

Abstract
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT.
Randomized controlled trials are the gold standard of drug efficacy assessment, but have limited power to assess adverse drug reactions.
Observational data derived from larger populations of patients are often affected by confounding variables, many of which are difficult to measure or simply not known.
WHAT THIS STUDY ADDS.
This study demonstrates the utility of the self-controlled case-series method of analysing the safety profile of drugs, which reduces confounding because comparisons are intraperson.
Although such data should not be viewed in isolation, the efficiency and versatility of this method suggest that it could become a future standard in drug safety assessment.
AIMS
Post licensing, the evaluation of drug safety relies heavily on the collation of sporadic, spontaneous reports on adverse effects. The aim was to assess the potential utility of a more systematic approach to the detection of adverse events that utilizes routinely collected clinical data from a large primary care population.
METHODS
We used the UK General Practice Research Database to assess the risk of several recently reported adverse events linked to the use of strontium ranelate for osteoporosis in postmenopausal women. The self-controlled case-series method was used to minimize the potential for biases in the quantification of risk estimates.
RESULTS
Age-adjusted rate ratios for venous thromboembolism, gastrointestinal disturbance, minor skin complaint and memory loss were 1.1 [95% confidence interval (CI) 0.2, 5.0], 3.0 (95% CI 2.3, 3.8), 2.0 (95% CI 1.3, 3.1) and 1.8 (95% CI 0.2, 14.1), respectively. No cases of osteonecrosis of the jaw, toxic-epidermal necrosis, Stevens–Johnson syndrome or drug rash with eosinophilia and systemic symptoms were found.
CONCLUSIONS
Although we confirmed the association between strontium ranelate and adverse events identified in the Phase III publications, there was no evidence of an association between strontium ranelate and the aforementioned potentially life-threatening adverse events. Our study demonstrates the relative ease with which this method can assess a variety of adverse events associated with a new drug in actual clinical practice. We believe this technique could be more widely adopted to assess the safety profile of new drugs.
Keywords: strontium, raloxifene case-series, adverse drug reactions, pharmacovigilance
Introduction
Although the randomized, double-blind, controlled clinical trial is the undisputed gold standard for assessing drug efficacy, such trials are usually insufficiently powered, or too brief, to detect rare adverse events or small but important increases in the rates of more common adverse events. Moreover, the ascertainment and reporting of information on adverse events in clinical trials achieve less prominence than data on efficacy. In addition, clinical trial protocols often exclude the elderly, and other patients with comorbidities, so limiting the external validity of data on adverse events. Therefore, at the time of product launch, there are often limited safety data of any new drug, in the short and longer term.
For this reason, new drugs undergo postmarketing surveillance. This system, used by the US Food and Drug Administration Adverse Event Reporting System and the European Medicines Evaluation Agency (EMEA) EudraVigilance Programme, relies heavily on spontaneous reports of adverse effects by physicians and other health workers. These analyses, at best, only create an uncontrolled case series that is low down the hierarchy of epidemiological evidence [1]. For example, in the UK all new medicines are flagged with an inverted black triangle that prompts healthcare professionals to report any suspected reaction through the Yellow Card Scheme. Although such systems have had some notable success, they have several important limitations. First, the onus on adverse event reporting is placed on health professionals, although the UK is about to extend this practice to patients. Second, it is biased towards the detection of rare and more unusual adverse effects, being limited in its potential to detect modest but important increases in the rates of more common clinical events with multifactorial causes, e.g. myocardial infarction. Third, although the regulatory authorities have increasingly required commitments to conduct postmarketing studies as a condition of approval, many of these commitments remain unmet [2]. Moreover, postmarketing observational studies remain unregistered and can be analysed in myriad ways [3]. Indeed, there is no way of knowing how many analyses are attempted before a particular result is reported. Fourth, since the information obtained from postmarketing surveillance is observational in nature, it can be prone to a number of biases that limit precise quantification of the risk of adverse events.
We therefore evaluated the potential of a novel approach to adverse event detection that overcomes some of the limitations of current approaches to evaluation of drug safety and pharmacovigilance. We used routinely collected information from the large UK General Practice Research Database (GPRD) to quantify the risks of adverse events that have been linked to the use of strontium ranelate for the treatment of postmenopausal osteoporosis. These include memory loss, dermatitis, gastrointestinal disturbance, venous thromboembolism (VTE) and severe skin reactions such as toxic-epidermal necrosis, Stevens–Johnson syndrome and drug rash with eosinophilia and systemic symptoms (DRESS). Osteonecrosis of the jaw, which has been reported with high-dose bisphosphonates used to treat osteoporosis [4], was also investigated. We used the self-controlled case-series method on the abovementioned longitudinal data.
Reasons for selecting strontium ranelate:
Strontium ranelate is a relatively new drug for the treatment of postmenopausal osteoporosis. Its safety profile is therefore not fully established in usual practice.
Strontium ranelate acts through an innovative mode of action (by both stimulating bone formation and inhibiting bone resorption) [5, 6].
Relative to many other new drugs, strontium ranelate has been studied in much larger Phase III studies for a longer duration. Therefore, its pre-marketing safety data are relatively robust (allowing for more accurate comparisons) [5, 6].
Strontium ranelate has been associated with both common Type I adverse events and rarer idiosyncratic Type II reactions [7, 8].
Different adverse event signals have been detected in Phase III publications (gastrointestinal and dermatological), the subsequent EMEA review of the data (VTE) and in postmarketing reports (DRESS) [7–9].
Methods
Participants
The GPRD is the world's largest computerized database of anonymized longitudinal medical records from primary care. Currently, data are being collected on >3.4 million patients from around 450 primary care practices throughout the UK [10]. Patients exposed to strontium ranelate between 1 December 2004 and 31 December 2006 were included in the study. This comprised >3000 person-years of observation from 35 general practices.
Assessment of VTE
Eligible participants were those who had a first-ever diagnosis of VTE within a pre-defined study window. Start dates were derived using the latter of the individual practice's up-to-standard date (GPRD-defined quality marker based on assessment of completeness, continuity and plausibility of data recording in key areas) or the patient's first registration date. End dates were derived using the minimum of the patient's transfer out date or the practice's last collection date. If patients had consulted their general practitioner in the 6 weeks before their diagnosed thromboembolism with symptoms likely to indicate a thromboembolic event [e.g. ‘calf pain’ for deep vein thrombosis (DVT)], their date of onset was altered to the date of first symptom. Individuals were excluded if they were male (as strontium ranelate is only licensed for use in postmenopausal women) or if they had documented trauma, major surgery or lower limb surgery in the 8 weeks prior to the event. Individuals were excluded if they had received hormone-replacement therapy within 1 year of their event due to the known risks of VTE. Patients prescribed warfarin prior to the event were also excluded, because this suggested that the thromboembolism was not a new event. Cases were also excluded if their medical records indicated that the venous thromboembolic event was likely to have been retrospectively recorded, e.g. if the patient's DVT or pulmonary embolism (PE) was recorded along with other diagnoses on the day of a ‘new-patient’ or ‘well-person’ screen. We also excluded those whose only diagnostic entry for their event appeared when the general practice received a post-mortem report, because we were concerned that the date recorded would not accurately reflect the date of the VTE. Finally, women were excluded if they had had a diagnosis of a cancer (except nonmelanoma skin cancer) within 1 year of the event date, as cancer is another known risk factor for VTE.
Analyses were repeated for raloxifene-exposed individuals to ensure that the methodology was sufficiently sensitive to detect the known association between raloxifene and VTE [11].
Assessment of gastrointestinal disturbance, dermatitis and memory loss
Gastrointestinal disturbance was investigated using a composite of nausea, vomiting, loose stools and diarrhoea. Patients with a diagnosis of cancer within 1 year of their gastrointestinal event, patients with recent surgery and all patients whose gastrointestinal event was recorded on the day of either a new-person screen or well-women check were excluded. No medical exclusion criteria were applied for assessment of dermatitis or memory loss.
Assessment of severe skin reactions
No medical exclusion criteria were applied for assessment of the composite outcome of toxic-epidermal necrosis, Stevens–Johnson syndrome and DRESS.
Approval for our study was given by the Independent Scientific Advisory Committee for Medicines and Healthcare Regulatory Agency database research.
Procedures
Adverse events were assessed using the self-controlled case-series method. This relies on intraperson comparisons in a population of individuals who have had the outcome of interest. This allows the incidence rate ratios (IRR) of events in defined intervals after an exposure to be determined relative to all other observed time periods for each person [12, 13].
Assessment of VTE
The start of the exposed period was defined as the date of first prescription of strontium ranelate. The end of the exposed period was defined as the earliest of the date of the last prescription plus the final prescription quantity or the first prescription date plus the entire total quantity of strontium ranelate prescribed for the patient. A 28-day wash-out period was then added to the end date to account for delays in obtaining prescriptions and pharmacy supplies and to allow for complete drug elimination. Strontium ranelate has a half-life of about 60 h [7], so after cessation of therapy, a negligible level of continued exposure would occur in about 13 days (based on five half-lives). In addition, a 28-day leeway between prescriptions was allowed to assume continuous exposure. Any gap of >28 days was regarded as a break in therapy and a new exposure period was designated. All other observation times within the study window were taken as the baseline (unexposed) period. Participants included had at least one prescription (exposure) for strontium ranelate and at least one VTE (event). Patients were also censored at their first event allowing for an analysis of incident VTE.
Figure 1 illustrates the assessment of individuals with a single period of exposure to strontium ranelate. The length of the exposed and baseline periods may vary for each participant.
Figure 1.
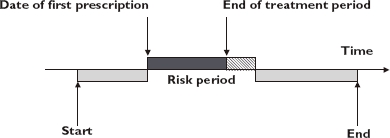
Pictorial representation of the self-controlled case-series method. Baseline period ( ); Wash-out period (
); Wash-out period ( )
)
Assessment of gastrointestinal disturbance, dermatitis and memory loss
As we were primarily concerned about adverse events warranting, or associated with, discontinuation of therapy, the start of the risk period was defined as the latest exposure date to strontium ranelate. The end of the risk period was designated according to the final prescription quantity plus 28-day wash-out. For example, if the final prescription date was 1 January 2006 (start of risk period) and a quantity sufficient for 28 days' dosing was issued, the end of the risk period would be 2 months later. A model for multiple outcomes was used, assuming each event was independent.
Assessment of severe skin reactions
As severe skin reactions usually occur at the outset of treatment (within 6 weeks), the risk period was defined as the first 10 weeks of therapy (again allowing for a 28-day leeway).
Assessment of osteonecrosis of the jaw
The general method utilized for assessment of VTE was employed for osteonecrosis of the jaw assessment, as no known risk periods are apparent.
Statistical analysis
We adjusted for age using ten 5-year age bands (45–49, 50–54, 55–59, etc). IRR and 95% confidence intervals (CIs) were calculated for events occurring within each stratum of the exposed period compared with baseline periods. Data were analysed with Stata (version 9.0; StataCorp LP, College Station, TX, USA).
Role of a funding source
Servier (Neuilly sur Seine, France) provided an unrestricted research grant but had no role in the study design, data collection, data analysis, data interpretation, writing of the report, or the decision to submit the paper for publication. The corresponding author had full access to all data in the study and took full responsibility for the decision to submit for publication.
Results
Patients exposed to strontium ranelate (n = 1574) were identified from the database. The median age of women in the study was 77 years [range 40–95; interquartile range (IQR) 69–83] and the mean total observation period was 12.6 years. A summary of the results is shown in Table 1.
Table 1.
Derived risk estimates of adverse events, using the self-controlled case-series method
| Adverse event (exposure) | N | n (baseline) | n (exposed) | IRR | 95% CI |
|---|---|---|---|---|---|
| Venous thromboembolism* (strontium) | 37 | 35 | 2 | 1.1 | 0.2, 5.0 |
| Venous thromboembolism* (raloxifene) | 55 | 40 | 15 | 4.7 | 1.6, 13.0 |
| Gastrointestinal effects† (strontium) | 676 | 1744 | 81 | 3.0 | 2.3, 3.8 |
| Dermatitis† (strontium) | 402 | 740 | 22 | 2.0 | 1.3, 3.1 |
| Memory loss† (strontium) | 20 | 22 | 1 | 1.8 | 0.2, 14.1 |
Model for first-ever event.
Model for multiple outcomes (independent assumption). CI, confidence interval; IRR, age-adjusted incidence rate ratio; N, number of individuals with exposure and outcome of interest; n, number of within-period events.
Assessment of VTE
No patients had a first-ever DVT within their exposed stratum. Two patients had a first-ever PE within their exposed stratum. After controlling for the confounding age variable, the corresponding adjusted IRR for VTE was 1.07 (95% CI 0.23, 5.03).
A retrospective analysis using 1575 patients exposed to raloxifene was used as a positive control to establish that the methodology was sufficiently sensitive to detect an increased incidence of VTE. The median age was 76 years (range 40–95; IQR 66–81) and the mean total observation period was 11 years. The corresponding age-adjusted IRR for VTE was 4.71 (95% CI 1.58, 13.00).
Assessment of gastrointestinal disturbance, dermatitis and memory loss
The corresponding age-adjusted IRRs for gastrointestinal disturbance, minor skin complaint and memory loss were 2.97 (95% CI 2.33, 3.79), 2.02 (95% CI 1.34, 3.05) and 1.81 (95% CI 0.23, 14.05), respectively. We conducted a sensitivity analysis for each of these end-points to determine if related chronic conditions affected the point estimate. There was no major effect on the point estimate by excluding chronic gastrointestinal disorders (ulcerative colitis, Crohn's disease, etc.), chronic skin complaints (psoriasis, atopic eczema, etc.) dementia or schizophrenia. The corresponding age-adjusted IRRs for gastrointestinal disturbance, minor skin complaint and memory loss after applying comorbid exclusions were 3.15 (95% CI 2.45, 4.05), 2.04 (95% CI 1.32, 3.16) and 1.89 (95% CI 0.23, 15.27), respectively.
Other adverse effects
There were no cases of toxic-epidermal necrosis, Stevens–Johnson syndrome, DRESS or osteonecrosis in women exposed to strontium ranelate, therefore no further analyses were necessary.
Discussion
Our study has demonstrated the utility of the self-controlled case-series method as an efficient method of pharmacovigilance and drug safety assessment. We used strontium ranelate as an example using adverse event signals in order to compare our results with the published Phase III studies, which are of similar statistical power (see Table 2). Thrombosis and serious skin reactions, such as DRESS, were not reported in the Phase III publications, but were subsequently highlighted during an EMEA review of the pooled data (odds ratio 1.5 at 3 years) [8] and from postmarketing surveillance, respectively. We found no association between these serious adverse events and exposure to strontium ranelate (VTE IRR 1.1). We validated our methodology by using it to detect the established relationship between raloxifene and VTE in a similar-sized population of raloxifene-exposed individuals, with a similar risk estimate (rate ratio of 4.6) to that reported in the published literature (rate ratio 6.6 in year 2) [11].
Table 2.
Comparison of risk estimates between randomized and observational data
| Randomized trial data risk ratio | Observational data risk ratio | |
|---|---|---|
| VTE (strontium) | 1.5 | 1.1 |
| VTE (raloxifene) | 6.6* | 4.7† |
| Gastrointestinal effects (strontium) | 1.5 | 3.0 |
| Dermatitis (strontium) | 1.2 | 2.0 |
| Memory loss (strontium) | 1.3 | 1.8 |
Year 2 risk ratio reported in Multiple Outcomes of Raloxifene Evaluation [11].
Raloxifene median exposure time in study: 2.3 years. VTE, venous thromboembolism.
Phase III clinical trials have identified gastrointestinal disturbance, dermatitis and memory loss as adverse events associated with strontium ranelate (risk ratio 1.5, 1.2 and 1.3, respectively); we also found an association between discontinuation of strontium ranelate and these events (IRR 3.0, 2.0 and 1.8, respectively). The stronger association that we identified might be explained by a higher discontinuation rate in observational studies (where it is difficult to distinguish adverse drug reactions from other adverse incidents) than in a clinical trial. This may exaggerate risk estimates derived from commonly presenting general practice consultations.
A limitation of our study is lack of statistical power, although even when eligible patient numbers are relatively small, the case-series method has statistical power equivalent to a much larger conventional cohort study [12]. However, this assumption applies only when the risk periods are short in relation to the observation period; and exposure is high. These circumstances retain high relative efficiency and thus this technique has been successfully employed in assessing acute adverse events secondary to vaccine administration [13–15]. More recently, it has been employed as a method of pharmacoepidemiology in assessing the risk of acute adverse events following initiation of drug therapy [16].
Other limitations of our study were the non-availability of secondary care prescriptions in the sampling frame, and thus allocation of the start of the exposure periods may be inaccurate. Moreover, accurate timing is crucial in the case-series method, and several days, or even weeks, may have elapsed before symptoms were evident and/or patients sought medical help. This may have resulted in inaccuracy in defining the exact date of an event.
A novel feature of our study was the use of the case-series method for a chronically administered drug to investigate the association of an adverse event without any known risk period (i.e. VTE analysis). Although this approach reduces the relative efficiency, it eliminates confounding by variables associated with both the outcome and avoidance of exposure by ensuring the comparisons were intraperson. The major time-varying confounder was age, and this was controlled for in this study; however, other unknown time-varying confounders may have affected the results. The case-series method requires only a sample of the cases (e.g. individuals exposed to strontium ranelate with a reported VTE), thus avoiding the need to follow large population cohorts or selecting controls. The case-series method was also used to assess a variety of adverse events associated with the discontinuation of therapy, and hence is more clinically significant.
The case-series method provides a quick and relatively simple way of assessing whether the association of a rare and potentially serious adverse effect, in actual clinical practice, is markedly divergent from the pre-marketing data. Hence, studies such as these provide unconfounded assessment of adverse events; in this particular study there was no evidence of an increased risk of VTE due to strontium ranelate. As well as having a useful role in the assessment of suspected adverse effects, recent extensions to the case-series method have been suggested to allow its use in the monitoring of previously unidentified adverse effects [17, 18].
Conclusion
The self-controlled case-series safety analyses of new drugs can be effectively utilized to inform on adverse events relatively soon after product launch. Such systematic large-scale studies can detect rare and potentially serious reactions in addition to small but important increases in the rate of common adverse events that are representative of real clinical practice. This method is amenable to assessment of adverse event rates for all new drugs and could be further modified to develop ‘stopping criteria’ as in pre-marketing clinical trials. Although such data should not be viewed in isolation, the efficiency and versatility of this method suggest that it could become a future standard in drug safety assessment.
Acknowledgments
We thank Tarita Murray-Thomas, and the GPRD research team, for supplying the data and providing advice and assistance. We also thank Heather Whittaker for her advice on the case-series method. L.S. is supported by a Wellcome Trust Senior Research Fellowship in Clinical Science. A.H. is a British Heart Foundation Senior Research Fellow.
REFERENCES
- 1.Schultz WB. Bolstering the FDA's drug-safety authority. N Engl J Med. 2007;357:2217–19. doi: 10.1056/NEJMp078212. [DOI] [PubMed] [Google Scholar]
- 2.Kazi D. Rosiglitazone and implications for pharmacovigilance. BMJ. 2007;334:1233–4. doi: 10.1136/bmj.39245.502546.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avorn J. In defense of pharmacoepidemiology – embracing the Yin and Yang of drug research. N Engl J Med. 2007;357:2219–21. doi: 10.1056/NEJMp0706892. [DOI] [PubMed] [Google Scholar]
- 4.Woo S, Hellstein J, Kalmar J. Systematic review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med. 2006;144:753–61. doi: 10.7326/0003-4819-144-10-200605160-00009. [DOI] [PubMed] [Google Scholar]
- 5.Meunier PJ, Roux C, Seeman E, Ortolani S, Badurski JE, Spector TD, Cannata J, Balogh A, Lemmel EM, Pors-Nielsen S, Rizzoli R, Genant HK, Reginster JY. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N Engl J Med. 2004;350:459–68. doi: 10.1056/NEJMoa022436. [DOI] [PubMed] [Google Scholar]
- 6.Reginster JY, Seeman E, De Vernejoul MC, Adami S, Compston J, Phenekos C, Devogelaer JP, Curiel MD, Sawicki A, Goemaere S, Sorensen OH, Felsenberg D, Meunier PJ. Strontium ranelate reduces the risk of nonvertebral fractures in postmenopausal women with osteoporosis: treatment of peripheral osteoporosis (TROPOS) study. J Clin Endocrinol Metab. 2005;90:2816–22. doi: 10.1210/jc.2004-1774. [DOI] [PubMed] [Google Scholar]
- 7.Protelos. Summary of Product Characteristics. Servier Labs. [last accessed: 21 August 2006]. Available at http://emc.medicines.org.uk/emc/assets/c/html/displaydoc.asp?documentid=15410.
- 8.EMEA Protelos Scientific Discussion 2005. [last accessed: 21 August 2006]. Available at http://www.emea.eu.int/humandocs/PDFs/EPAR/protelos/121604en6.pdf.
- 9.EMEA Press Release 2007. [last accessed: 30 December 2007]. Doc. Ref. EMEA/417458/2007). Available at http://www.emea.europa.eu/humandocs/PDFs/EPAR/protelos/PressRelease_Protelos_41745807en.pdf.
- 10.General Practice Research Database. [last accessed: 20 August 2007]. Available at http://www.gprd.com/home/
- 11.Grady D, Ettinger B, Moscarelli E, Plouffe L, Jr, Sarkar S, Ciaccia A, Cummings S Multiple Outcomes of Raloxifene Evaluation Investigators. Safety and adverse effects associated with raloxifene: multiple outcomes of raloxifene evaluation. Obstet Gynecol. 2004;104:837–44. doi: 10.1097/01.AOG.0000137349.79204.b8. [DOI] [PubMed] [Google Scholar]
- 12.Farrington CP, Nash J, Miller E. Case series analysis of adverse reactions to vaccines: a comparative evaluation. Am J Epidemiol. 1996;143:1165–73. doi: 10.1093/oxfordjournals.aje.a008695. [DOI] [PubMed] [Google Scholar]
- 13.Farrington CP. Relative incidence estimation from case series for vaccine safety evaluation. Biometrics. 1995;51:228–35. [PubMed] [Google Scholar]
- 14.Farrington P, Pugh S, Colville A, Flower A, Nash J, Morgan-Capner P, Rush M, Miller E. A new method for active surveillance of adverse events from diphtheria/ tetanus/pertussis and measles/mumps/rubella vaccines. Lancet. 1995;345:567–69. doi: 10.1016/s0140-6736(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 15.Farrington CP. Control without separate controls: evaluation of vaccine safety using case-only methods. Vaccine. 2004;22:2064–70. doi: 10.1016/j.vaccine.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard R, Lewis S, West J, Smith C, Godfrey C, Smeeth L, Farrington P, Britton J. Bupropion and the risk of sudden death: a self-controlled case series analysis using the Health Improvement Network. Thorax. 2005;60:848–50. doi: 10.1136/thx.2005.041798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hocine MN, Musonda P, Andrews NJ, Farrington PC. Sequential case-series analysis for pharmacovigilance. Journal of the Royal Statistical Society, Series A. in press. [Google Scholar]
- 18.Musonda P, Hocine MN, Andrews NJ, Tubert-Bitter P, Farrington PC. Monitoring vaccine safety using case series CUSUM charts. Vaccines. doi: 10.1016/j.vaccine.2008.08.010. in press. [DOI] [PubMed] [Google Scholar]