 to 14, and 200 mg of flecainide on day 15. Blood samples for PK evaluation were collected at 0 (predose), 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48 and 72 h after flecainide administration, and additional safety evaluations, including serial 12-lead electrocardiograms, were performed.
CYP2D6 genotyping
Genomic DNA was extracted from the peripheral whole blood of each subject using a QIAamp DNA Blood mini kit (Qiagen GmbH, Hilden, Germany). In brief, the presence of the CYP2D6*5 (CYP2D6 gene deletion), *2N (CYP2D6 gene multiplication) or *36 (CYP2D6 gene conversion to CYP2D7P in exon 9) allele was determined using amplification by polymerase chain reaction (PCR) [14, 15]. The presence of the CYP2D6*10 (100 C→T) allele was determined using amplification by PCR-restriction fragment length polymorphism [16]. The presence of the CYP2D6*2 (285 C→T), *4 (1846 G→A), *14 (1758 G→A),*21 (2573 C insertion) or *41 (2988 G→A) allele was determined using single base extension by SNaPshot analysis using an ABI PRISM® SNaPshot™ Multiplex Kit (Applied Biosystems, Foster city, CA, USA) [17].
Assay of flecainide levels in plasma
Following liquid–liquid extraction using methyl-t-butyl ether, plasma concentrations of flecainide were measured by tandem mass spectrometry (MS; API 4000, Applied Biosystems/MDS Sciex, Toronto, Canada) coupled with high-performance liquid chromatography (Agilent 1100 series; Agilent Technologies, Wilmington, DE, USA). Chromatographic separation was achieved under gradient conditions on a Luna CN 100A column (100 × 2.0 mm, 3 µm; Phenomenex, Torrance, CA, USA) with a mobile phase consisting of 10 mmol ammonium formate and acetonitrile. The MS/MS system was operated using an electrospray in positive ionization mode. For flecainide and the internal standard, haloperidol, the precursor-to-product ion reactions monitored were m/z 415.2→398.4 and 376.0→123.1, respectively. The lower limit of quantification for flecainide was 1.0 ng ml−1. The day-to-day coefficient of variation was 4.40% at 2.5 ng ml−1, 4.36% at 20 ng ml−1 and 5.77% at 400 ng ml−1.
Pharmacokinetics
Individual PK parameters were calculated by a noncompartmental method using WinNonlin® (version 5.1; Pharsight Co., Mountain View, CA, USA). The maximum drug concentrations in plasma (Cmax) and the time at Cmax (Tmax) were determined directly from the observed values. Area under the concentration–time curve from time 0 to infinity (AUC0–∞) was calculated using the linear-up and log-down trapezoidal method in plasma concentration–time curves. Apparent clearance was calculated as dose administered over AUC0–∞. Elimination half-life was calculated from linear regression of log-transformed plasma concentration and time. Mean residence time (MRT) was calculated as area under the first-moment time curve over AUC.
Statistical analysis
The Kruskal–Wallis test was used for comparisons of demography and PK parameters among the three genotypic groups. Two-sided paired t-tests were used to compare PK parameters of the inhibited state with those of the basal state. For overall analysis, linear mixed models were used, including genotype and drug interaction as fixed effects, and with an interaction term between these fixed effects. S-PLUS® (version 6.2; Insightful Corp., Seattle, WA, USA) was used for statistical analysis, and the level of significance used was 0.05.
Results
Because the original plan was to enrol subjects who had the genotypes CYP2D6*1/*1 or *1/*2, CYP2D6*1/*10 or CYP2D6*10/*10, we had planned to determine the presence or absence of CYP2D6*2, *2N, *4, *5, *10, *14, *21 and *41 alleles among more than 80 alleles of CYP2D6, considering the frequencies already known in Koreans [4, 18]. However, after conducting the clinical study, it was reported that CYP2D6*36 had been previously misclassified as CYP2D6*10[14]. After re-examination, one subject previously classified as CYP2D6*10/*10 was revealed actually to be CYP2D6*10/*36. Therefore, the CYP2D6*10/*10 group included one subject with the genotype CYP2D6*10/*36. None of the demographic characteristics was significantly different in the three CYP2D6 genotypic groups (Table 1); group 1, CYP2D6*1/*1 (n = 5) or *1/*2 (n = 2); group 2, CYP2D6*1/*10 (n = 7); group 3, CYP2D6*10/*10 (n = 6) or *10/*36 (n = 1).
Table 1.
Demographic characteristics of subjects
| CYP2D6 genotype | Group 1 (n = 7) | Group 2 (n = 7) | Group 3 (n = 7) | P-value* |
|---|---|---|---|---|
| Age (years) | 24.3 ± 3.3 | 25.3 ± 3.3 | 23.9 ± 2.5 | 0.647 |
| Height (cm) | 173.0 ± 4.5 | 172.4 ± 4.3 | 175.3 ± 5.0 | 0.401 |
| Weight (kg) | 66.3 ± 7.3 | 67.7 ± 8.2 | 73.3 ± 6.1 | 0.168 |
Kruskal–Wallis test. Group 1, CYP2D6*1/*1 (n = 5) or *1/*2 (n = 2); group 2, CYP2D6*1/*10 (n = 7); group 3, CYP2D6*10/*10 (n = 6) or *10/*36 (n = 1). Data are presented as arithmetic mean ± standard deviation.
Statistically significant differences were found in the elimination half-life and MRT of flecainide among the CYP2D6 genotypic groups in the basal state (P = 0.021, 0.011, respectively, Table 2 and Figure 1). The results were similar even with the subject who had the CYP2D6*10/*36 allele excluded from the analysis (P = 0.034, 0.015, respectively). There was an increasing tendency of the AUC as the number of variant alleles (CYP2D6*10) increased; however, the difference was not statistically significant (Table 2). Apparent clearance showed a decreasing tendency in groups 2 and 3 compared with group 1, but this also was not significant. Cmax and Tmax values showed no differences among the three genotypic groups.
Table 2.
Pharmacokinetic parameters of flecainide according to CYP2D6 genotypes and periods
| CYP2D6 genotype | *1/*1 (n = 5) or *1/*2 (n = 2) | *1/*10 (n = 7) | *10/*10 (n = 6) or *10/*36 (n = 1) |
|---|---|---|---|
| Period 1 (basal) | |||
| AUC (h ng ml−1) | 5717.5 ± 1795.1 | 6719.2 ± 1478.5 (120.7; 91.3, 159.5) | 7034.9 ± 1939.0 (123.6; 93.5, 163.4) |
| CL/F (l h−1) | 38.6 ± 13.9 | 31.0 ± 6.5 (82.9; 62.7, 109.5) | 31.3 ± 12.6 (80.9; 61.2, 106.9) |
| t1/2 (h) | 10.0 ± 1.0 | 11.6 ± 1.0 (115.5; 97.7, 136.3) | 12.5 ± 3.0 (120.9; 102.4, 142.9) |
| MRT (h) | 15.2 ± 1.2 | 17.6 ± 1.1 (116.2; 100.5, 134.2) | 18.4 ± 4.1 (118.8; 108.1, 137.2) |
| Cmax (ng ml−1) | 391.2 ± 97.4 | 425.4 ± 123.7 (108.2; 85.5, 137.0) | 402.6 ± 89.1 (104.0; 82.1, 131.7) |
| Tmax (h) | 1.5 [1.0, 3.0] | 1.0 [1.0, 3.0] | 2.0 [1.0, 3.0] |
| Period 2 (paroxetine-inhibited) | |||
| AUC (h ng ml−1) | 7317.5 ± 2230.6 (128.5; 122.2, 135.2) | 7828.2 ± 1592.3 (116.6; 107.3, 126.8) | 6925.7 ± 1472.9 (100.6; 90.2, 112.2) |
| CL/F (l h−1) | 29.8 ± 9.8 (77.8; 74.0, 81.8) | 26.6 ± 5.9 (85.7; 78.9, 93.2) | 30.1 ± 6.9 (99.4; 89.2, 110.9) |
| t1/2 (h) | 12.1 ± 2.0 (119.2; 108.6, 130.7) | 13.5 ± 2.3 (115.8; 100.4, 133.7) | 12.1 ± 2.5 (98.1; 89.8, 107.1) |
| MRT (h) | 18.0 ± 2.3 (118.0; 111.3, 125.0) | 19.8 ± 2.6 (112.0; 103.2, 121.7) | 17.4 ± 2.9 (95.7; 88.9, 103.1) |
| Cmax (ng ml−1) | 477.3 ± 121.4 (122.5; 97.1, 154.6) | 474.0 ± 130.0 (111.9; 99.9, 125.4) | 467.6 ± 105.1 (115.9; 104.9, 128.0) |
| Tmax (h) | 1.0 [1.0, 4.0] | 1.0 [1.0, 1.5] | 1.5 [1.0, 3.0] |
Data are presented as arithmetic mean ± standard deviation, except for Tmax, median [range]; values in parentheses indicate percentages compared with genotypic group 1 (period 1) or compared with basal values (period 2), expressed as geometric mean ratio and 90% confidence interval. AUC, area under the concentration–time curve from 0 to infinity; CL/F, apparent clearance; MRT, mean residence time; t1/2, elimination half-life.
Figure 1.
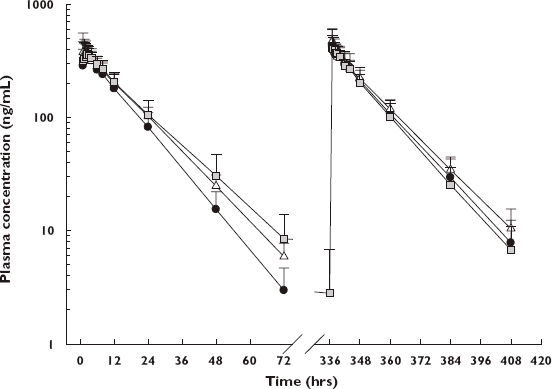
Plasma concentration–time profiles of flecainide by three genotypic groups. Values are presented as arithmetic mean + standard deviation. Left, basal state; right, paroxetine-inhibited state. *1/*1 or *1/*2 (N = 7) ( ); *1/*10 (N = 7) (
); *1/*10 (N = 7) ( ); *10/*10 or *10/*36 (N = 7) (
); *10/*10 or *10/*36 (N = 7) ( )
)
During the paroxetine administration period, the AUCs of flecainide were increased to 128.5% (122.2–135.2%) and 116.6% (107.3–126.8%) of the basal values in groups 1 and 2, respectively (Table 2 and Figure 2). The apparent clearances were reduced to 77.8% (74.0–81.8%) and 85.7% (78.9–93.2%) of the basal values in groups 1 and 2, respectively. However, the AUC and apparent clearance in group 3 did not exhibit any changes after paroxetine administration (Table 2 and Figure 2). The changes of elimination half-life and MRT in the paroxetine-inhibited state displayed similar results (Table 2 and Figure 2). Cmax and Tmax values showed no remarkable changes in any of the three genotypic groups. No differences were found among the three genotypic groups in the paroxetine-inhibited state in terms of AUC, apparent clearance, elimination half-life, MRT, Cmax or Tmax.
Figure 2.
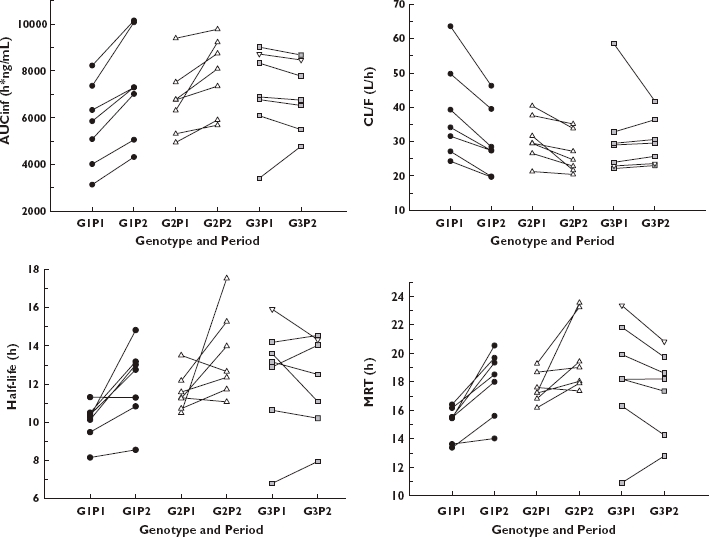
Area under the concentration–time curve (left upper panel), apparent clearance (right upper panel), elimination half-life (left lower panel) and mean residence time (right lower panel) of flecainide according to CYP2D6 genotypes and periods. G1P1, group 1 (CYP2D6*1/*1 or *1/*2) and period 1; G1P2, group 1 and period 2; G2P1, group 2 (CYP2D6*1/*10) and period 1; G2P2, group 2 and period 2; G3P1, group 3 (CYP2D6*10/*10 or *10/*36) and period 1; G3P2, group 3 and period 2. Reverse triangle represents *10/*36 subject.
Discussion
In this study, we have shown that elimination half-life and MRT were significantly different among the three genotypic groups; elimination half-life and MRT of CYP2D6*1/*10 subjects were longer than in those with the CYP2D6*1/*1 or *1/*2 allele, and those with CYP2D6*10/*10 were longer than in those with the CYP2D6*1/*10 allele. Also, AUC, elimination half-life and MRT were increased and apparent clearance was decreased after paroxetine co-administration in only groups 1 and 2. To our knowledge, this is the first report that paroxetine inhibited metabolism of flecainide and that this interaction was different among CYP2D6 genotypes, namely in subjects with the CYP2D6*10 allele. Interestingly, one subject with CYP2D6*10/*10 exhibited a remarkable decrease in apparent clearance after paroxetine administration (Figure 2). This subject showed a relatively high clearance in the basal state, allowing for the effect of paroxetine on the inhibition of CYP2D6 metabolism to be great. Findling et al. observed similar phenomena in children and adolescents with various CYP2D6 genotypes; the largest decreases in paroxetine clearance were seen in those patients with the greatest clearance at the initial dose level [19].
However, although subjects with CYP2D6*1/*10 or *10/*10 allele did show a trend for increased AUC and decreased apparent clearance compared with subjects with the wild-type alleles, AUC and apparent clearance in the basal state did not exhibit statistically significant differences among three genotypic groups. This may be explained by the fact that approximately 30% of flecainide is excreted in urine in an unchanged form, and although R-flecainide is metabolized mainly by CYP2D6 [7], the metabolic pathway of S-flecainide is not yet clarified. Therefore, these factors are also thought to contribute to the individual variability in flecainide PK.
In a previous study with Caucasian subjects, Funck-Brentano et al. have reported that flecainide PK was not significantly different between seven EM and five PM subjects with CYP2D6, and also reported that co-administration of amiodarone decreased the apparent total clearance and nonrenal clearance of both the EM and PM groups [9]. The same investigators reported that there was no significant difference of flecainide PK between 20 EM and four PM subjects with CYP2D6 [20]. However, in these studies the subjects' genotypes were not reported; only phenotypes based on the metabolic ratio of urinary dextromethorphan (DM) were used. It is known that the DM metabolic ratio displays consistency in distinguishing CYP2D6 PM from EM subjects [2]. However, Evans et al. have reported that DM metabolic ratios overlapped extensively between the homozygous EMs and heterozygous EMs of the CYP2D6 genotype, so it did not distinguish these two groups reliably [21]. Gaedigk A et al. have replicated these findings in a separate analysis using African-American and Caucasian populations [22]. It might be possible that subjects classified as EMs using DM metabolic ratios in the above-mentioned studies comprised heterozygous EMs in addition to homozygous wild-type subjects. Furthermore, they did find some tendencies of PK differences (e.g. P = 0.08 for nonrenal clearance [9]), although statistically significant differences were not detected, probably due to the small number of subjects studied (four to five PMs). Doki et al. also found that CYP2D6 genotype affected the apparent clearance of flecainide significantly (11, 16, 21 and 27% reduction in EM/IM, EM/PM, IM/IM and IM/PM, respectively) [12].
There are several limitations of the current study that suggest caution in how these results are interpreted. First, this was a single-dose study in healthy volunteers, and genotypic effects by CYP2D6 might be different after repeated dosing of flecainide, because a small extent (approximately 10–15%) of nonlinearity was seen in the multiple-dose study in patients with ventricular arrhythmia [7, 23]. Because this study was conducted in healthy volunteers, a usual daily dosage (200 mg) of flecainide was used, resulting in plasma drug concentrations within therapeutic range for all genotypic groups. However, situations may be considered in which higher doses might be administered to patients with arrhythmia, and drug exposure may be even greater in patients with hepatic or renal impairment. Second, dosing of paroxetine was stopped on day 14, whereas dosing of the substrate flecainide was done on day 15. This could potentially result in underestimation of the magnitude of interaction, although paroxetine has a relatively long half-life (longer than that of flecainide) and is importantly a mechanism-based inactivator of CYP2D6. In addition, the differences in magnitudes of drug interaction among genotypic groups might be greater when using other substrates that are metabolized entirely by CYP2D6. Third, although the therapeutic range of flecainide is relatively narrow, it needs to be further evaluated whether a 30% increase of flecainide exposure after paroxetine co-administration would influence the efficacy or safety of flecainide in the clinical setting. Not including pharmacodynamic analysis obtained from serial electrocardiographic measurements may also be a limitation of this study. However, some cases of flecainde-induced QT prolongation and torsade de pointes have been reported in clinical settings even with daily flecainide doses of 200 mg [24, 25]. Also, in some cases therapeutic drug monitoring is being performed for flecainide due to large interindividual PK variability of flecainide [12]. Therefore, caution should be exercised in patients with higher doses of flecainide, with hepatic or renal impairments or in patients using CYP2D6 inhibitors including paroxetine.
In summary, this study reports that the extent of drug interaction between flecainide and paroxetine is influenced by the CYP2D6*10 allele in healthy subjects, which is frequent in Asians. The clinical significance of possible associations between increases in the QTc interval and the CYP2D6*10 allele should be further evaluated in relevant patient populations.
Competing interests
None to declare.
Acknowledgments
Presented in part as a poster at the 108th annual meeting of the American Society for Clinical Pharmacology and Therapeutics, Anaheim, CA, 21–24 March 2007. An abstract has been published in Clin Pharmacol Ther 2007; 81 (Suppl. 1), S81. This study was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (AO30001). We thank Hwa-Sook Kim for skilful determinations of the drug plasma concentrations, and Seon-Jeong Kim for genotyping of CYP2D6 alleles.
REFERENCES
- 1.Sachse C, Brockmoller J, Bauer S, Root I. Cytochrome p450 2d6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–95. [PMC free article] [PubMed] [Google Scholar]
- 2.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 3.Furman KD, Grimm DR, Mueller T, Holley-Shanks RR, Bertz RJ, Williams LA, Spear BB, Katz DA. Impact of CYP2D6 intermediate metabolizer alleles on single-dose desipramine pharmacokinetics. Pharmacogenetics. 2004;14:279–84. doi: 10.1097/00008571-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Yoon YR, Cha IJ, Shon JH, Kim KA, Cha YN, Jang IJ, Park CW, Shin SG, Flockhart DA, Shin JG. Relationship of paroxetine disposition to metoprolol metabolic ratio and CYP2D6*10 genotype of Korean subjects. Clin Pharmacol Ther. 2000;67:567–76. doi: 10.1067/mcp.2000.106128. [DOI] [PubMed] [Google Scholar]
- 5.Lim HS, Ju Lee H, Seok Lee K, Sook Lee E, Jang IJ, Ro J. Clinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancer. J Clin Oncol. 2007;25:3837–45. doi: 10.1200/JCO.2007.11.4850. [DOI] [PubMed] [Google Scholar]
- 6.Gross AS, Mikus G, Fischer C, Hertrampf R, Gundert-Remy U, Eichelbaum M. Stereoselective disposition of flecainde in relation to the sparteine/debrisoquine metabolizer phenotype. Br J Clin Pharmacol. 1989;28:555–66. doi: 10.1111/j.1365-2125.1989.tb03542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.3M Pharmaceuticals. 3M product information: Tambocor™. [last accessed: 25 September 2007]. Available at http://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?id=1423&type=display.
- 8.Morganroth J, Horowitz LN. Flecainide: its proarrhythmic effect and expected changes on the surface electrocardiogram. Am J Cardiol. 1984;53:89B–94B. doi: 10.1016/0002-9149(84)90509-5. [DOI] [PubMed] [Google Scholar]
- 9.Funck-Brentano C, Becquemont L, Kroemer HK, Buhl K, Knebel NG, Eichelbaum M, Jaillon P. Variable disposition kinetics and electrocardiographic effects of flecainide during repeated dosing in humans: contribution of genetic factors, dose-dependent clearance, and interaction with amiodarone. Clin Pharmacol Ther. 1994;55:256–69. doi: 10.1038/clpt.1994.26. [DOI] [PubMed] [Google Scholar]
- 10.Munafo A, Buclin T, Tuto D, Biollaz J. The effect of a low dose of quinidine on the disposition of flecainide in healthy volunteers. Eur J Clin Pharmacol. 1992;43:441–3. doi: 10.1007/BF02220625. [DOI] [PubMed] [Google Scholar]
- 11.Birgersdotter UM, Wong W, Turgeon J, Roden DM. Stereoselective genetically-determined interaction between chronic flecainide and quinidine in patients with arrhythmias. Br J Clin Pharmacol. 1992;33:275–80. doi: 10.1111/j.1365-2125.1992.tb04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doki K, Homma M, Kuga K, Kusano K, Watanabe S, Yamaguchi I, Kohda Y. Effect of CYP2D6 genotype on flecainide pharmacokinetics in Japanese patients with supraventricular tachyarrhythmia. Eur J Clin Pharmacol. 2006;62:919–26. doi: 10.1007/s00228-006-0188-x. [DOI] [PubMed] [Google Scholar]
- 13.Venkatakrishnan K, Obach RS. In vitro–in vivo extrapolation of CYP2D6 inactivation by paroxetine: prediction of nonstationary pharmacokinetics and drug interaction magnitude. Drug Metab Dispos. 2005;33:845–52. doi: 10.1124/dmd.105.004077. [DOI] [PubMed] [Google Scholar]
- 14.Gaedigk A, Bradford LD, Alander SW, Leeder JS. CYP2D6*36 gene arrangements within the CYP2D6 locus: association of CYP2D6*36 with poor metabolizer status. Drug Metab Dispos. 2006;34:563–9. doi: 10.1124/dmd.105.008292. [DOI] [PubMed] [Google Scholar]
- 15.Gaedigk A, Gotschall RR, Forbes NS, Simon SD, Kearns GL, Leeder JS. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics. 1999;9:669–82. doi: 10.1097/01213011-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Fukuda T, Yamamoto I, Nishida Y, Zhou Q, Ohno M, Takada K, Azuma J. Effect of the CYP2D6*10 genotype on venlafaxine pharmacokinetics in healthy adult volunteers. Br J Clin Pharmacol. 1999;47:450–3. doi: 10.1046/j.1365-2125.1999.00913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heller T, Kirchheiner J, Armstrong VW, Luthe H, Tzvetkov M, Brockmoller J, Oellerich M. AmpliChip CYP450 GeneChip: a new gene chip that allows rapid and accurate CYP2D6 genotyping. Ther Drug Monit. 2006;28:673–7. doi: 10.1097/01.ftd.0000246764.67129.2a. [DOI] [PubMed] [Google Scholar]
- 18.Dahl ML, Yue QY, Roh HK, Johansson I, Sawe J, Sjovist F, Bertilsson L. Genetic analysis of the CYP2D6 locus in relation to debrisoquine hydroxylation capacity in Korean, Japanese and Chinese subjects. Pharmacogenetics. 1995;5:159–64. doi: 10.1097/00008571-199506000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Findling RL, Nucci G, Piergies AA, Gomeni R, Bartolic EI, Fong R, Carpenter DJ, Leeder JS, Gaedigk A, Danoff TM. Multiple dose pharmacokinetics of paroxetine in children and adolescents with major depressive disorder or obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31:1274–85. doi: 10.1038/sj.npp.1300960. [DOI] [PubMed] [Google Scholar]
- 20.Tenneze L, Tarral E, Ducloux N, Funck-Brentano C. Pharmacokinetics and electrocardiographic effects of a new controlled-release form of flecainide acetate: comparison with the standard form and influence of the CYP2D6 polymorphism. Clin Pharmacol Ther. 2002;72:112–22. doi: 10.1067/mcp.2002.125946. [DOI] [PubMed] [Google Scholar]
- 21.Evans WE, Relling MV. Concordance of P450 2D6 (debrisoquine hydroxylase) phenotype and genotype: inability of dextromethorphan metabolic ratio to discriminate reliably heterozygous and homozygous extensive metabolizers. Pharmacogenetics. 1991;1:143–8. [PubMed] [Google Scholar]
- 22.Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–42. doi: 10.1038/sj.clpt.6100406. [DOI] [PubMed] [Google Scholar]
- 23.Boriani G, Strocchi E, Capucci A, Calliva R, Frabetti L, Ambrosioni E, Magnani B. Flecainide: evidence of non-linear kinetics. Eur J Clin Pharmacol. 1991;41:57–9. doi: 10.1007/BF00280107. [DOI] [PubMed] [Google Scholar]
- 24.Nogales Asensio JM, Moreno Sanchez N, Doncel Vecino LJ, Villar Mariscal C, Lopez-Minguez JR, Merchan Herrera A. Torsade-de-pointes in a patient under flecainide treatment, an unusual case of proarrhythmicity. Int J Cardiol. 2007;114:E65–7. doi: 10.1016/j.ijcard.2006.07.124. [DOI] [PubMed] [Google Scholar]
- 25.Thevenin J, Da Costa A, Roche F, Romeyer C, Messier M, Isaaz K. Flecainide induced ventricular tachycardia (torsades de pointes) Pacing Clin Electrophysiol. 2003;26:1907–8. doi: 10.1046/j.1460-9592.2003.00290.x. [DOI] [PubMed] [Google Scholar]