

Abstract
Tyr122-hydrophobic cluster (Y122-HC) is an interaction network formed by the top part of the second transmembrane helix and the cytoplasmic actuator and phosphorylation domains of sarcoplasmic reticulum Ca2+-ATPase. We have previously found that Y122-HC plays critical roles in the processing of ADP-insensitive phosphoenzyme (E2P) after its formation by the isomerization from ADP-sensitive phosphoenzyme (E1PCa2) (Wang, G., Yamasaki, K., Daiho, T., and Suzuki, H. (2005) J. Biol. Chem. 280, 26508–26516). Here, we further explored kinetic properties of the alanine-substitution mutants of Y122-HC to examine roles of Y122-HC for Ca2+ release process in E2P. In the steady state, the amount of E2P decreased so that of E1PCa2 increased with increasing lumenal Ca2+ concentration in the mutants with K0.5 110–320 μm at pH 7.3. These lumenal Ca2+ affinities in E2P agreed with those estimated from the forward and lumenal Ca2+-induced reverse kinetics of the E1PCa2-E2P isomerization. K0.5 of the wild type in the kinetics was estimated to be 1.5 mm. Thus, E2P of the mutants possesses significantly higher affinities for lumenal Ca2+ than that of the wild type. The kinetics further indicated that the rates of lumenal Ca2+ access and binding to the transport sites of E2P were substantially slowed by the mutations. Therefore, the proper formation of Y122-HC and resulting compactly organized structure are critical for both decreasing Ca2+ affinity and opening the lumenal gate, thus for Ca2+ release from E2PCa2. Interestingly, when K+ was omitted from the medium of the wild type, the properties of the wild type became similar to those of Y122-HC mutants. K+ binding likely functions via producing the compactly organized structure, in this sense, similarly to Y122-HC.
Sarcoplasmic reticulum Ca2+-ATPase (SERCA1a)2 of the P-type ion-transporting ATPase family catalyzes Ca2+ transport coupled with ATP hydrolysis from the cytoplasm to lumen against a concentration gradient of ∼10,000-fold (1–8). In the initial steps (steps 1 and 2 in Scheme 1), the enzyme is activated by binding of two cytoplasmic Ca2+ ions at the transport sites with a submicromolar high affinity (E2 to E1Ca2). The activated enzyme is then auto-phosphorylated at Asp351 by ATP and forms a phosphoenzyme intermediate (EP) (step 3), thereby the bound Ca2+ ions are occluded in the transport sites. This EP is rapidly dephosphorylated by ADP in the reverse reaction reproducing ATP, therefore “ADP-sensitive EP” (E1P). In the next step (step 4), E1PCa2 is isomerized to the ADP-insensitive form, E2PCa2. Upon this change at the catalytic site, the Ca2+ sites are deoccluded and opened to the lumenal side, and the Ca2+ affinity is largely reduced, releasing the bound Ca2+ ions into the lumen (step 5). The Ca2+ release process is thought to be very rapid with the wild-type Ca2+-ATPase, and the accumulation of E2PCa2 intermediate had actually never been found until we recently identified and trapped successfully this intermediate by a mutation study (9). In the final step, the Asp351-acylphosphate of E2P is hydrolyzed to reproduce the dephosphorylated and inactive E2 form (step 6). The transport cycle is totally reversible, e.g. E2P can be formed from E2 by Pi in the absence of Ca2+, and the subsequent lumenal Ca2+ binding to E2P produces E1PCa2.
SCHEME 1.

Three-dimensional structures in several intermediate states and their
analogs have been solved
(10–18).
The Ca2+-ATPase has three cytoplasmic domains, P (phosphorylation),
N (nucleotide binding), and A (actuator or anchor), and ten transmembrane
helices (M1–M10). The two Ca2+ binding sites consist of
residues on M4, M5, M6, and M8
(10). The P domain possesses
the phosphorylation site (Asp351) and is directly linked to the
long helices M4 and M5. The ATP binding site is on the N domain connected to
the P domain. The A domain is linked to M1, M2, and M3 via the A/M1-, A/M2-,
and A/M3-linkers. The cytoplasmic three domains largely move and change their
organization states during the Ca2+-transport cycle
(19–21),
and these changes are linked with the rearrangements in the transmembrane
helices for the Ca2+ transport. As a most remarkable change, in the
EP isomerization (loss of ADP sensitivity) and Ca2+
release, the A domain largely rotates and the P domain largely inclines toward
the A domain, and these domains produce their tight association (see
Fig. 1 for the change
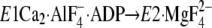 as the model for the overall process
E1∼PCa2·ADP →
E2·Pi, including the EP isomerization and
Ca2+ release). These structural changes therefore involve distinct
events in distinct regions, yet they are coordinated; namely 1) the loss of
ADP sensitivity at the cytoplasmic region, 2) the decrease in the
Ca2+ affinity at the transmembrane region, and 3) the opening of
the Ca2+-releasing pathway (lumenal gating).
as the model for the overall process
E1∼PCa2·ADP →
E2·Pi, including the EP isomerization and
Ca2+ release). These structural changes therefore involve distinct
events in distinct regions, yet they are coordinated; namely 1) the loss of
ADP sensitivity at the cytoplasmic region, 2) the decrease in the
Ca2+ affinity at the transmembrane region, and 3) the opening of
the Ca2+-releasing pathway (lumenal gating).
FIGURE 1.

Structure of SERCA1a and formation of Tyr122-hydrophobic
cluster. The coordinates for the structures
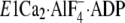 (the analog for the transition state of the phosphoryl transfer
E1∼PCa2·ADP, left panel) and
(the analog for the transition state of the phosphoryl transfer
E1∼PCa2·ADP, left panel) and
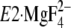 (E2·Pi analog
(21), right panel) of
Ca2+-ATPase were obtained from the Protein Data Bank (PDB accession
codes 1T5T and 1WPG, respectively
(12,
14)). The arrows
indicate approximate movements of the A and P domain and the top part of M2
(Leu119/Tyr122) in the change from
(E2·Pi analog
(21), right panel) of
Ca2+-ATPase were obtained from the Protein Data Bank (PDB accession
codes 1T5T and 1WPG, respectively
(12,
14)). The arrows
indicate approximate movements of the A and P domain and the top part of M2
(Leu119/Tyr122) in the change from
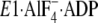 to
to
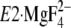 . The seven hydrophobic
residues, Ile179/Leu180/Ile232 on the A
domain, Leu119/Tyr122 on the A/M2-linker,
Val705/Val726 on the P domain are depicted as van der
Waals spheres. They gather to form a hydrophobic cluster around
Tyr122 in the change
. The seven hydrophobic
residues, Ile179/Leu180/Ile232 on the A
domain, Leu119/Tyr122 on the A/M2-linker,
Val705/Val726 on the P domain are depicted as van der
Waals spheres. They gather to form a hydrophobic cluster around
Tyr122 in the change
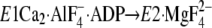 (Y122-HC, surrounded by the red dotted circle). The top part of M2,
including Leu119/Tyr122, is unwound in
(Y122-HC, surrounded by the red dotted circle). The top part of M2,
including Leu119/Tyr122, is unwound in
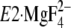 ,
,
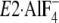 , and
, and
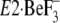 with bound thapsigargin
(TG), and thus becomes the A/M2-linker loop (see the region of M2 colored by
pink).
with bound thapsigargin
(TG), and thus becomes the A/M2-linker loop (see the region of M2 colored by
pink).
Recently, we found that mutations in a specific hydrophobic interaction network, “Tyr122-hydrophobic cluster” (Y122-HC), at the A-P domain interface disrupt markedly the processing of ADP-insensitive EP formed from ATP with Ca2+ and also the hydrolysis of E2P formed from Pi without Ca2+, thus causing nearly complete inhibition of the Ca2+-ATPase activity (22, 23). In these Y122-HC mutants, the high affinity binding of cytoplasmic Ca2+, the resulting E1PCa2 formation, and the loss of the ADP sensitivity were all found to occur normally as in the wild type (22, 23). Y122-HC is formed by gathering of the seven residues of the three regions upon their motions; i.e. the largely rotated A domain (Ile179, Leu180, and Ile232), the inclined P domain (Val705 and Val726), and the top part of the largely inclined M2 (or the A/M2-linker) (Leu119 and Tyr122). Thus Y122-HC produces the compactly organized structure of E2P. Our previous analyses indicate that, in the Y122-HC mutants, there is a kinetic limit after the loss of ADP sensitivity and before the hydrolysis of the Ca2+-free E2P, therefore the Ca2+ release from E2PCa2 is likely retarded (22, 23). Almost the same kinetic results were found with the mutations in another A-P domain interaction network at the Val200 loop of the A domain (24). Notably, E2PCa2, the ADP-insensitive EP with two Ca2+ ions occluded at the transport sites was recently identified and trapped successfully by the elongation of the A/M1-linker with two or more amino acid insertions (9). In the elongation mutants, Y122-HC is not formed properly yet in E2PCa2 trapped, but it is properly formed in the Ca2+-released form of E2P produced by Pi without Ca2+. Thus the observation is consistent with the involvement of Y122-HC in the Ca2+ release process from E2PCa2.
In the present study, to further clarify roles of Y122-HC in the
Ca2+ deocclusion/release processes and thus in the long range
communication between the cytoplasmic and transmembrane regions, we explored
kinetic features of the alanine-substitution mutants of Y122-HC. The results
revealed that the mutations cause a marked increase in the apparent affinity
of E2P for lumenal Ca2+ and also a substantial retardation
of the lumenal Ca2+ access to E2P. Therefore, the
formation of Y122-HC is critical for decreasing the affinity for
Ca2+, for lumenal gating (opening of the release pathway), and thus
for Ca2+ release into lumen. Importantly, the assembling manner of
the seven residues in Y122-HC in the very recently revealed crystal structure
 (17,
18) somewhat differs from that
in
(17,
18) somewhat differs from that
in 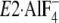 and
and
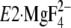 . Therefore, we
discussed the significance of this difference in terms of the possible
sequential gathering of the seven residues into Y122-HC on the basis of the
observed difference in the extents of their mutational effects. In addition,
we found with the wild type that its kinetic behavior became similar to that
of Y122-HC mutants when K+ was omitted from the medium of the wild
type. Results revealed for the first time the critical role of K+
binding in the wild type for Ca2+ deocclusion/release from
E2PCa2.
. Therefore, we
discussed the significance of this difference in terms of the possible
sequential gathering of the seven residues into Y122-HC on the basis of the
observed difference in the extents of their mutational effects. In addition,
we found with the wild type that its kinetic behavior became similar to that
of Y122-HC mutants when K+ was omitted from the medium of the wild
type. Results revealed for the first time the critical role of K+
binding in the wild type for Ca2+ deocclusion/release from
E2PCa2.
EXPERIMENTAL PROCEDURES
Mutagenesis and Expression—Mutations were created by the QuikChange™ site-directed mutagenesis kit (Stratagene) and plasmid pGEM7-Zf(+) or pGEM3-Zf(+) (Promega, Madison, WI) containing ApaI-KpnI or KpnI-SalI fragments of rabbit SERCA1a cDNA as a template. The ApaI-KpnI or KpnI-SalI fragments were then excised from the products and used to replace the corresponding region in the full-length SERCA1a cDNA in the pMT2 expression vector (25). The pMT2 DNA was transfected into COS-1 cells by the liposome-mediated transfection method. Microsomes were prepared from the cells as described previously (26). The “control microsomes” were prepared from COS-1 cells transfected with the pMT2 vector containing no SERCA1a cDNA.
ATPase Activity—The rate of ATP hydrolysis was determined at 25 °C in a mixture containing 20 μg/ml microsomal protein, 0.1 mm [γ-32P]ATP, 3 μm A23187, 0.1 m KCl, 7 mm MgCl2, various concentrations of CaCl2 up to 3 mm, 0.01 mm EGTA, and 50 mm MOPS/Tris (pH 7.3).
Formation and Hydrolysis of EP—Phosphorylation of SERCA1a in microsomes with [γ-32P]ATP or 32Pi and dephosphorylation of 32P-labeled SERCA1a were performed under conditions described in the figure legends. The reactions were quenched with ice-cold trichloroacetic acid containing Pi. Rapid kinetics measurements of phosphorylation and dephosphorylation were performed with a handmade rapid mixing apparatus (27), otherwise the method was as above. The precipitated proteins were separated at pH 6.0 by 5% SDS-PAGE, according to Weber and Osborn (28). The radioactivity associated with the separated Ca2+-ATPase was quantitated by digital autoradiography as described previously (29). The amount of EP formed with the expressed SERCA1a was obtained by subtracting the background radioactivity with the control microsomes. This background was <1% of the radioactivity of EP formed with the expressed wild-type SERCA1a.
Miscellaneous—Protein concentrations were determined by the method of Lowry et al. (30) with bovine serum albumin as the standard. Free Ca2+ concentrations were calculated by the Calcon program. Data were analyzed by nonlinear regression using the program Origin (MicroCal Software, Inc., Northampton, MA). Three-dimensional models of the enzyme were reproduced by using the program VMD (31).
RESULTS
Ca2+-induced Change in Accumulation of ADP-insensitive EP in the Presence of 0.1 m K+ at Steady State—We first determined the steady state Ca2+-ATPase activity in the presence of increasing Ca2+ and ionophore A23187 with the alanine-substitution mutants of the seven residues of Y122-HC and the wild type. The Ca2+-ATPase activity was nearly completely inhibited in all the mutants in agreement with our previous observation (22, 23), and the complete inhibition was found at all the Ca2+ concentrations examined (see supplemental Fig. S1 for the representative mutant Y122A). Thus the possible lumenal Ca2+ effect was not revealed by this type of measurements. Therefore in Fig. 2, to assess the affinity of the lumenally oriented Ca2+ transport site of E2P (known as the low affinity sites with the mm over ∼10 mm Kd value), the amounts of ADP-insensitive EP were determined with the representative mutant Y122A at steady state at various Ca2+ concentrations and pH values in the presence of A23187 and KCl. The total amounts of EP (ADP-sensitive EP plus ADP-insensitive EP) were nearly the same under all the sets of conditions.
FIGURE 2.

Ca2+ dependence of accumulation of ADP-insensitive EP in the steady state in mutant Y122A. Microsomes expressing the mutant Y122A were phosphorylated with [γ-32P]ATP at various Ca2+ concentrations and pHs as indicated at 0 °C for 5 min in a mixture containing 20 μg/ml microsomal protein, 50 mm MOPS/Tris, 0.1 m KCl, 7 mm MgCl2, 0.01 mm EGTA, 3 μm A23187, 10 μm [γ-32P]ATP, and various concentrations of CaCl2. For the determination of the accumulated ADP-insensitive EP, an equal volume (50 μl) of a mixture containing 10 mm ADP, 7 mm MgCl2, 10 mm EGTA, 50 mm MOPS/Tris (pH 6.8, 7.3, or 7.8 as indicated), and 0.1 m KCl was added to the above phosphorylation mixture. At 1 s after this addition, the reaction was quenched with trichloroacetic acid. ADP-sensitive EP disappeared entirely within 1 s after the addition of ADP. The total amounts of EP were nearly the same under all the conditions (data not shown). The amount of ADP-insensitive EP is shown as a percentage of the total amount of EP. Solid lines show the least squares fit to the Hill equation. Apparent Ca2+ affinities and Hill coefficients thus obtained were 360 μm and 2.1 (pH 6.8), 160 μm and 1.9 (pH 7.3), and 76 μm and 2.0 (pH 7.8).
In the mutant Y122A, the fraction of the ADP-insensitive EP was very high at the low Ca2+ concentrations at all pHs (Fig. 2). This agrees with the property of this mutant (22, 23) that the hydrolysis of E2P is nearly completely inhibited thus causing its accumulation. The fraction of ADP-insensitive EP in the mutant markedly decreased, and it was converted to the ADP-sensitive EP with increasing Ca2+ concentration at several tens of micromolar to the sub-millimolar range. The apparent Ca2+ affinity in this Ca2+-induced change increased with increasing pH, and the Hill coefficients were found to be 2 in all pH values (see the legend to Fig. 2). In the wild type, the fraction of ADP-insensitive EP was low at pH 7.3 and 7.8 being ∼10% or less, and was a significant level, 35% at pH 6.8 (supplemental Fig. S2). These levels were not changed at 1 μm to 3 mm Ca2+. Consistently, the lumenal Ca2+ affinity of E2P of the wild type is known to be in the millimolar to 10 mm range (see Ref. 32–34). The results suggested that the lumenal Ca2+ affinity of transport sites of E2P in the mutant may be significantly higher than that in the wild type.
Time Courses of Forward and Ca2+-induced Reverse Conversions between E1PCa2 and E2P—In Fig. 3 with Y122A, the ADP-insensitive EP, and the ADP-sensitive EP was first accumulated at steady state at 10 μm Ca2+ and 1 mm Ca2+, respectively, at pH 7.3. Then the Ca2+ concentration jump was made from 10 μm to 1 mm or from 1 mm to 80 nm, and the change in the fraction of the ADP-insensitive EP was followed. Because the hydrolysis of E2P was nearly completely blocked in Y122A (with the rate << 0.01 s-1) (22, 23), the time courses represent the forward and reverse isomerization between E1PCa2 and E2P. When Ca2+ was increased from 10 μm to 1 mm, the fraction of ADP-insensitive EP rapidly decreased from 80% to 10% (i.e. it was converted to the ADP-sensitive EP) with a rate 0.4 s-1. On the other hand, when the Ca2+ concentration was decreased from 1 mm to 80 nm, i.e. virtually Ca2+ was removed, the ADP-sensitive EP was converted to the ADP-insensitive EP with a rate 0.022 s-1.
FIGURE 3.

Time course of the change in the fraction of ADP-insensitive EP upon Ca2+-concentration jump. Microsomes expressing the mutant Y122A (20 μg/ml) were phosphorylated with [γ-32P]ATP at pH 7.3 for 5 min in the presence of 10 μm (○) or 1 mm CaCl2 (•) without EGTA in the phosphorylation solution otherwise as described in Fig. 2. Then an equal volume of the solution containing 2 mm CaCl2 or 2 mm EGTA (otherwise as in the phosphorylation solution) was added to give final free Ca2+ concentrations 1 mm and 80 nm, respectively. At the indicated times after this Ca2+ jump, the total amount of EP and the amount of ADP-insensitive EP was determined as in Fig. 2. Solid lines show the least squares fit to a single exponential. The total amount of EP was not changed during the period of observation in both cases (data not shown). The amount of ADP-insensitive EP is shown as a percentage of the total amount of EP.
Effect of Ca2+ Ionophore A23187 in Accumulation of ADP-insensitive EP—To ascertain that the Ca2+-dependent changes in the fraction of ADP-insensitive EP (Figs. 2 and 3) are caused by lumenal Ca2+, we examined also in the absence of A23187 the Ca2+ dependence of the steady-state fraction of ADP-insensitive EP (Fig. 4). The Ca2+-dependent change was rather small in the absence of A23187, in contrast to the very large change in its presence. Therefore, the observed Ca2+-induced conversion from the ADP-insensitive EP to ADP-sensitive one in Y122A is due to the Ca2+ binding to the lumenally oriented transport sites.
FIGURE 4.

Effect of calcium ionophore on the Ca2+ dependence of ADP-insensitive EP accumulation in the steady state. Microsomes expressing the mutant Y122A (20 μg/ml) were phosphorylated at 0 °C for 5 min in the medium containing 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, 10 μm CaCl2, 10 μm [γ-32P]ATP, and 50 μm Ruthenium Red (to block Ca2+ channels as much as possible) in the presence (○) or absence (•) of 3 μm A23187. Then the free Ca2+ concentration was changed as indicated on the abscissa by mixing with an equal volume of a solution containing 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, 2 mm EGTA, and various concentrations of CaCl2. After 10 s of this Ca2+ jump, the total amount of EP and the fraction of the ADP-insensitive EP were determined as described in Fig. 2. The inclusion of Ruthenium Red in the presence of A23187 caused a slight decrease in the ADP-insensitive EP fraction at the Ca2+ concentrations below ∼100 μm (cf. Fig. 2) for unknown reasons.
Ca2+-induced Change in Accumulation of ADP-insensitive EP of Seven Y122-HC Mutants in the Presence of 0.1 m K+—In Fig. 5, each of the other six residues involved in the Y122-HC (Leu119/Ile179/Leu180/Ile232/Val705/Val726) was substituted with alanine, and its effect on the ADP-insensitive EP level was examined as in Fig. 2 in the presence of A23187 and K+. All the Y122-HC mutants exhibited the marked Ca2+-dependent change in the ADP-insensitive EP fraction.3 The apparent affinities for lumenal Ca2+ in the Y122-HC mutants were found to be between 110 and 320 μm with a Hill coefficient of ∼2 (Table 1). The results showed that all these Y122-HC mutants possess the lumenally oriented transport sites with the affinities as high as that of Y122A.
FIGURE 5.

Ca2+ dependence of accumulation of ADP-insensitive EP in mutants for Tyr122-hydrophobic cluster. Microsomes expressing each of the seven Y122-HC mutants (indicated in the figure) were phosphorylated with [γ-32P]ATP at various concentrations of Ca2+ and pH 7.3, otherwise as described in Fig. 2. Solid lines show the least squares fit to the Hill equation. The fitting parameters, including the apparent Ca2+ affinity and the Hill coefficient, thus obtained are listed in Table 1.
TABLE 1.
Parameters obtained for Ca2+ dependence of accumulation of ADP-insensitive EP in Y122-HC mutants As shown in Fig. 5, the lumenal Ca2+-induced change in the steady-state accumulation of ADP-insensitive EP of the seven Y122-HC mutants in the presence of 0.1 m K+ were fitted to the Hill equation. The parameters thus obtained by the least squares fit are listed here. K0.5 is the Ca2+ concentration giving the half-maximum change in the fraction of ADP-insensitive EP among the total amount of EP, therefore the apparent affinity for lumenal Ca2+. The highest fraction of the ADP-insensitive EP at the low Ca2+ concentration range (0–10 μm) and its lowest fraction at the high Ca2+ concentration range (over ∼mm) are also listed as the obtained parameters in the fitting (see Fig. 5). The value nH is the Hill coefficient.
|
Mutant
|
Fraction of ADP-insensitive EP at highest and lowest
Ca2+ concentration ranges
|
K0.5
|
nH
|
||
|---|---|---|---|---|---|
| High Ca2+ | Low Ca2+ | ||||
| % of total amount of EP | mm | ||||
| L119A | 11 | 78 | 0.12 | 2.1 | |
| Y122A | 6 | 86 | 0.16 | 1.9 | |
| I179A | 12 | 92 | 0.25 | 2.3 | |
| L180A | 9 | 44 | 0.11 | 1.6 | |
| I232A | 10 | 89 | 0.32 | 2.0 | |
| V705A | 39 | 80 | 0.27 | 1.6 | |
| V726A | 23 | 65 | 0.28 | 2.0 | |
Ca2+-induced Change in Accumulation of ADP-insensitive EP of Mutants and Wild Type in the Absence of K+—In Fig. 6, the same sets of steady-state analysis as in Fig. 2 were done with the wild type and Y122A but here in the absence of K+. It is well known (35, 36) that, in the absence of K+, the E2P hydrolysis of the wild type is markedly slowed, and therefore the ADP-insensitive EP significantly accumulates. The fraction of ADP-insensitive EP in the wild type in the absence of K+ decreased with increasing Ca2+ concentration as in Y122A with the Hill coefficient ∼2. The apparent affinity for lumenal Ca2+ increased with increasing pH in the wild type as in Y122A. The pH-dependent changes are consistent with the fact that the residues for Ca2+ ligation at the transport sites are also involved in the proton binding (and its counter transport); the observed Ca2+-induced changes reflect the Ca2+ binding to the lumenally oriented transport sites of E2P. At each pH, the affinity of the wild type was similar to or slightly lower than that of Y122A. Thus in the absence of 0.1 m K+, the property of the wild type became similar to that of Y122A. In Y122A, elimination of K+ exhibited no significant effect on the apparent affinity for lumenal Ca2+ (cf. Fig. 2).
FIGURE 6.

Ca2+ dependence of accumulation of ADP-insensitive EP in the absence of K+. Microsomes expressing the wild type (A) or Y122A (B) SERCA1a were phosphorylated with [γ-32P]ATP at various Ca2+ concentrations and pH (6.8 (○), 7.3 (•), and 7.8 (▵)) in the presence of 0.1 m LiCl in place of KCl, otherwise under exactly the same conditions as those described in Fig. 2. The amount of ADP-insensitive EP was determined by addition of the ADP solution that contains 0.1 m LiCl in place of KCl otherwise as in Fig. 2. The total amount of EP was nearly constant under all the conditions (data not shown). The amount of ADP-insensitive EP is shown as a percentage of the total amount of EP. Solid lines show the least squares fit to the Hill equation. Apparent Ca2+ affinities and Hill coefficients thus obtained with the wild type (panel A) were 930 μm and 1.8 (pH 6.8), 400 μm and 1.5 (pH 7.3), and 310 μm and 1.9 (pH 7.8), and those obtained with Y122A (panel B) were 1000 μm and 1.9 (pH 6.8), 220 μm and 2.2 (pH 7.3), and 130 μm and 1.7 (pH 7.8).
The observed effect of K+ on the wild type is probably due to its binding in the cytoplasmic region. In crystallographic as well as mutational studies (12, 37), the K+ binding site of the Ca2+-ATPase was identified to be in the cytoplasmic region but not in the lumenal or transmembrane regions (see Fig. 11). Actually, we found experimentally that, when K+ at 0.1 m was added without any K+-ionophore to the Ca2+-ATPase in SR vesicles phosphorylated in the absence of K+, the Ca2+-dependence of the ADP-insensitive EP fraction observed as in Fig. 6A became immediately (within 10 s after the K+ addition) that in the presence of 0.1 m K+ as in supplemental Fig. S2 (data not shown).
FIGURE 11.

Bound K+ and Tyr122-hydrophobic cluster in crystal
structures. The part of structures
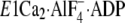 (E1∼PCa2·ADP analog, left) and
(E1∼PCa2·ADP analog, left) and
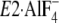 (E2∼P
analog, right) around Y122-HC and the bound K+ ion are
shown in schematic models (PDB codes: 1T5T and 1XP5
(12,
15)). The two structures were
manually aligned with M8–M10 helices, which do not move virtually in the
two. K+ bound in these structures is shown by a yellow van
der Waals sphere. Gln244 on the A/M3-linker at the
immediate vicinity of the bound K+ in
(E2∼P
analog, right) around Y122-HC and the bound K+ ion are
shown in schematic models (PDB codes: 1T5T and 1XP5
(12,
15)). The two structures were
manually aligned with M8–M10 helices, which do not move virtually in the
two. K+ bound in these structures is shown by a yellow van
der Waals sphere. Gln244 on the A/M3-linker at the
immediate vicinity of the bound K+ in
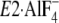 is indicated by ball
and stick model.
is indicated by ball
and stick model.
Kinetics of Lumenal Ca2+-induced E2P to E1PCa2 Reverse Transition Followed by Its ADP-induced Rapid Decay to E1Ca2 in the Presence of 0.1 m K+—Then with the representative mutant Y122A, we explored kinetically the lumenal Ca2+ accessibility to the lumenally oriented transport sites of E2P formed from Pi without Ca2+ and the resulting lumenal Ca2+-induced E2P to E1PCa2 reverse transition. In Fig. 7, we included ADP and thereby followed the Ca2+- and ADP-induced decay of E2P to E1Ca2 via E1PCa2 in the reverse reaction. The E2P hydrolysis in the absence of Ca2+ was extremely slow (as previously demonstrated with the Y122-HC mutants (22, 23)), and the E2P decay was dramatically accelerated by the addition of Ca2+ and ADP (Fig. 7A). For example, the rate in the presence of 1 mm Ca2+ was 200-times faster than that of the forward E2P hydrolysis in the absence of Ca2+. ADP alone without Ca2+ or Ca2+ alone without ADP did not accelerate the EP decay (data not shown). As shown in Fig. 3, the increase of Ca2+ to 1 mm converted the ADP-insensitive EP (E2P) to the ADP-sensitive one (E1PCa2), and E1PCa2 thus formed was not decomposed in the absence of ADP. Therefore the Ca2+- and ADP-induced decay of E2P in Fig. 7A obviously occurred in the reverse reaction by the lumenal Ca2+ binding; E2P + 2Ca2+ → E1PCa2, then E1PCa2 + ADP → E1Ca2 + ATP (Scheme 1). This view agrees with the previous demonstration with SR Ca2+-ATPase (34). The rate of the EP decay in the presence of Ca2+ and ADP increased almost linearly with increasing Ca2+ concentrations and was not saturated even at 3 mm (Fig. 7B).4 Here, note that the steady-state level of ADP-sensitive EP (E1PCa2) in the conversion from the ADP-insensitive EP (E2P) upon the lumenal Ca2+ binding in Y122A was almost fully saturated at 1 mm Ca2+ (see Fig. 2 at pH 7.3). The results indicate that the lumenal Ca2+ binding to E2P and formation of E2PCa2 (E2P + 2Ca2+ → E2PCa2) is likely the rate-limiting for the overall Ca2+- and ADP-induced reverse decay via the E2P to E1PCa2 conversion with subsequent extremely rapid ADP-induced E1PCa2 decay to E1Ca2. This means that the slope in Fig. 7B of the Ca2+ dependence probably reflects the rate constant for the lumenal Ca2+ access and binding to the lumenally oriented transport sites of E2P and resulting E2PCa2 formation.
FIGURE 7.

Lumenal Ca2+- and ADP-induced reverse E2P decay of Y122A in the presence of K+. A, microsomes (100 μg/ml) expressing the mutant Y122A were phosphorylated with 32Pi for 10 min at room temperature in the absence of Ca2+ in a medium containing 50 mm MOPS/Tris (pH 7.3), 7 mm MgCl2, 1 mm EGTA, 15 μm A23187, 0.1 mm 32Pi, and 20% Me2SO (that favors extremely the E2P formation (38)), and then the reaction mixture was chilled on ice. Subsequently, the phosphorylated sample was diluted at 0 °C with a 20-fold volume of a chase solution containing 50 mm MOPS/Tris (pH 7.3), 105 mm KCl, 7 mm MgCl2, 1 mm EGTA, 0.1 mm non-radioactive Pi, and 0.105 mm ADP without or with various concentrations of CaCl2 to give the final free Ca2+ concentrations as indicated. At the indicated periods, the chase reaction was terminated by trichloroacetic acid and the amount of EP was determined. Solid lines show the least squares fit to a single exponential decay. The decay rates thus obtained were plotted versus the Ca2+ concentration in panel B.
The Ca2+- and ADP-dependent acceleration of the reverse E2P decay was assayed also with all the other Y122-HC mutants (supplemental Fig. S3). The rates of the reverse E2P decay increased almost linearly with increasing Ca2+ concentrations even at 3 mm, except those of I232A and V705A5 over ∼1 mm Ca2+. Nevertheless, the slope of the Ca2+ dependence below 1 mm Ca2+ was estimated to be ∼0.2 s-1mm-1 in all the mutants as in Y122A. Therefore, the rate of the lumenal Ca2+ access and binding to the transport sites is similar in all the mutants of Y122-HC.
Kinetics of Lumenal Ca2+ Access to E2P of Wild Type and Y122A with and without K+—Then in Fig. 8, with the wild type and the representative mutant Y122A in the presence and absence of 0.1 m K+, we analyzed the Ca2+- and ADP-dependent acceleration of the reverse decay of E2P formed from Pi without Ca2+. As the well characterized property of the wild type, the forward hydrolysis of E2P without bound Ca2+ is very slow in the absence of K+, but markedly accelerated and thus very rapid in the presence of 0.1 m K+ (35, 36) (see the rates without Ca2+ in Fig. 8A). Nevertheless, even with the wild type in the presence of K+, we observed an apparently single exponential decay of E2P after the addition of Ca2+ and ADP at all the Ca2+ concentrations examined (time courses are not shown for simplicity). This is consistent with the kinetics described in the textbook by Fersht (39) that, in the parallel reactions in which a compound undergoes two or more single-step reactions simultaneously, its disappearance rate is described by a single exponential decay. In our case, the two reactions are the forward E2P hydrolysis and the Ca2+-/ADP-induced reverse E2P decay. The single decay rates thus obtained are plotted in Fig. 8A.
FIGURE 8.

Lumenal Ca2+- and ADP-induced reverse E2P decay of wild type and Y122A in the absence and presence of K+. A, microsomes (100 μg/ml) expressing wild type (WT) or Y122A were phosphorylated with 32Pi in the presence of A23187 and absence of Ca2+, and then chilled on ice, as described in Fig. 7. Subsequently, the phosphorylated sample was diluted at 0 °C with a 20-fold volume of a chase solution containing non-radioactive Pi, various concentrations of CaCl2, and ADP in the presence of 105 mm KCl (open symbols) or 105 mm LiCl in place of KCl (closed symbols), otherwise as described in Fig. 7. The time courses of EP decay were fitted to single exponential (data not shown, see Fig. 7A as an example). The rates thus obtained were plotted versus the Ca2+ concentrations. B, the fraction ADP-insensitive EP (FE2P) in the total amount of EP at steady state was simulated by using the rate of E2P decay (v2) and the rate of the E1PCa2 to E2P isomerization (loss of ADP sensitivity, v1) with an equation FE2P = v1/(v1 + v2). Here, v2 is the E2P decay rate obtained above in panel A at each Ca2+ concentrations. The v1 was obtained as described in Fig. 3 by the Ca2+ jump experiments from high (1 mm) to low (80 nm, virtually Ca2+ removal) for Y122A with 0.1 m K+ or Li+ (without K+) and for the wild type with 0.1 m Li+ (without K+). For the wild type with 0.1 m K+, the forward decay rate of E1PCa2 formed from ATP was used as the v1 value, because the E1PCa2 to E2P transition (the loss of ADP sensitivity) is rate-limiting for the E1PCa2 decay via E2P and its subsequent rapid hydrolysis. The v1 values actually used for the calculation were 0.049 s-1 (wild type with K+), 0.071 s-1 (wild type with Li+), 0.021 s-1 (Y122A with K+), and 0.034 s-1 (Y122A with Li+). The fraction of ADP-insensitive EP thus calculated was plotted versus the Ca2+ concentrations. The solid lines show the least squares fit to the Hill equation. In the inset, the ordinate is in a magnified scale for wild type with K+. In Table 2, the affinities and the Hill coefficients of the wild type and the Y122-HC mutants thus “estimated by kinetic analyses” in the absence and presence of K+ at pH 7.3 are summarized together with those actually “determined by the steady-state analyses” of the lumenal Ca2+-induced change of the fraction of ADP-insensitive EP otherwise under the same conditions in Figs. 2 and 6.
In the wild type in the presence of 0.1 m K+, the Ca2+ dependence of the EP decay rate was complicated because of the rapid E2P hydrolysis without Ca2+ (∼0.4 s-1), no change in the rate at 0–0.6 mm Ca2+, and the gradual increase above 0.6 mm. On the other hand, in the wild type in the absence of K+ in which the E2P hydrolysis without bound Ca2+ is markedly slowed, the nearly linear increase in the rate of Ca2+-/ADP-induced reverse E2P decay was observed at least up to ∼3 mm Ca2+ as in the Y122-HC mutants in the presence of K+. The slope of the wild type without K+ was actually close to that of Y122A with K+. Therefore, the rate of lumenal Ca2+ access and binding to the transport sites of E2P of the wild type in the absence of K+ is similar to that of Y122A. With the wild type in the presence of K+, evaluation of the lumenal Ca2+ access rate by this approach was not possible because of the complicated Ca2+-dependence curve. In Y122A, little effect was seen when K+ was omitted at 0–1 mm Ca2+, although the slope became gradually less steep at the higher Ca2+ concentration in the absence of K+.
In Fig. 8B, by using the rates of the E2P decay in the presence of added Ca2+ and ADP (determined in Fig. 8A) and the rates of the forward E1PCa2 to E2P transition (as determined in Fig. 3), we simulated the fraction of the steady-state level of the ADP-insensitive EP (E2P) in the total amount of EP at each Ca2+ concentration. Note that this simulation was made possible by the fact that nearly all the phosphorylation sites are phosphorylated at steady state (in either E1P or E2P form) under the conditions used for the steady-state and kinetic analyses at all the Ca2+ concentrations in this study. Namely, the E2 to E1Ca2 transition and the E1PCa2 formation from E1Ca2 with ATP are rapid enough to be ignored from the simulation. Therefore, the fraction of ADP-insensitive EP (E2P) in the steady state will be determined by the rate of its formation in the forward E1PCa2 to E2P transition, v1, and by the rate of its decay, v2 that includes both the forward hydrolysis of Ca2+-unbound E2P to E2 and the Ca2+-induced reverse transition to E1PCa2 with the subsequent ADP-induced decay. This means that the simulation can be made even with the wild type in the presence of K+ (as v2 can include the forward E2P hydrolysis). In the steady-state conditions, the decay rate (v2) and formation rate (v1) should be equal, therefore the fraction of ADP-insensitive EP (FE2P) in the total amount of EP will be estimated by an equation: FE2P = v1/(v1 + v2). Here, the E2P decay rate (v2) was obtained in Fig. 8A at each Ca2+ concentration. The E1PCa2 to E2P transition rate (v1) was estimated from the Ca2+ jump experiments from high (1 mm) to low (80 nm) for Y122A with and without 0.1 m K+ and the wild type without K+, as described in Fig. 3. With the wild type in the presence of 0.1 m K+, the forward decay rate of E1PCa2 formed by ATP was used as v1, because the E1PCa2 to E2P transition (the loss of ADP sensitivity) is rate-limiting for the E1PCa2 decay via E2P and its hydrolysis.
The Ca2+-dependent curves thus obtained by the simulation for the steady-state level of ADP-insensitive EP for Y122A with and without K+ and the wild type without K+ agreed very well with the respective ones determined at the steady state (cf. Figs. 2 (with K+) and 6 (without K+) at pH 7.3). The affinities for lumenal Ca2+ estimated from the simulated curves are in fact almost the same as those actually determined at steady state (Table 2). The agreements assure the validity of the simulation and further allow us to estimate the lumenal Ca2+ affinity of E2P of the wild type in the presence of K+. In the simulation for the wild type in the presence of K+ (open circles and inset in Fig. 8B), the fraction of ADP-insensitive EP was very low, and the extent of its change was extremely small as expected from the steady-state measurements (cf. supplemental Fig. S2 (pH 7.3)). The apparent affinity of wild type for lumenal Ca2+ in the presence of K+ was thus estimated by the small change to be 1.5 mm (see Table 2). This affinity was ∼3.5-times lower than that of wild type without K+ and 10-times lower than that of Y122A with and without K+. Thus, by omitting K+, the lumenal Ca2+ affinity of E2P in the wild type became higher and similar to that in Y122A.
TABLE 2.
Affinities of E2P for lumenal Ca2+ estimated by kinetic analyses and those determined at steady-state analyses As described in Fig. 8B, the lumenal Ca2+ affinities (K0.5) of E2P of the wild type and the mutant Y122A were estimated by the kinetic analyses of the lumenal Ca2+-induced E2P to E1PCa2 reverse transition and of the forward E1PCa2 to E2P transition. The K0.5 values and the Hill coefficients (nH) thus estimated kinetically in Fig. 8 in the absence and presence of 0.1 m K+ at pH 7.3 are summarized here. Listed together are those determined by the steady-state analyses of the lumenal Ca2+-induced change in the accumulated fraction of the ADP-insensitive EP (E2P) under otherwise the same conditions in Figs. 2 and 6 for the mutant Y122A with and without K+ and the wild type without K+.
|
K+ (0.1 m)
|
Estimated by kinetic analyses
|
Determined by steady-state analyses
|
|||
|---|---|---|---|---|---|
| K0.5 | nH | K0.5 | nH | ||
| mm | mm | ||||
| WT | + | 1.48 | 2.2 | –a | –a |
| – | 0.45 | 1.7 | 0.40 | 1.5 | |
| Y122A | + | 0.15 | 1.9 | 0.16 | 1.9 |
| – | 0.22 | 1.7 | 0.22 | 2.2 | |
Not determined because the accumulation of ADP-insensitive EP was very low at all the Ca2+ concentrations examined, and therefore possible change was not revealed
Kinetics of Lumenal Ca2+-induced E2P to E1PCa2 Reverse Transition of E2P of Wild Type in the Presence of 0.1 m K+ Was Revealed by the Absence of ADP—Unfortunately, in the above experimental design and approach of Fig. 8A, we were not able to estimate the lumenal Ca2+ access rate in E2P of the wild type in the presence of K+ because of the observed complexity of the Ca2+-dependent curve. In Fig. 9, we therefore employed a modified and thus different approach to examine the lumenal Ca2+-induced reverse conversion from E2P to E1PCa2. Namely, E2P was formed with Pi, and then a medium containing various concentrations of Ca2+ but without ADP (in contrast to its presence in Fig. 8) was added to E2P, and the subsequent EP decay was followed (Fig. 9A). In the absence of Ca2+, E2P was all hydrolyzed rapidly to E2 in a single exponential function. The E2P hydrolysis was inhibited gradually with increasing Ca2+ concentrations (over 0.1 mm), and the decay time course became biphasic as typically seen with 1 mm Ca2+. With increasing Ca2+ concentration, the fraction of the first and rapid phase decreased, that of the second phase increased, and the rate of the second phase became slower. The observation agrees with the previous kinetics analysis (9, 40). The first phase corresponds to the rapid and forward hydrolysis of the Ca2+-unbound E2P to E2. The EP species in the second phase was all ADP-sensitive (data not shown), therefore E1PCa2 formed from E2P by the lumenal Ca2+ binding. E1PCa2 decayed very slowly in the absence of ADP, because the E1PCa2 to E2P transition is much slower than the E2P hydrolysis, and this transition is retarded by the Ca2+ replacement of Mg2+ at the catalytic site of E1PCa2 at the approximately millimolar high Ca2+ concentrations (41, 42).
FIGURE 9.

Kinetics of lumenal Ca2+-induced change of E2P to E1PCa2 in the wild type in the presence of K+ without ADP. A, the microsomes expressing the wild type were phosphorylated with 32Pi in the presence of A23187 and absence of Ca2+ and chilled on ice, as described in Fig. 7. Subsequently, the phosphorylated sample was mixed at 0 °C with a 20-fold volume of chase solution containing 105 mm KCl and various concentrations of CaCl2 without ADP, otherwise as described in Fig. 7. The final free Ca2+ concentrations were indicated in the figure. At the indicated time periods after this addition, the chase reaction was terminated by trichloroacetic acid, and the amount of EP was determined. Solid lines show the least squares fit to a double exponential decay. The EP remaining in the second and slow phase was all in the ADP-sensitive form (thus E1PCa2, data not shown), and the first and rapid phase is the forward hydrolysis of E2P without bound Ca2+. B, the fraction of EP in the second phase in the total amount of EP was obtained by extrapolating to the zero time in the double exponential decay fitting, and plotted versus the Ca2+ concentration. The data were fitted well with the Hill equation (solid line), and the Ca2+ concentration giving the 50% saturation and the Hill coefficient were found to be 750 μm and 1.5. Here note that the EP amount in the second phase is dependent on the ratio between the rate of the forward E2P hydrolysis and that of the reverse E2P to E1PCa2 conversion upon the lumenal Ca2+ binding to E2P: the plot reflects the relative values between these forward and reverse rates of E2P rather than the lumenal Ca2+ affinity of E2P. C, by using the data obtained in A and B with the wild type in the presence of K+, the rate of the lumenal Ca2+-induced E1PCa2 formation from E2P (krev (○)) was calculated at each Ca2+ concentration by the equation, krev = khFs/(1 - Fs). Here, Fs is the fraction of EP in the second phase, and kh is the forward hydrolysis rate of E2P without Ca2+. For comparison with the wild type in the absence of K+ (•) and Y122A in the presence of K+ (▵), their lumenal Ca2+ access rates (the rates of the lumenal Ca2+-induced reverse E2P decay via E1PCa2 in the presence of ADP) obtained in Fig. 8A are plotted.
The fraction of the second and slow phase of the EP decay was obtained by extrapolating to the zero time and plotted versus the Ca2+ concentration (Fig. 9B). The plot showed saturation at 5–10 mm Ca2+. Here it is critical to note that, as previously discussed in detail (9), the fraction of EP of the second phase (the fraction remaining after the first phase) is dependent on the ratio between the rates of the forward E2P hydrolysis and of the reverse E2P to E1PCa2 conversion upon the lumenal Ca2+ binding to E2P. Namely, the plot in Fig. 9B reflects the relation between these forward and reverse rates of E2P rather than the lumenal Ca2+ affinity of E2P. For example, at the 50% saturation of the curve, the rate of the Ca2+-induced E1PCa2 formation from E2P is equal to that of the E2P hydrolysis to E2. Then in Fig. 9C for the wild type in the presence of K+, the rate of the E1PCa2 formation from E2P by the lumenal Ca2+ binding to E2P was calculated (krev, open circles) at each Ca2+ concentration by using the fraction of the slow and second phase (Fs) and the E2P hydrolysis rate (kh) with the equation, krev = khFs/(1 - Fs). The rate increased largely with increasing Ca2+ concentration.
In this kinetics, we eliminated the contribution of forward E2P hydrolysis on the overall E2P decay kinetics, and thereby revealed the rate of reverse E2P transition to E1PCa2 induced by the lumenal Ca2+ binding of the wild type in the presence of K+. For comparison in Fig. 9C, the rates of the lumenal Ca2+-induced reverse E2P decay estimated for the wild type without K+ and Y122A with K+ in Fig. 8A were replotted. Note again that, in these cases, the hydrolysis of Ca2+-unbound E2P was very slow and retarded; therefore, the observed Ca2+-/ADP-induced decay rates in their linear regions up to 3 mm Ca2+ reflect mostly the rates of the lumenal Ca2+ access and binding to E2P in the reverse E2P decay. Note also that the experimental design in Fig. 9A employed for the wild type with K+ was not applicable to the wild type without K+ and Y122A, because the E2P hydrolysis is very slow and almost completely retarded in these cases, and therefore the E2P decay upon the Ca2+ addition cannot be described as the biphasic decay. Conversely, the experimental design employed in Fig. 8A to estimate the rates of the lumenal Ca2+ access was not applicable to the wild type in the presence of K+ because of the complexity of the Ca2+-dependent curve as described above in Fig. 8A.
Thus in Fig. 9C, employing the inevitably different but most suitable experimental designs depending on the different kinetic properties, we were able to compare the rates of the E2P to E1PCa2 reverse transition induced by the lumenal Ca2+ binding to the transport sites of E2P at the limited Ca2+ concentration range up to 3 mm. In the wild type in the presence of K+, the rate was Ca2+-dependent and not saturated even at 3 mm, thus reflecting at least the Ca2+-dependent and rate-limiting process; i.e. the lumenal Ca2+-induced change from E2P to E2PCa2. This reverse transition rate in the wild type in the presence of K+ was significantly faster than those in the wild type in the absence of K+ and in Y122A (as well as in the other Y122-HC mutants (supplemental Fig. S3)) especially at the high Ca2+ concentration over 1 mm.
Here it is also interesting to note that the affinity of E2P for the lumenal Ca2+ in the wild type without K+ and the Y122-HC mutants is significantly higher than in the wild type with K+ (see Fig. 8B). If the rate of lumenal Ca2+ access and binding to E2P is solely slowed in the wild type without K+ and Y122-HC mutants, a decrease in the affinity is rather the consequence, which is in contrast to the observed increase. Therefore the rates of the Ca2+ release from E2PCa2 in the wild type without K+ and in the Y122-HC mutants are also presumably retarded significantly as compared with that in the wild type in the presence of K+. Namely, the mutations of Y122-HC and the lack of K+ binding affect the energy levels of Ca2+-free and -bound E2P states, as well as that of the transition state for lumenal gating (opening), and favor the Ca2+-bound state E2PCa2 and the closed lumenal gate.
DISCUSSION
Roles of Y122-HC in Ca2+ Release from E2PCa2 and in E2P Hydrolysis—In this study, we found that the mutations of any of the seven residues in Y122-HC increase the lumenal Ca2+ affinity and retard the lumenal Ca2+ access to the transport sites in E2P. These mutations also retard markedly the hydrolysis of the Ca2+-released form of E2P (22, 23). Thus, the proper formation of Y122-HC from the seven residues is critical for both Ca2+ release into lumen from E2PCa2 (reducing the Ca2+ affinity and opening the lumenal gate), and formation of the E2P catalytic site for the subsequent Asp351-acylphosphate hydrolysis. The formation of Y122-HC therefore functions critically for realizing and stabilizing the compactly organized and thus distorted structure of the Ca2+-released form of E2P. The stabilization of this state is certainly important for making the time period long enough for Ca2+ release into lumen and likely for proton bindings to the empty Ca2+ sites, and for the fine rearrangement of the catalytic site for the subsequent Asp351-acylphosphate hydrolysis.
As shown in Fig. 10 and supplemental Fig. S4, the extents of the mutational effects on the lumenal Ca2+ affinities and on the E2P hydrolysis rates varied significantly among the seven Y122-HC mutants and depended on their positions. The residues of which mutation exhibited the strongest effects on increasing lumenal Ca2+ affinity were Leu119, Tyr122, and Leu180. This agrees with the critical role of M2 for rearrangement of the transmembrane helices for the Ca2+ release; i.e. the tight association of the top part of M2 with the largely rotating A domain in Y122-HC functions for the lever-like inclination of M2 to push the lumenal part of M4 to open the lumenal gate (14). In fact, in E2PCa2 trapped by the elongation of the A/M1-linker, the Leu119/Tyr122 region on the top part of M2 is not involved fully in Y122-HC (9).
FIGURE 10.

Strength of the mutational effects of seven residues in
Tyr122-hydrophobic cluster on E2P hydrolysis and lumenal
Ca2+ affinity. The detailed structure at Y122-HC is shown with
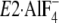 (the analog for
E2∼P, the transition state of the E2P hydrolysis
(21), PDB code: 1XP5
(15)). The seven residues
involved in Y122-HC (Tyr122/Leu119,
Ile179/Leu180, Ile232, and
Val705/Val726),
(the analog for
E2∼P, the transition state of the E2P hydrolysis
(21), PDB code: 1XP5
(15)). The seven residues
involved in Y122-HC (Tyr122/Leu119,
Ile179/Leu180, Ile232, and
Val705/Val726),
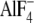 bound at the phosphorylation site
Asp351, and the bound potassium ion are shown by van der Waals
spheres. The seven residues in Y122-HC are colored
differently based on the strength of the retardation of the E2P
hydrolysis rate (lower panel) and that of the increase in the lumenal
Ca2+ affinity (upper panel). The color changes
gradually from red for the strongest effects to blue for
weakening.
bound at the phosphorylation site
Asp351, and the bound potassium ion are shown by van der Waals
spheres. The seven residues in Y122-HC are colored
differently based on the strength of the retardation of the E2P
hydrolysis rate (lower panel) and that of the increase in the lumenal
Ca2+ affinity (upper panel). The color changes
gradually from red for the strongest effects to blue for
weakening.
The residues of which mutations exhibited the strongest retardation of the
E2P hydrolysis were Ile232 at the top part of the
A/M3-linker and, again, Leu119 and Tyr122 on the
A/M2-linker (top part of M2). Thus these residues on the linkers seem to
contribute most critically to produce the proper configuration of the
catalytic site. Consistently, the proteolytic cleavage at Leu119 on
the A/M2-linker causes a marked inhibition of the E2P hydrolysis
(43). The structural changes
producing the Ca2+ release may be transmitted to the catalytic site
via these residues of Y122-HC on the linkers, thereby ensuring the
E2P hydrolysis to occur after the Ca2+ release. In any
case, the different degree of the contributions of the seven residues of
Y122-HC to the Ca2+ release and subsequent formation of the
E2P catalytic site may suggest a possible sequential gathering of the
seven residues. This possibility will be discussed more in the last section of
“Discussion” in relation to the crystal structure
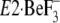 (17,
18).
(17,
18).
Structural Mechanism Involving Y122-HC and Other Critical
Elements—In
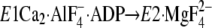 as an overall structural change, including the EP isomerization and
Ca2+ release (supplemental Fig. S5A), the A domain largely
rotates and M2 largely inclines. Also the P domain markedly inclines toward
the lower side of the A domain and rotates by ∼20° around the
phosphorylation site (Asp351) parallel to the membrane and in the
opposite direction of the A-domain rotation. These motions involve (can be
dissected into) the horizontal and vertical factors, parallel and
perpendicular to the membrane plane. As a consequence of the motions, the A
and P domains and M2 will come to their appropriate positions producing their
tight association at Y122-HC. At the A-P domain interface in the E2P
analog structures, there is another interaction network between these domains
at the Val200 loop, Asp196–Asp203 of
the A domain (Fig. 1, and see
supplemental Fig. S3 in Ref.
23 for the details of the
interactions and central role of Val200). Our previous mutations of
Val200 showed (24)
that this A-P domain interaction is critical for Ca2+ release from
E2PCa2 and for formation of the E2P catalytic
site, thus very similarly to Y122-HC. Then note that, in the E2P
analog structures (see supplemental Fig. S5A for
as an overall structural change, including the EP isomerization and
Ca2+ release (supplemental Fig. S5A), the A domain largely
rotates and M2 largely inclines. Also the P domain markedly inclines toward
the lower side of the A domain and rotates by ∼20° around the
phosphorylation site (Asp351) parallel to the membrane and in the
opposite direction of the A-domain rotation. These motions involve (can be
dissected into) the horizontal and vertical factors, parallel and
perpendicular to the membrane plane. As a consequence of the motions, the A
and P domains and M2 will come to their appropriate positions producing their
tight association at Y122-HC. At the A-P domain interface in the E2P
analog structures, there is another interaction network between these domains
at the Val200 loop, Asp196–Asp203 of
the A domain (Fig. 1, and see
supplemental Fig. S3 in Ref.
23 for the details of the
interactions and central role of Val200). Our previous mutations of
Val200 showed (24)
that this A-P domain interaction is critical for Ca2+ release from
E2PCa2 and for formation of the E2P catalytic
site, thus very similarly to Y122-HC. Then note that, in the E2P
analog structures (see supplemental Fig. S5A for
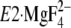 ), the two networks at
Y122-HC and at Val200 are located at each side of the A-P domain
interface on its top view and at the bottom and upper parts of the
interface, respectively on its side view. Thus the two are situated
horizontally and vertically with the specific relative positioning. It is very
likely that this positioning of the two is most efficiently functioning to
realize and stabilize the compactly organized and distorted structure of the
Ca2+-released E2P: i.e. the interactions at the
two positions are most appropriate to produce the horizontal and vertical
motions of the P and A domains and M2 required for Ca2+ release
from E2PCa2 and to stabilize the Ca2+-released
E2P state. Certainly these motions cause the rearrangements in the
transmembrane helices for Ca2+ release: e.g. the P-domain
inclination with slight rotation is directly associated with the bending and
slight rotation of connected M4/M5 and downward movement of M4, thus their
twisting-like motion. The largely inclining M2 pushes the lumenal part of M4
(supplemental Fig. S5B). Hence the Ca2+ sites are
destroyed, and the lumenal gate is opened.
), the two networks at
Y122-HC and at Val200 are located at each side of the A-P domain
interface on its top view and at the bottom and upper parts of the
interface, respectively on its side view. Thus the two are situated
horizontally and vertically with the specific relative positioning. It is very
likely that this positioning of the two is most efficiently functioning to
realize and stabilize the compactly organized and distorted structure of the
Ca2+-released E2P: i.e. the interactions at the
two positions are most appropriate to produce the horizontal and vertical
motions of the P and A domains and M2 required for Ca2+ release
from E2PCa2 and to stabilize the Ca2+-released
E2P state. Certainly these motions cause the rearrangements in the
transmembrane helices for Ca2+ release: e.g. the P-domain
inclination with slight rotation is directly associated with the bending and
slight rotation of connected M4/M5 and downward movement of M4, thus their
twisting-like motion. The largely inclining M2 pushes the lumenal part of M4
(supplemental Fig. S5B). Hence the Ca2+ sites are
destroyed, and the lumenal gate is opened.
It should be noted that, for the loss of the ADP sensitivity E1PCa2 → E2PCa2, the large rotation of the A domain and its docking onto the P domain should occur so as to bring the T181GES loop above Asp351-acylphosphate to block the ADP access from the N domain. As the motive force of this large A-domain rotation approximately parallel to membrane plane, the strain imposed on the A/M3-linker in E1PCa2 was predicted to be critical (13, 14, 20, 44). Also, the sufficiently long length of the A/M1-linker was revealed to be critical for this EP isomerization, in this case, probably for realizing the E2PCa2 structure, in which the A domain is positioned above the P domain (9, 45). For the subsequent Ca2+ release in E2PCa2 → E2P + 2Ca2+, the A/M1-linker with its appropriately short length (therefore its strain) is critical (9). Actually, the elongation of this linker blocks completely Ca2+ deocclusion/release from E2PCa2, thus trapping this E2PCa2 state in which Y122-HC is not properly formed yet in contrast to its proper formation in the Ca2+-released form of E2P with the lumenally opened normal Ca2+ release pathway (9). The results clearly demonstrated that the native and appropriately short length of A/M1-linker functions critically in inducing the motions from the E2PCa2 state, especially inclination of the A and P domains and M2, to accomplish the Y122-HC formation and the Ca2+ deocclusion/release from E2PCa2. During the Y122-HC formation, the interaction force being produced in Y122-HC will likely function to induce the final process of the vertical and horizontal motions of the P and A domain and M2 to realize and stabilize the Ca2+-released E2P structure (supplemental Fig. S6). Importantly also, the E2P catalytic site is produced by these rearrangements. In this mechanism, a possible hydrolysis of Asp351-acylphosphate without releasing Ca2+ will be avoided; thereby the ordered reaction sequence of the Ca2+ release from E2PCa2 and the subsequent E2P hydrolysis will be accomplished for the energy coupling.
Possible Structural Role of K+ for Reducing
Ca2+ Affinity and Lumenal
Gating—K+ is known to markedly accelerate the
E2P hydrolysis (35,
36) and also to modulate the
E2 to E1Ca2 transition in the non-phosphorylated
Ca2+-ATPase (46,
47). In the present study, we
further found that the K+ binding is important for reducing the
affinity for Ca2+ and lumenal gating thus for Ca2+
release from E2PCa2. In the crystal structure
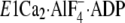 ,
K+ is situated at the bottom part of the P domain and coordinated
by the backbone carbonyls of the loop
Leu711–Glu715 and by the Glu732 side
chain (Fig. 11). The
K+ binding at this site was indeed previously found by the
mutations to be critical for the stimulation of the E2P hydrolysis
(37). In the structures
E2P analogs and E2(TG), this K+ site of the P
domain comes very close to the A/M3-linker, and actually K+ at this
site is further coordinated by the Gln244 side chain on the
A/M3-linker (see
,
K+ is situated at the bottom part of the P domain and coordinated
by the backbone carbonyls of the loop
Leu711–Glu715 and by the Glu732 side
chain (Fig. 11). The
K+ binding at this site was indeed previously found by the
mutations to be critical for the stimulation of the E2P hydrolysis
(37). In the structures
E2P analogs and E2(TG), this K+ site of the P
domain comes very close to the A/M3-linker, and actually K+ at this
site is further coordinated by the Gln244 side chain on the
A/M3-linker (see 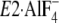 in
Fig. 11). Because the alanine
substitution of Gln244 and those of Glu-Gln-Asp245 gave
virtually no effect on Ca2+ transport activity
(48), K+ at this
region may be coordinated by their neighboring residues or backbone carbonyls
on the A/M3-linker and thereby perform a structural function. In the present
study, we found that the lack of K+ binding has the consequences
very similar to those of the mutations at Y122-HC. It is therefore possible
that the K+ binding functions with similar structural effects as
Y122-HC to produce the proper structure of the Ca2+-released form
of E2P.
in
Fig. 11). Because the alanine
substitution of Gln244 and those of Glu-Gln-Asp245 gave
virtually no effect on Ca2+ transport activity
(48), K+ at this
region may be coordinated by their neighboring residues or backbone carbonyls
on the A/M3-linker and thereby perform a structural function. In the present
study, we found that the lack of K+ binding has the consequences
very similar to those of the mutations at Y122-HC. It is therefore possible
that the K+ binding functions with similar structural effects as
Y122-HC to produce the proper structure of the Ca2+-released form
of E2P.
Then note that the K+ site of the P domain in
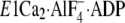 is situated at much higher position from the membrane plane than the
Gln244 region on the A/M3-linker
(Fig. 11) and that, in the
change
is situated at much higher position from the membrane plane than the
Gln244 region on the A/M3-linker
(Fig. 11) and that, in the
change
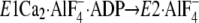 (or
(or 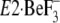 and
and
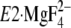 ), the P domain with
connected M4/M5 largely inclines toward the A domain, hence the K+
site with bound K+ on the P domain moves down to the
Gln244 region on the A/M3-linker to make contact. The interactions
between the bottom part of the P domain and the A/M3-linker via bound
K+ thus produced would likely cross-link them and hence contribute
to formation and stabilization of this compactly organized
Ca2+-released structure of E2P with the reduced
Ca2+ affinity and lumenally opened gate. Alternatively, it is also
possible that the appropriate P-domain structure produced by K+
binding on this domain solely contributes to the formation of the
Ca2+-released E2P structure.
), the P domain with
connected M4/M5 largely inclines toward the A domain, hence the K+
site with bound K+ on the P domain moves down to the
Gln244 region on the A/M3-linker to make contact. The interactions
between the bottom part of the P domain and the A/M3-linker via bound
K+ thus produced would likely cross-link them and hence contribute
to formation and stabilization of this compactly organized
Ca2+-released structure of E2P with the reduced
Ca2+ affinity and lumenally opened gate. Alternatively, it is also
possible that the appropriate P-domain structure produced by K+
binding on this domain solely contributes to the formation of the
Ca2+-released E2P structure.
Y122-HC in Crystal Structure of
 —The crystal structures of
—The crystal structures of
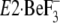 , the analog for the
E2P ground state
(21), were solved at the
atomic level very recently with and without bound thapsigargin, TG
(
, the analog for the
E2P ground state
(21), were solved at the
atomic level very recently with and without bound thapsigargin, TG
(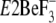 and
and
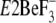 (17,
18)). Surprisingly, in this
crystallized
(17,
18)). Surprisingly, in this
crystallized 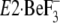 , the side
chains of Ile119 and Tyr122 are somewhat pointing away
from the clustered other five residues on the A and P domains
(Ile179/Leu180/Ile232 and
Val705/Val726), although all these seven residues are
closely located in the
, the side
chains of Ile119 and Tyr122 are somewhat pointing away
from the clustered other five residues on the A and P domains
(Ile179/Leu180/Ile232 and
Val705/Val726), although all these seven residues are
closely located in the 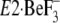 structures of both 2ZBE (17)
and 3B9B (18). On the other
hand, Y122-HC is formed fully from all these seven residues in
structures of both 2ZBE (17)
and 3B9B (18). On the other
hand, Y122-HC is formed fully from all these seven residues in
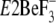 as well as in the other
E2P analogous structures,
as well as in the other
E2P analogous structures,
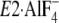 and
and
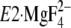 . Thus, the assembling
manner of the seven residues in the crystal structure
. Thus, the assembling
manner of the seven residues in the crystal structure
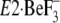 seemingly conflicts with
our results that the gathering of all the seven residues, including
Tyr122/Ile119 in Y122-HC, is required for producing the
Ca2+-released E2P. Furthermore, Tyr122 and
Ile119 on the top part of M2 (A/M2-linker) are likely most critical
in Y122-HC and play central roles (Fig.
10). Our previous biochemical structural analysis of SR
Ca2+-ATPase in solution by the proteolysis and the lumenal
Ca2+ accessibility demonstrated
(21) that, in
seemingly conflicts with
our results that the gathering of all the seven residues, including
Tyr122/Ile119 in Y122-HC, is required for producing the
Ca2+-released E2P. Furthermore, Tyr122 and
Ile119 on the top part of M2 (A/M2-linker) are likely most critical
in Y122-HC and play central roles (Fig.
10). Our previous biochemical structural analysis of SR
Ca2+-ATPase in solution by the proteolysis and the lumenal
Ca2+ accessibility demonstrated
(21) that, in
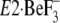 without TG,
Leu119/Tyr122 are surely gathered and involved in
Y122-HC, and thereby the lumenal gate is opened and the lumenal
Ca2+ is accessible to the transport sites. Thus the crystal
structure
without TG,
Leu119/Tyr122 are surely gathered and involved in
Y122-HC, and thereby the lumenal gate is opened and the lumenal
Ca2+ is accessible to the transport sites. Thus the crystal
structure 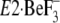 seems to
conflict also with these biochemical results obtained in solution.
seems to
conflict also with these biochemical results obtained in solution.
Nevertheless, as a comprehensive idea, the crystal structure of
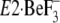 may be consistent with
(or indicative of) the view that the gathering of the seven residues to form
Y122-HC upon motions of the A and P domains and M2 (A/M2-linker) occurs in
some ordered sequence but not necessarily at once (see
Fig. 10 and under
“Discussion”). Most peculiar to us is that Tyr122 and
Leu119, of which mutations exhibited the most inhibitory effects,
are not involved yet in the hydrophobic cluster in the crystal structure
may be consistent with
(or indicative of) the view that the gathering of the seven residues to form
Y122-HC upon motions of the A and P domains and M2 (A/M2-linker) occurs in
some ordered sequence but not necessarily at once (see
Fig. 10 and under
“Discussion”). Most peculiar to us is that Tyr122 and
Leu119, of which mutations exhibited the most inhibitory effects,
are not involved yet in the hydrophobic cluster in the crystal structure
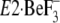 . Here note that, in the
structure
. Here note that, in the
structure  , a
Mg2+ ion is bound near the Ca2+ binding sites in the
transmembrane domain because an extremely high Mg2+ concentration
employed for crystallization
(18), or protonation on the
residues of transmembrane helices, including Ca2+ ligands, must
have occurred as in low pH for crystallization
(17). Thus, these ligations
are probably involved critically in the stabilization of the transmembrane
helices for the crystallization. This might mean that the transmembrane
structure thus stabilized differs from that without any ligations,
i.e. the state immediate after the Ca2+ release (the empty
Ca2+ sites) that is realized by the contribution of Y122-HC.
Therefore, Y122-HC is, in return, disrupted or not properly produced yet in
the crystal structure
, a
Mg2+ ion is bound near the Ca2+ binding sites in the
transmembrane domain because an extremely high Mg2+ concentration
employed for crystallization
(18), or protonation on the
residues of transmembrane helices, including Ca2+ ligands, must
have occurred as in low pH for crystallization
(17). Thus, these ligations
are probably involved critically in the stabilization of the transmembrane
helices for the crystallization. This might mean that the transmembrane
structure thus stabilized differs from that without any ligations,
i.e. the state immediate after the Ca2+ release (the empty
Ca2+ sites) that is realized by the contribution of Y122-HC.
Therefore, Y122-HC is, in return, disrupted or not properly produced yet in
the crystal structure 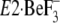 as
if it occurs with the lumenal Ca2+ binding in the
E2PCa2 state as postulated in this study. Therefore the
following sequential gathering of the seven residues to produce Y122-HC can be
speculated: The five hydrophobic residues on the A domain
(Ile179/Leu180/Ile232) and P domain
(Val705/Val726) are first gathered through the motions
of the A and P domains and top part of M2 (A/M2-linker), and subsequently, the
top part of M2, including Ile119 and Tyr122, makes
further motions during the final process of the M2 inclination to join them
and produce the fully assembled Y122-HC, thereby to realize and stabilize
fully the gathered state of the A and P domains and top part of M2
(A/M2-linker) as in the Ca2+-released form of E2P. Being
in agreement with this view, in E2PCa2 trapped by the
elongation of the A/M1-linker, Leu119/Tyr122 on the top
part of M2 is not fully involved yet in Y122-HC
(9). The
Ca2+-released and empty Ca2+ sites (without any
protonation and stabilization immediately after the Ca2+ release)
will be subsequently protonated producing the E2P ground state for
its hydrolysis.
as
if it occurs with the lumenal Ca2+ binding in the
E2PCa2 state as postulated in this study. Therefore the
following sequential gathering of the seven residues to produce Y122-HC can be
speculated: The five hydrophobic residues on the A domain
(Ile179/Leu180/Ile232) and P domain
(Val705/Val726) are first gathered through the motions
of the A and P domains and top part of M2 (A/M2-linker), and subsequently, the
top part of M2, including Ile119 and Tyr122, makes
further motions during the final process of the M2 inclination to join them
and produce the fully assembled Y122-HC, thereby to realize and stabilize
fully the gathered state of the A and P domains and top part of M2
(A/M2-linker) as in the Ca2+-released form of E2P. Being
in agreement with this view, in E2PCa2 trapped by the
elongation of the A/M1-linker, Leu119/Tyr122 on the top
part of M2 is not fully involved yet in Y122-HC
(9). The
Ca2+-released and empty Ca2+ sites (without any
protonation and stabilization immediately after the Ca2+ release)
will be subsequently protonated producing the E2P ground state for
its hydrolysis.
Alternatively, if a possible contribution of such ligation in the
transmembrane domain (Mg2+ or protonation) should not be concerned
in the crystallization of
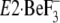 , the followings might be
possible: first of all, the arrangements of helices of the
Ca2+-released empty transport sites must be unstable, for example,
due to possible repulsions between the negative charges of the Ca2+
ligands. Then to relieve the instability, the most effective gathering of
Tyr122/Leu119 in Y122-HC, which produces and stabilizes
the Ca2+-released empty state, might possibly be disrupted; thereby
the helices may be rearranged so as to form the more stabilized arrangements
that can be crystallized.
, the followings might be
possible: first of all, the arrangements of helices of the
Ca2+-released empty transport sites must be unstable, for example,
due to possible repulsions between the negative charges of the Ca2+
ligands. Then to relieve the instability, the most effective gathering of
Tyr122/Leu119 in Y122-HC, which produces and stabilizes
the Ca2+-released empty state, might possibly be disrupted; thereby
the helices may be rearranged so as to form the more stabilized arrangements
that can be crystallized.
Supplementary Material
Acknowledgments
We thank Dr. David H. MacLennan, University of Toronto, for his generous gift of SERCA1a cDNA and Dr. Randal J. Kaufman, Genetics Institute, Cambridge, MA, for his generous gift of the expression vector pMT2. We are also grateful to Dr. Chikashi Toyoshima, University of Tokyo, for helpful discussions.
This work was supported by a grant-in-aid for scientific research (C) (to K. Y.) and (B) (to H. S.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
Footnotes
The abbreviations used are: SERCA1a, adult fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase; EP, phosphoenzyme; E1P, ADP-sensitive phosphoenzyme; E2P, ADP-insensitive phosphoenzyme; MOPS, 3-(N-morpholino)propanesulfonic acid; TG, thapsigargin; Y122-HC, Tyr122-hydrophobic cluster.
The maximal level of the ADP-insensitive EP of L180A at low Ca2+ concentrations (44% of total amount of EP) was significantly lower than those of the other mutants. It may be due to the significantly faster E2P hydrolysis rate in L180A as compared with the rates in the others, as shown in supplemental Fig. S4.
Its slope was approximately 0.2 s-1mm-1 at 0–2 mm Ca2+. In Fig. 3, the rate of the forward E1PCa2 to E2P conversion was estimated to be 0.02 s-1. Therefore the calculation with these values, 0.02 s-1 divided by 0.2 s-1 mm-1, gave the apparent affinity for lumenal Ca2+ in E2P of Y122A as 100 μm. This value agreed very well with that (160 μm) obtained in Fig. 2 under the same conditions (pH 7.3) at the steady state.
As a possible reason for the less steep Ca2+-dependent curve observed with I232A and V705A over ∼1 mm, it might be possible that the rate in the transition from E2PCa2 to E1PCa2 is slower in these mutants than in the other mutants, and this step became rate-limiting at the high Ca2+ concentrations where the lumenal Ca2+-induced change to E2PCa2 became fast.
References
- 1.Hasselbach, W., and Makinose, M. (1961) Biochem. Z. 333 518-528 [PubMed] [Google Scholar]
- 2.Ebashi, S., and Lipmann, F. (1962) J. Cell Biol. 14 389-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inesi, G., Sumbilla, C., and Kirtley, M. E. (1990) Physiol. Rev. 70 749-776 [DOI] [PubMed] [Google Scholar]
- 4.Møller, J. V., Juul, B., and le Maire, M. (1996) Biochim. Biophys. Acta 1286 1-51 [DOI] [PubMed] [Google Scholar]
- 5.MacLennan, D. H., Rice, W. J., and Green, N. M. (1997) J. Biol. Chem. 272 28815-28818 [DOI] [PubMed] [Google Scholar]
- 6.McIntosh, D. B. (1998) Adv. Mol. Cell. Biol. 23A 33-99 [Google Scholar]
- 7.Toyoshima, C., and Inesi, G. (2004) Annu. Rev. Biochem. 73 268-292 [DOI] [PubMed] [Google Scholar]
- 8.Toyoshima, C. (2008) Arch. Biochem. Biophys. 476 3-11 [DOI] [PubMed] [Google Scholar]
- 9.Daiho, T., Yamasaki, K., Danko, S., and Suzuki, H. (2007) J. Biol. Chem. 282 34429-34447 [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000) Nature 405 647-655 [DOI] [PubMed] [Google Scholar]
- 11.Toyoshima, C., and Nomura, H. (2002) Nature 418 605-611 [DOI] [PubMed] [Google Scholar]
- 12.Sørensen, T. L.-M., Møller, J. V., and Nissen, P. (2004) Science 304 1672-1675 [DOI] [PubMed] [Google Scholar]
- 13.Toyoshima, C., and Mizutani, T. (2004) Nature 430 529-535 [DOI] [PubMed] [Google Scholar]
- 14.Toyoshima, C., Nomura, H., and Tsuda, T. (2004) Nature 432 361-368 [DOI] [PubMed] [Google Scholar]
- 15.Olesen, C., Sørensen, T. L.-M., Nielsen, R. C., Møller, J. V., and Nissen, P. (2004) Science 306 2251-2255 [DOI] [PubMed] [Google Scholar]
- 16.Takahashi, M., Kondou, Y., and Toyoshima, C. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 5800-5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toyoshima, C., Norimatsu, Y., Iwasawa, S., Tsuda, T., and Ogawa, H. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 19831-19836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olesen, C., Picard, M., Winther, A.-M. L., Gyrup, C., Morth, J. P., Oxvig, C., Møller, J. V., and Nissen, P. (2007) Nature 450 1036-1042 [DOI] [PubMed] [Google Scholar]
- 19.Danko, S., Daiho, T., Yamasaki, K., Kamidochi, M., Suzuki, H., and Toyoshima, C. (2001) FEBS Lett. 489 277-282 [DOI] [PubMed] [Google Scholar]
- 20.Danko, S., Yamasaki, K., Daiho, T., Suzuki, H., and Toyoshima, C. (2001) FEBS Lett. 505 129-135 [DOI] [PubMed] [Google Scholar]
- 21.Danko, S., Yamasaki, K., Daiho, T., and Suzuki, H. (2004) J. Biol. Chem. 279 14991-14998 [DOI] [PubMed] [Google Scholar]
- 22.Yamasaki, K., Daiho, T., Danko, S., and Suzuki, H. (2004) J. Biol. Chem. 279 2202-2210 [DOI] [PubMed] [Google Scholar]
- 23.Wang, G., Yamasaki, K., Daiho, T., and Suzuki, H. (2005) J. Biol. Chem. 280 26508-26516 [DOI] [PubMed] [Google Scholar]
- 24.Kato, S., Kamidochi, M., Daiho, T., Yamasaki, K., Wang, G., and Suzuki, H. (2003) J. Biol. Chem. 278 9624-9629 [DOI] [PubMed] [Google Scholar]
- 25.Kaufman, R. J., Davies, M. V., Pathak, V. K., and Hershey, J. W. B. (1989) Mol. Cell. Biol. 9 946-958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maruyama, K., and MacLennan, D. H. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 3314-3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanazawa, T., Saito, M., and Tonomura, Y. (1970) J. Biochem. (Tokyo) 67 693-711 [DOI] [PubMed] [Google Scholar]
- 28.Weber, K., and Osborn, M. (1969) J. Biol. Chem. 244 4406-4412 [PubMed] [Google Scholar]
- 29.Daiho, T., Suzuki, H., Yamasaki, K., Saino, T., and Kanazawa, T. (1999) FEBS Lett. 444 54-58 [DOI] [PubMed] [Google Scholar]
- 30.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951) J. Biol. Chem. 193 265-275 [PubMed] [Google Scholar]
- 31.Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graphics 14 33-38 [DOI] [PubMed] [Google Scholar]
- 32.Coan, C., Verjovski-Almeida, S., and Inesi, G. (1979) J. Biol. Chem. 254 2968-2974 [PubMed] [Google Scholar]
- 33.Prager, R., Punzengruber, C., Kolassa, N., Winkler, F., and Suko, J. (1979) Eur. J. Biochem. 97 239-250 [DOI] [PubMed] [Google Scholar]
- 34.de Meis, L., and Inesi, G. (1982) J. Biol. Chem. 257 1289-1294 [PubMed] [Google Scholar]
- 35.Shigekawa, M., and Pearl, L. J. (1976) J. Biol. Chem. 251 6947-6952 [PubMed] [Google Scholar]
- 36.Shigekawa, M., and Dougherty, J. P. (1978) J. Biol. Chem. 253 1451-1457 [PubMed] [Google Scholar]
- 37.Sørensen, T. L.-M., Clausen, J. D., Jensen, A.-M. L., Vilsen, V., Møller, J. V., Andersen, J. P., and Nissen, P. (2004) J. Biol. Chem. 279 46355-46358 [DOI] [PubMed] [Google Scholar]
- 38.de Meis, L., Martins, O. B., and Alves, E. W. (1980) Biochemistry 19 4252-4261 [DOI] [PubMed] [Google Scholar]
- 39.Fersht, A. R. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, pp. 132-168, W. H. Freeman and Co., New York
- 40.Sato, K., Yamasaki, K., Daiho, T., Miyauchi, Y., Takahashi, H., Ishida-Yamamoto, A., Nakamura, S., Iizuka, H., and Suzuki, H. (2004) J. Biol. Chem. 279 35595-35603 [DOI] [PubMed] [Google Scholar]
- 41.Shigekawa, M., Wakabayashi, S., and Nakamura, H. (1983) J. Biol. Chem. 258 8698-8707 [PubMed] [Google Scholar]
- 42.Wakabayashi, S., and Shigekawa, M. (1987) J. Biol. Chem. 262 11524-11531 [PubMed] [Google Scholar]
- 43.Lenoir, G., Picard, M., Gauron, C., Montigny, C., Le Maréchal, P., Falson, P., le Maire, M., Møller, J. V., and Champeil, P. (2004) J. Biol. Chem. 279 9156-9166 [DOI] [PubMed] [Google Scholar]
- 44.Møller, J. V., Lenoir, G., Marchand, C., Montigny, C., le Maire, M., Toyoshima, C., Juul, B. S., and Champeil, P. (2002) J. Biol. Chem. 277 38647-38659 [DOI] [PubMed] [Google Scholar]
- 45.Daiho, T., Yamasaki, K., Wang, G., Danko, S., and Suzuki, H. (2003) J. Biol. Chem. 278 39197-39204 [DOI] [PubMed] [Google Scholar]
- 46.Lee, A. G., Baker, K., Khan, Y. M., and East, J. M. (1995) Biochem. J. 305 225-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Champeil, P., Henao, F., and de Foresta, B. (1997) Biochemistry 36 12383-12393 [DOI] [PubMed] [Google Scholar]
- 48.Clarke, D. M., Maruyama, K., Loo, T. W., Leberer, E., Inesi, G., and MacLennan, D. H. (1989) J. Biol. Chem. 264 11246-11251 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.