

I believe that the genetic code was the greatest discovery of the 20th century (Fig. 1). The code was worked out during the period that I was a student: a medical student at Tufts University (I dropped out after a year), a graduate student at MIT and Cornell (with Gordon Hammes), and a postdoctoral student at Stanford University (with Paul Flory). My research as a student was centered on theoretical and experimental physical chemistry, as applied to biological systems. With Hammes, I had done a spectroscopic and thermodynamic analysis of ribonuclease and its interaction with one of its ligands. I also had done both experimental and theoretical analyses of kinetic relaxation spectroscopy, inspired in part by the pioneering work of Eigen, who himself had been a mentor of Hammes. (Eigen gave lectures at Cornell that I attended, and we spent time in conversation as well. In more recent years, we have seen each other on a regular basis and continued our conversations.) With Flory at Stanford, I worked on statistical mechanics of polypeptides. All of that graduate and postdoctoral work was exciting, particularly the mathematical side that I especially loved because of its elegance, rigor, and abstraction. However, while this sort of mental activity was going on, I kept thinking about the newly discovered genetic code and aminoacyl-tRNA synthetases.
FIGURE 1.

Flow of genetic information from DNA to the tRNA-mRNA on the ribosome.
I have now spent my entire academic career working on the tRNA synthetase family of enzymes. It is these enzymes that establish the rules of the code through their aminoacylation reactions, whereby each amino acid is matched with a triplet nucleotide anticodon embedded in the cognate tRNA. In the first reaction, an amino acid (AA) is condensed with ATP to give a tightly bound aminoacyl adenylate (Equation 1). In the second reaction (Equation 2), the aminoacyl group is transferred to the 3′-end of the accepting tRNA.
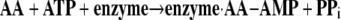 |
(Eq. 1) |
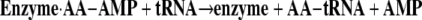 |
(Eq. 2) |
For each of the 20 amino acids, there is a separate enzyme and a family of isoaccepting tRNAs. Once these facts (Equations 1 and 2 and the existence of tRNA isoacceptors) were established, many people felt that the field was “stale,” and they moved on to other things. For me and a few others, it was only beginning. Today, I feel that the opportunities and questions are even more profound and exciting than they were when I started. Here, I sketch just one part of the work that has excited me from the beginning and that today has gone far beyond my or anyone else's expectations.
Fig. 2 shows cultured mammalian cells going into an apoptosis-like state because of the dominance of a mutation in a transgene encoding a tRNA synthetase (1). This enzyme, murine valyl-tRNA synthetase, has a normal active site and fuses valine to tRNAVal with the same efficiency as does the wild-type enzyme. It has a subtle mutational defect, however, in a second active site, a site that clears errors of aminoacylation. Here, the problem is the occasional confusion of threonine for valine. The confusion arises because the hydroxyl methyl side chain of threonine is isosteric with the isopropyl side chain of valine.
FIGURE 2.

Mouse 3T3 fibroblasts transfected with genes encoding either wild-type or T535P editing-defective ValRS. Expression of the transgenes is controlled by addition of an inducer. The induction of mutant, but not wild-type (WT), ValRS drives the cells into an apoptosis-like state. The figure is adapted from Nangle et al. (1).
Fig. 3 shows the “sticky” (sti) mouse studied by Susan Ackerman, our collaborator at The Jackson Laboratory. This mouse is diminutive and has ataxia. It harbors a mutation in the editing site of alanyl-tRNA synthetase. This site clears errors due to the confusion of glycine and serine for alanine (2).
FIGURE 3.

The diminutive sti mouse (foreground) has ataxia and harbors a gene encoding a mild defect in editing by AlaRS (2).
In both examples, subtle errors of aminoacylation lead to pathologies caused by mistranslation (Fig. 4), i.e. the insertion into growing polypeptides of the wrong amino acid at a codon for valine (Fig. 2) or alanine (Fig. 3). (The erroneous insertion of amino acids into proteins synthesized in cells bearing an editing-defective tRNA synthetase was established by direct chemical and mass spectrometric analysis.) Significantly, the connection of defects in editing to pathology and disease came from years of work in simpler bacterial systems. We eventually realized that, from the work in bacteria, even mild defects in editing would be challenging in the more complex environment of a mammalian cell.
FIGURE 4.

Mistranslation through editing defects results in the production of statistical proteins, where more than one kind of amino acid is incorporated at a specific codon.
That some sort of editing system was needed was first pointed out by Linus Pauling (3). When the code was elucidated, Pauling figured that tRNA synthetases would have difficulty discriminating closely similar amino acids. He used the examples of valine and isoleucine (Fig. 5). He realized that a pocket for the isobutyl side chain of Ile would accommodate the isopropyl side chain of Val. The van der Waals interactions of the pocket with the extra methylene group of isoleucine would only be enough to assure discrimination of the order of a factor of 100–200 or so. (Indeed, in the amino acid activation reaction of Equation 1, we found a discrimination ratio of about the predicted amount.) During that era, Anne Norris in Paul Berg's laboratory showed that, indeed, IleRS misactivated valine, but when the IleRS·Val-AMP complex was challenged with tRNA, Val-AMP broke down, thereby creating an abortive reaction cycle that consumes ATP (Equations 3 and 4) (4).
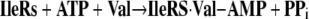 |
(Eq. 3) |
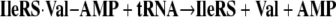 |
(Eq. 4) |
FIGURE 5.

Discrimination problem for IleRS. The discrimination of valine from isoleucine was first seen to be a problem by Pauling, who pointed out that the code is far more accurate than could be explained by having a single active site differentiate between the two amino acids, which differ by a single methylene group.
While I was at Stanford as a postdoctoral student, doing pure theoretical work, I talked to Berg about this result and about other work in his laboratory that related to tRNA synthetases. Those conversations further stimulated my interest in the tRNA synthetase family of enzymes. I decided that, upon taking an assistant professorship at MIT, I would give up my theoretical work and become an enzyme biochemist working on tRNA synthetases.
It was a great struggle. Mathematical physics and chemistry were natural to me and uplifted my spirits. Although I had some biochemical experience in the Hammes laboratory, I had never done any of the serious kinds of enzyme purifications and biochemical reaction analyses involving nucleic acids such as transfer RNAs. The tRNAs were themselves newly discovered and had to be isolated to study the aminoacylation reactions. Although Vernon Ingram at MIT was purifying individual tRNA amino acid acceptors using countercurrent distribution methods, we ourselves used purified bulk tRNA that was not partitioned into the different acceptors. For tRNA purifications and many other methods, we were greatly helped by Tom RajBhandary, who arrived at MIT from Wisconsin (where he had been trained with Gobind Khorana) a year after me and who generously gave me and my laboratory his time and advice. He also did not object to the simplicity and naïveté of my questions. (A few years later, to learn bacterial genetics, I worked side by side with postdoctoral student Nancy Kleckner in David Botstein's laboratory at MIT and, shortly later, learned “hands-on” recombinant DNA technology from John Carbon at Santa Barbara. Here again, as a trained theoretical physical chemist, I had all kinds of simple and naïve questions that were patiently answered by these experts in genetics and molecular biology.) I also began to have contact with Alexander Rich and Sunghou Kim, who were vigorously working on obtaining the three-dimensional structure of tRNA. These contacts and the many conversations with Alex Rich over the years since then gave me a much broader perspective of the possibilities and grandeur of the field I had entered.
My mind was set on figuring out how the synthetases avoided errors of aminoacylation, in an era that had none of the sophisticated techniques that are prevalent today. I figured that the key experiment was to make a mischarged tRNA species and then see if any of the enzymes had an activity that cleared the mischarged amino acid. For example, in Equation 5,
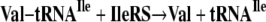 |
(Eq. 5) |
I chose IleRS because of the prior published work on this enzyme and because of the Berg laboratory's work showing the breakdown of IleRS-bound Val-AMP when confronted with tRNAIle. Rumor had it that the Berg group was attempting to use chemical methods to attach Val to tRNAIle to test whether the chemically misacylated tRNAIle could be cleared by IleRS. Meanwhile, we went in a different direction.
Alan Schreier, a graduate student in my laboratory, was encouraged to focus on the deacylation of tRNAs that were charged with their own cognate amino acids. We observed a weak activity with a number of the synthetases that he studied. For example, two of several deacylation reactions he uncovered and investigated are shown in Equations 6 and 7 (5).
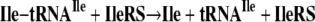 |
(Eq. 6) |
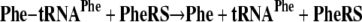 |
(Eq. 7) |
Although the rates of these reactions were low, they were faster than the spontaneous chemical rate of deacylation, and they occurred in the absence of AMP and PPi. The latter observation meant that the reactions were not the reverse of aminoacylation (Equations 1 and 2) and suggested a distinct activity that had not previously been studied. Most interesting, he could show that, by using inhibitors, the reactions were at a site distinct from that for aminoacylation, the first concrete evidence for a second active site. We speculated that this reaction was the long-sought editing activity for deacylating mischarged tRNA, such as tRNAIle or tRNAPhe. Our reasoning was that the slow activity seen in Equations 6 and 7 was speeded up when the wrong amino acid was put onto a tRNA (5).
We wrote up our results and submitted them to Biochemistry in late 1971, but the paper was held up by a reviewer. When our paper eventually came back, the reviewer had marked it up and, perhaps inadvertently, highlighted in pen or pencil our point about the activity being the one needed for correcting errors of aminoacylation. We knew that this point had struck home and would probably be soon tested by others working in the field.
Meanwhile, we wanted to show that, indeed, mischarged tRNA would be cleared rapidly by the newly discovered deacylase activity. Emmet Eldred, another graduate student in the laboratory, had done important work on the mechanism of transfer of the isoleucyl moiety of Ile-AMP from the adenylate to the 3′-end of tRNA. After finishing this work, with Schreier's work going on side by side in the laboratory, I emphasized to Eldred that the single most important thing to achieve next was the production of Val-tRNAIle. At this point, Tom RajBhandary called our attention to some work of Giegé and collaborators. In that work, the powerful Strasbourg group at the CNRS (headed by Jean Pierre Ebel) showed that addition of organic solvents to an aminoacylation mixture caused a synthetase to misacylate their tRNAs (6). Eldred seized on this observation and used their methods to produce Val-tRNAIle and then went on to figure out a way to isolate the mischarged species.
Armed with the long-sought substrate, Eldred showed that IleRS rapidly cleared Val-tRNAIle. We were elated and submitted the paper to the Journal of Biological Chemistry in early 1972. The work was rapidly published as a Communication (7), on the heels of the Schreier Biochemistry paper. Later that year, we were gratified to see the Yarus group confirm and extend Schreier's observations on PheRS by showing that Ile-tRNAPhe was rapidly deacylated by that enzyme (8). In that era (1970s), the Yarus group went on to make additional contributions, as did the Cramer laboratory in Germany and the Strasbourg group in France, while Alan Fersht did powerful kinetic and mechanistic studies, especially with ValRS and IleRS. Many years later, some of Fersht's mechanistic ideas from that period (especially the distinction between “pre-transfer” and “post-transfer” editing) were found to be consistent with results from entirely new experimental approaches that eventually became possible.
In our laboratory, after the initial Schreier and Eldred discoveries, work on editing ceased for about two decades. My interest at the time was mainly in establishing the deacylation activity itself, and not in highly mechanistic studies. Once we had discovered the activity, we moved on to other problems in the field, such as the overall design of the tRNA synthetase family of enzymes and how sequencing, genetics, and molecular biology, combined with biochemistry and structural analysis, could teach us more about that design. We also took on the challenge of understanding how a synthetase picked out its tRNA partner, using nucleotide determinants in the tRNA, but as the awareness of the RNA world emerged from the pioneering work of Cech, Altman, Pace, and others, we began thinking once again about editing. We knew that the discrimination of cognate versus non-cognate amino acid for editing was typically in the context of tRNA. With that in mind, we speculated that early synthetases may have been ribozymes for aminoacylation that eventually became ribonucleoproteins, analogous to the RNase P particle for tRNA processing (Fig. 6) (9). Thus, we imagined that amino acid recognition for editing may have evolved out of a ribonucleoprotein-like state, where protein and nucleic acid determinants afforded the discrimination of cognate from non-cognate amino acid.
FIGURE 6.

Schematic illustration of the evolution of aminoacylation (charging), starting with a ribozyme. AA, aminoacyl; RNP, ribonucleoprotein.
That idea in itself stimulated motivation for returning to the problem of editing. At the same time, our work on the architectural design and modular assembly of the enzymes created a greater awareness of the possibilities for identifying an active site for editing, and yet despite crystallographic analyses by several laboratories, the identification of a site for editing remained elusive.
The breakthrough came from genetic work of Eric Schmidt, a graduate student in the laboratory (10). Schmidt mutationally isolated the site for editing by obtaining mutations in IleRS that had little or no effect on aminoacylation but affected the activity for editing (Fig. 7). The converse mutations were also identified, i.e. mutations that altered aminoacylation but did not disrupt editing. The editing site identified by mutational analysis was in an insertion, previously identified and studied by another student, Ruth Starzyk (11). Laura Lin and Steve Hale then cloned the insertion as a separate domain and showed that it, by itself, could clear Val-tRNAIle. They also cloned the analogous insertion from ValRS and showed that it deacylated Thr-tRNAVal (12).
FIGURE 7.

Location of mutations in isoleucyl-tRNA synthetase that distinguish the active site from the site for editing. The actual identification of the two sites was achieved before the first x-ray structure of IleRS was reported (14).
Once again, I learned that science, in the words of Vannevar Bush, is an endless frontier. The discovery of the deacylation activity eventually led to the discovery and cloning of the site for editing, but that, in turn, only prompted more questions, such as the mechanism of translocation from the active site for aminoacylation to the site for editing (Fig. 8, A–C). This question and many others were taken up in the next era. As we learned more and more, we returned to genetics to investigate the effects of editing defects in bacteria. This work showed that defects in editing were conditionally lethal and that the genetic code could be invaded and rendered ambiguous unless tight control was maintained on the centers for editing in the various synthetases. On the other hand, ambiguity from editing defects could also afford, under some conditions, an advantage for organisms that had a limited supply of a canonical amino acid, such as a limited supply of valine for ValRS. (An editing defect enables the organism to occasionally “fill in” at a position in a nascent polypeptide with a non-canonical surrogate (like α-amino butyrate) amino acid that was not cleared by a defective editing machinery.) We also found that, in aging bacteria, defects in editing were mutagenic because of mistranslation of proteins in the replication apparatus that, over time, made errors that were fixed in the genome. Gradually, we worked our way into mammalian systems, which seem far more sensitive than bacteria to editing defects. This progression led to the results shown in Figs. 3 and 4 in mammalian cells and the mouse.
FIGURE 8.

A, shown is a schematic illustration of the two sites for amino acid recognition: the active site for aminoacylation and the site for editing. The editing site is a steric sieve, which cannot accommodate isoleucine. B, translocation from the active site to the center for editing is rate-determining (for IleRS) and is controlled by the bound tRNA. Val‡ is activated valine, as Val-AMP or Val-tRNAIle. When present as Val-tRNAIle, the tRNAIle remains bound, but its 3′-end can toggle between the active site for aminoacylation and the editing site. C, the “toggling” of the 3′-end of Val-tRNAIle is illustrated explicitly.
The story continues into the future (13). Now we are focused on the idea that editing defects may be associated with a variety of diseases, and specific experiments with specific diseases in mind have been designed to investigate this question. Although we are convinced that strong germ line mutational disruptions of editing are lethal, a mild mutation, such as that seen with the sti mouse, could be vertically transmitted in the population. Also, environmentally generated somatic mutations may, over time, engender pathologies in specific tissues. These are questions that keep me awake and thinking, when I might otherwise be getting a good night's sleep. Yet, it is no different now than what I felt in the very beginning, when I was enthralled, as a young theoretician at Stanford, by the idea of working on tRNA synthetases and, shortly later, as a junior faculty member at MIT, by the idea of finding a way to make mischarged tRNA.
Acknowledgments
The work on editing activity has been supported over the years by grants from the National Institutes of Health, by a fellowship from the National Foundation for Cancer Research, and by The Skaggs Foundation.
References
- 1.Nangle, L. A., Motta, C. M., and Schimmel, P. (2006) Chem. Biol. 13 1091–1100 [DOI] [PubMed] [Google Scholar]
- 2.Lee, J. W., Beebe, K., Nangle, L. A., Jang, J., Longo-Guess, C. M., Cook, S. A., Davisson, M. T., Sundberg, J. P., Schimmel, P., and Ackerman, S. L. (2006) Nature 443 50–55 [DOI] [PubMed] [Google Scholar]
- 3.Pauling, L. (1958) Festschrift fuer Prof. Dr. Arthur Stoll, Birkhauser Verlag, Basel, Switzerland
- 4.Baldwin, A. N., and Berg, P. (1966) J. Biol. Chem. 241 831–838 [PubMed] [Google Scholar]
- 5.Schreier, A. A., and Schimmel, P. R. (1972) Biochemistry 11 1582–1589 [DOI] [PubMed] [Google Scholar]
- 6.Giegé, R., Kern, D., Ebel, J. P., and Taglang, R. (1971) FEBS Lett. 15 281–285 [DOI] [PubMed] [Google Scholar]
- 7.Eldred, E. W., and Schimmel, P. R. (1972) J. Biol. Chem. 247 2961–2964 [PubMed] [Google Scholar]
- 8.Yarus, M. (1972) Proc. Natl. Acad. Sci. U. S. A. 69 1915–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schimmel, P., and Schmidt, E. (1995) Trends Biochem. Sci. 20 1–2 [DOI] [PubMed] [Google Scholar]
- 10.Schmidt, E., and Schimmel, P. (1994) Science 264 265–267 [DOI] [PubMed] [Google Scholar]
- 11.Starzyk, R. M., Webster, T. A., and Schimmel, P. (1987) Science 237 1614–1618 [DOI] [PubMed] [Google Scholar]
- 12.Lin, L., Hale, S. P., and Schimmel, P. (1996) Nature 384 33–34 [DOI] [PubMed] [Google Scholar]
- 13.Beebe, K., Mock, M., Merriman, E., and Schimmel, P. (2008) Nature 451 90–93 [DOI] [PubMed] [Google Scholar]
- 14.Nureki, O., Vassylyev, D. G., Tateno, M., Shimada, A., Nakama, T., Fukai, S., Konno, M., Hendrickson, T. L., Schimmel, P., and Yokoyama, S. (1998) Science 280 578–582 [DOI] [PubMed] [Google Scholar]