

Abstract
Proteins destined to circulate in the blood are first folded and assembled in the endoplasmic reticulum of secretory cells. For antibodies, like many other serum proteins, the folding and assembly steps involve the formation of disulfide bonds. Such bonds have been thought to be static features of proteins, stabilizing domains, and linking polypeptide chains, although some cases of extracellular disulfide bond cleavage have been noted. Recently, the human IgG2 antibody subclass was found to possess multiple structures differing in specific disulfide linkages. These structures are naturally occurring and can, in some cases, affect the activity of the antibody. Here we show that these IgG2 disulfide linkages interconvert while circulating in humans. Secretory cells initially produce primarily one form (IgG2-A), which is rapidly converted to a second form (IgG2-A/B) while circulating in the blood, followed by a slower conversion to a third form (IgG2-B). This work demonstrates that the disulfide structure of the IgG2 antibody is dynamic in vivo, on a time scale similar to that of the protein's lifetime. Thus, changes to the IgG2 disulfide structure provide a marker of the protein's age and may alter its activity over its lifetime.
Disulfide bonds form in the endoplasmic reticulum (ER)2 as part of the folding and assembly of the newly synthesized secretory protein. Systems within the ER are designed to catalyze the formation of disulfide bonds (EroIp/protein disulfide isomerase) (1, 2) and to repair incorrectly formed disulfide bonds through catalytic reshuffling (protein disulfide isomerase) (7, 8). Because the enzymes involved in disulfide quality control are solely residents of the ER, it has generally been assumed that the disulfides formed in the ER are permanent features of secreted proteins.
The main function of disulfide bonds is considered to be stabilizing globular proteins, but other functions have been proposed (3, 4). The stabilizing function may be achieved through lowering the unfolded state entropy of disulfide linked regions. Alternatively, the links may serve to reduce domain flexibility and restrict mobility to specific regions of the molecule. Antibodies contain multiple interchain and intrachain disulfide bonds, which could play roles in either stabilizing or limiting flexibility.
The IgG antibody class possesses a single intrachain disulfide bond within each antiparallel β barrel domain (9), 12 in all, and an interchain disulfide bond between the light chain (LC) and the heavy chain (HC) (5, 10). In addition, each IgG subclass contains a specific number of HC to HC disulfide links in the hinge region: two for IgG1 and IgG4, four for IgG2, and 11 for IgG3. By stabilizing each domain, the intrachain disulfide bonds may restrict flexibility to the interdomain regions. Interchain disulfide bonds could also limit mobility. However, interheavy chain disulfide bonds in IgG4 antibodies seem to not prevent the chain dissociation, as demonstrated by the observation that half-molecules can shuffle between different antibodies in vivo (11).
Recently published studies describe disulfide bonds within the IgG2 antibody class that can form more than one pair combination in vivo (5, 6). These alternate linkages have been classified by whether the interchain links in the Fab arms are restricted to the arm or linked to the hinge region (see Fig. 1). In the IgG2-A form, the cysteine near or at the C terminus of the LC is linked to the Fab arm of the HC. This is the structure previously described in the literature. Two new structures were shown that involve Fab arm linkages to the hinge regions. Both Fab arms have links to the hinge region in the IgG2-B structure, and only one is linked to the hinge in the IgG2-A/B structure. Relatively mild redox conditions can change the distribution of A, B, and A/B isoforms in vitro, and the changes alter the antibody binding and activity for some of the monoclonal antibodies tested. Conditions that promote IgG2 disulfide exchange for these specific linkages are similar to those found in serum, suggesting that disulfide shuffling could occur on endogenous IgG2s in vivo. Such disulfide conversion would differ from the traditional view of disulfide bonds linking the protein into its final static structure.
FIGURE 1.

Simplified models of IgG2 disulfide isoforms. IgG2 antibodies represented here show the polypeptide chains and the interchain disulfide bonds. Intrachain disulfide bonds have been removed for simplicity. On the IgG2-A structure (top), each LC is connected to the HC Fab arm. On the IgG2-A/B structure (middle), one LC is connected to the Fab arm, whereas the other LC is connected to the hinge region. In the IgG2-B structure (bottom), both LC arms are connected to the hinge region. For each LC-hinge connection, there is a corresponding hinge-HC Fab arm linkage. White segments, LC; gray segments, HC. Note that the some of the specific disulfide connectivities have not been determined. These linkages are shown to illustrate the class differences.
EXPERIMENTAL PROCEDURES
Materials—HiTrap A columns (1 ml) were purchased from GE Healthcare. Actigel ALD Superflow was from Sterogene Bioseparations. Monoclonal antibody (mAb)-ligand resin was prepared using Actigel ALD according to the manufacturer's protocol. Monobromobimane (Thiolyte) was purchased from Calbiochem. The Jupiter C18 (2 × 250 mm, 300 Å, 5 μm) reversed-phase column was from Phenomenex. HT protein express kits, including chips and reagents, were purchased from Caliper Life Sciences (Mountain View, CA). Iodoacetamide was from Sigma. All organic solvents were of analytical or HPLC grade.
Cell Culture and Purification—The recombinant human IgG2 mAb used in this study was expressed in suspended Chinese hamster ovary cell batch cultures and purified using well established chromatography procedures at Amgen (5). To obtain intracellular mAb, 50 ml of actively growing cell culture was chilled on ice and then sedimented (∼1200 × g for 15 min at 4 °C). To wash the cells, the resulting supernatant was removed, and the cell pellet was resuspended in 5 ml of ice-cold phosphate-buffered saline (PBS). The cells were sedimented as before. The washed cell pellet was resuspended into 3 ml of ice-cold 50 mm Tris, 150 mm NaCl, pH 7.0, and disrupted with brief pulsing with a sonic probe. Afterward, the detergent Triton X-100 was added to a final volume of 1% (w/v), and the resulting mixture was rocked at room temperature for 30 min. Unbroken cells and debris were removed by sedimentation as before. The mAb from the detergent-disrupted cells and the original supernatant were affinity-purified using a 1-ml protein A HiTrap column.
Measurement of Free Thiols by Monobromobimane—Free cysteine in serum (or cell culture) was measured using labeling with monobromobimane as described previously (12, 13) except that a Phenomenex C18 column and a water (0.1% trifluoroacetic acid)/methanol gradient (0–40% methanol, 40 min, 0.2 ml/min) were used.
Microchip Capillary Electrophoresis-SDS and Glycan Mapping—Capillary electrophoresis-SDS was performed on the Caliper LC90 under nonreducing conditions. Concentrations of the antibody samples were determined using the Lab-Chip 90 instrument (Caliper Life Sciences) (14). Some modifications were made from the manufacturer's suggested protocol. Briefly, a 5-μl antibody sample was mixed with 35 μl of denaturation solution. The denaturation solution was prepared by mixing 700 μl of HT protein express sample buffer with 70 μl of 250 mm iodoacetamide (to block free thiols). The samples were incubated at 70 °C for 15 min, after which 70 μl of water was added to each sample before loading onto the instrument for analysis. The concentration of each sample was calculated by comparing the main peak area with a standard curve established from antibody standards of a series of concentrations. Glycan mapping was performed as described previously (15).
Human pK Study of mAb—A 1000-mg mAb dose was administered to adult human patients in a single intravenous injection. Blood samples were collected over several weeks at selected times. After allowing time to clot, the clot was separated from serum by centrifugation (2000 × g for 15 min). Serum was stored in cryotubes at -20 °C or colder until used. mAb concentrations in serum were determined by a sandwich enzyme-linked immunosorbent assay using two anti-idiotypic antibodies against mAb.
mAb Ligand Affinity Purification—A 0.5-ml aliquot of freshly clarified human serum containing mAb was mixed with 4.5 ml of PBS and 0.2 ml of mAb-ligand resin. The tube containing the mixture was rocked at room temperature for 4 h. At the end of the incubation, the bound mAb was removed by sedimentation (350 × g for 5 min). Sedimented resin, containing the bound mAb, was gently resuspended in PBS and transferred to a small plastic column. After washing with 3 × 5 ml of PBS containing 0.5 m NaCl, the mAb was eluted with 0.5 ml of 10 mm glycine, pH 1.5. The pH of the eluted material was adjusted to ∼5 with 1 m Tris-HCl, pH 8.
Disulfide Exchange in Vitro—mAb was incubated in whole blood at 37 °C as described (6). Preliminary experiments indicated that the addition of 5 μm cysteine at the initiation of the experiment and at the end of Day 1 could maintain the free cysteine concentration between 6 and 10 μm. At specific time points, the mAb was affinity-purified from the blood and analyzed by nonreducing reversed-phase HPLC (RP-HPLC).
Nonreducing RP-HPLC—Purified mAb samples were analyzed by nonreducing RP-HPLC as described previously (16), except that column temperature, flow rate, mobile phase composition, and gradient were modified to achieve better resolution for the low amounts analyzed. The column was maintained at 65 °C, with a flow rate of 0.75 ml/min. Mobile phase A was 2% n-propyl alcohol and 0.1% trifluoroacetic acid. Mobile phase B was 80% n-propyl alcohol, 10% acetonitrile, and 0.1% trifluoroacetic acid. Samples were injected at a loading condition of 10% B for 1 min, followed by a 2-min gradient from 10% B to 22.5% B. Elution was achieved with a linear gradient of 22.5–29% B over 42 min. The column was then flushed for 5 min with 100% B, followed by re-equilibration with the loading condition for 5 min.
Mathematical Modeling and Fitting Procedures—The time evolution of the relative levels of three species can be modeled by kinetic Equations 1, 2, 3,
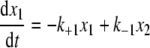 |
(Eq. 1) |
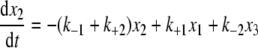 |
(Eq. 2) |
 |
(Eq. 3) |
where x1, x2, and x3 are time-dependent relative concentrations of the three species, IgG2-A, IgG2-A/B, and IgG2-B, respectively, and kx values are kinetic rate constants shown in chemical Equation 4 under “Results.”
This set of equations is used to fit the in vivo data to solve for the kinetic rate constant kx values with in vivo data, where only conversions between species are applicable. The terms x1, x2, and x3 are percent concentrations of each species relative to the total concentration of all three of them, and x1 + x2 + x3 is a conserved quantity. This assumes that clearance is not the cause of the changes in relative levels. Initial conditions are treated as unknown parameters in addition to all the kinetic rate constants. We used in vivo data from the average of three patients (see Fig. 8A) and the nonlinear least square fit routine lsqnonlin in combination with the ordinary differential equation solver ode23 from Matlab™ to perform the fitting. The unknown parameters were first estimated based on the available data. Random noise within predefined intervals was added to these estimates as initial guesses for the fitting procedure. More specifically, 500 sets of initial guesses were used. The resulting parameters were the set that generated the smallest sum of squares of the residuals.
FIGURE 8.

Mathematical modeling of conversion kinetics. A, modeled kinetic plots (dashed lines) have been superimposed on the actual in vivo disulfide conversion data (solid lines). Data shown are for 0, 0.04, 1, 2, 4, 10, and 13 days. The symbols represent the averaged data from three patients. Day 10 and 13 data are based on two patients because of serum availability. Error bars represent ± 1 S.D. The error bars for IgG2-A data are difficult to see because they are smaller than the symbol size. B, theoretical kinetic plots. Using the kinetic rate constants obtained from modeling shown in A, a kinetic simulation was performed assuming 100% A in the starting material. The line colors are for the same isoforms shown in A.
RESULTS
Disulfide Conversion in Culture—Isolated recombinant human IgG2s expressed and secreted from Chinese hamster ovary tissue culture cells contain a mixture of three structurally distinct disulfide forms, IgG2-A, IgG2-B, and IgG2-A/B (Fig. 1). Composition of the disulfide isoforms changes in media over time, either during active cell growth or when added to warm cell-depleted media, suggesting that the cysteine- or other thiol-containing compounds in the media promoted conversion between the forms. Because the percentage of A/B and B isoforms increases over time under these conditions, the isoform or isoforms originally secreted from the cells must be enriched in the A isoform. To determine the disulfide structure initially formed in the cell, cells from actively growing batch cultures expressing mAb were sedimented, washed, and disrupted with detergent. Samples were then analyzed by a nonreducing reversed-phase method developed specifically to measure these IgG2 disulfide isoforms (5, 6). The B isoform elutes in peak 1, the A/B in peak 2, and the A isoform in peaks 3 and 4 (Fig. 2, bottom trace). The structural basis for the A isoform separation into peaks 3 and 4 is not understood. Nonreduced Lys-C peptide maps used to identify the three isoform classes do not show differences in the material obtained from these two peaks (5).
FIGURE 2.

RP-HPLC analysis of mAb in cell culture media and disrupted cells. Samples were taken from a Day 7 batch culture. Top trace, supernatant; bottom trace, detergent-disrupted washed cells. Chromatographic profiles with labeled peaks corresponding to the IgG2-A (A), IgG2-A/B (A/B) and IgG2-B (B) isoforms are shown. A* is an isoform that cannot be distinguished from A through peptide mapping. mAU, milli absorbance units.
Affinity-purified IgG2 from these cells was enriched in the IgG2-A isoform as compared with the culture material. High mannose forms Man8 and Man9 were the main glycan forms on this material (Fig. 3), as opposed to mainly complex glycans in the secreted material, suggesting that most of the mAb was obtained from the ER (17) of the disrupted cells. In batch cultures, secreted antibodies remain in the media until the material is harvested, and this population is composed of a mixture of antibodies differing in age. Disulfide isoform composition of a Day 7 batch culture supernatant is shown for comparison (Fig. 2, top trace). These results suggest that the cells initially produce the mAb enriched in the A isoform, which is converted to the A/B and B isoforms over time in the cell culture media.
FIGURE 3.

Glycan map of mAb taken from disrupted cells (A) and media (B). 2-aminobenzamide-labeled N-linked glycans were analyzed by RP-HPLC as described previously (15). Certain 2-aminobenzamide-labeled glycans are indicated. M, high mannose glycans listed with the corresponding number of mannose residues. G0F, biantennary complex glycan with fucose and no terminal galactose; G1F, similar to G0F but with one terminal galactose; G2F, similar to G0F but with both antennae terminating in galactose; LU, light units.
Based on the cell culture experiments, it would be plausible that similar conversion occurs in blood. Cell culture medium during growth is near neutral pH at 37 °C and contains 20–30 μm cysteine. Likewise, serum pH is close to neutral and has been determined to contain 13–20 μm low molecular weight thiol-containing compounds, such as cysteine and glutathione (18, 19). Because the media environment during cell culture, where disulfide conversion occurs, is not significantly different from that of physiological blood serum, one might expect disulfide isoforms to convert in vivo. Moreover, experiments attempting to mimic physiological serum conditions in vitro found a conversion of the IgG2 disulfide isoforms (6) and that physiological thiol levels were sufficient to catalyze disulfide exchange.
IgG2 Isolation from Human Serum and Analysis—Conditions were developed to recover mAb drug from clinical patients' serum using a single antibody ligand affinity purification step. Under these conditions, ∼70% of mAb (25 μg/ml) added to human serum was recovered. The isolation procedure did not affect the disulfide isoform composition (Fig. 4) when mAb was only briefly incubated in the serum. mAb isolated from serum in this manner was >95% pure, as determined by microchip-based capillary electrophoresis (data not shown).
FIGURE 4.

Effect of mAb affinity purification from serum on disulfide variants. A, mAb profile of starting material. B, mAb affinity-purified from human serum supplemented with 25 μg/ml mAb in vitro. Note that the RP-HPLC method is as originally published (6) and was not optimized for the low load. mAU, milli absorbance units.
Following administration of a single 1000-mg intravenous mAb dose, blood was collected from patients at selected time points. mAb isolated from these time point samples was analyzed by RP-HPLC to measure the disulfide isoforms. Injection volumes were adjusted to account for the known mAb concentration differences in the samples. Fig. 5 shows an example of the changes observed in RP-HPLC profile over time in vivo. The peaks containing isoforms A (peak 3) and A/B (peak 2) diminish over time relative to peak 1 (B form). A plot showing changes to the relative areas of these peaks is shown in Fig. 6. Both peak 2 (A/B) and peak 3 (A) decrease with time after injection, whereas peak 1 increases (B). The minor peak 4, which contains an isoform structurally indistinguishable from the A isoform, does not appear to change with time and therefore has been omitted in this study.
FIGURE 5.

RP-HPLC analysis of mAb disulfide variants over time in a single patient. Peaks are labeled as in Fig. 2. Each chromatogram is labeled with the time between dosing and blood withdrawal from this patient.
FIGURE 6.

Plot of disulfide variant levels versus circulation time in a single patient. Data from Fig. 5 were plotted. Solid triangles represent the relative integrated area from peak 3 (IgG2-A), squares from peak 2 (IgG2-A/B), and diamonds from peak 1 (IgG2-B). Open triangles represent peak 4 (lgG2-A*).
Mechanism of Redistribution—Two likely mechanisms can account for this change in composition. In addition to disulfide exchange, differential clearance could result in changes in the relative levels of the disulfide forms. More than half of the originally injected antibody was cleared during the 13-day testing period. If forms A and A/B clear faster than the B isoform, changes in composition would be observed. Because the disulfide isoforms can affect ligand-binding activity, it is easy to envisage how such differences could result in changes in clearance rates.
Multiple lines of evidence support disulfide exchange as the mechanism for disulfide redistribution in vivo over differential clearance. One argument that can be made against differential clearance is that the overall clearance rates do not correlate well with the changes in composition. As seen in Fig. 7, patients showing significant differences in initial rates in overall mAb clearance (Fig. 7A) do not differ significantly in the rates of isoform A (Fig. 7B) or other isoform (Fig. 7, C and D) compositional changes. Another, maybe more significant, argument is that the disulfide isoform composition changes seen in vivo follow those observed in vitro. Isoform conversion occurs in vitro in the presence of cysteine, either in buffered saline or whole blood (6). Loss of IgG2-A and enrichment of IgG2-B were obtained in the presence of cysteine when incubating a mAb sample at 37 °C containing a mixture of the disulfide isoforms. The known concentrations of small molecular weight thiols in the blood appear to be sufficient to drive disulfide redistribution in vitro.
FIGURE 7.

Clearance and conversion plots. A, total mAb clearance from serum. Three patients are shown. Serum samples for Patient 2 were available only for Days 0–3. B, relative levels of the IgG2-A isoform remaining in the serum samples. The patients listed in B are the same ones listed in A. C, relative levels of the IgG2-A/B isoform remaining in the serum samples. D, relative levels of the IgG2-B isoform remaining in the serum samples. The key for C and D is shown in B. E, the relative levels of the IgG2-A isoform remaining following incubation in serum at 37 °C in vitro. The value was normalized to the time 0 sample.
In an attempt to mimic physiological conditions in vitro, mAb was added to whole blood samples and incubated at 37 °C for 2 days in the presence of low concentrations of cysteine (18, 19). Loss of the A isoform in blood in vitro mimics the redistribution seen in vivo (Fig. 7E). Because it is difficult to maintain stable cysteine concentrations in the in vitro incubations, direct comparisons of the conversion kinetics were not made. However, the results show that isoform conversion occurs in serum with low concentrations of free thiols. Therefore, disulfide exchange can explain the isoform compositional changes in vivo.
Calculated Reaction Rates—The rates of isoform conversion in vivo were calculated from the RP-HPLC data. Isoform composition data from three patients were averaged and plotted. To arrive at a model, certain assumptions were made. 1) Peaks 1, 2, and 3 represent isoforms B, A/B, and A, respectively. The small peak 4, which appears to not convert under these conditions, was ignored for this modeling. 2) Conversion is a unimolecular reaction. Free thiol most likely plays a catalytic role in disulfide exchange and the physiological concentration does not change, so it was not relevant to the modeling. 3) More significant is the assumption that the rate is unimolecular with respect to antibody concentration. In a unimolecular reaction, the rate is proportional to absolute reactant concentration, but not the relative concentration. Therefore, the antibody concentration changes occurring as a result of clearance should not affect the rate of conversion defined as a percent of each isoform. Using Equation 4,
 |
(Eq. 4) |
and the averaged data of the peaks 3, 2, and 1 from the three patients, kinetic rate constants were calculated from fitted plots. Fig. 8A contains plots of the clinical data overlaid with the modeled equation. Good alignments of the actual and theoretical curves were obtained.
Additional information gleaned from these plots further supports a conversion mechanism over differential clearance. In a differential clearance mechanism, the A/B isoform is not an intermediate in the A to B reaction. Faster A isoform clearance would result in increases in relative levels of both of the slower clearing isoforms. Although a fairly small effect, the modeled curves and the data appear to show that the A/B rate curves are specifically affected by the loss of the A form. The A/B levels show slower loss rates for the first 2 days after mAb administration and then dip (Fig. 8A), consistent with expectations of disulfide conversion and A/B as a chemical intermediate in the A → B pathway.
The following rate constants were calculated from the model k+1 is 1.51, k-1 is 0.498, k+2 is 0.142, and k-2 is 0.0436, all in units of day-1. It is apparent from these values that the forward reaction rate constants for A → A/B are about 10 times faster than those for A/B → B, so A/B to B conversion is rate-limiting in the A → B pathway.
The mAb used in these clinical studies contained relatively low levels of the A isoform. For a theoretical sample containing solely IgG2-A, conversion to A/B and B can be plotted using these rate reactions (Fig. 8B). Isoform A would be rapidly lost within 2 days after expression or injection. As isoform A is lost, isoform A/B is rapidly formed and then slowly converted to B over several days.
From these individual rate constants, the equilibrium constants for each partial reaction A → A/B and A/B → B can be calculated (i.e. k+1/k-1 and k+2/k-2, respectively). For the A → A/B reaction, the equilibrium constant, K1, is calculated to be 3.03, and for the A/B → B reaction, K2 is calculated to be 3.25. The overall equilibrium constant, calculated as a product of the individual partial reactions, K1K2, would be ∼9.8. Thus, at equilibrium, the predicted percentages of the three forms would be 7.2, 21.8, and 70.8% of A, A/B, and B, respectively. Values of the dosed material are nearing the predicted equilibrium values by Day 13, further supporting the kinetic modeling. The mAb lot used in the clinical experiments contained relatively low levels of the A isoform, only about 2-fold higher than the predicted equilibrium values. Better modeling would likely be achieved with an antibody sample highly enriched in the A isoform.
The physiological change in Gibbs free energy (ΔG) can be calculated from the predicted equilibrium constants (at 37 °C). For the A → A/B reaction, the equilibrium constant, ΔG1, is calculated to be -0.68 kcal/mol, and for the A/B → B reaction, ΔG2 is calculated to be -0.73 kcal/mol. Adding the individual free energies together provides the overall physiological ΔG for the A → B reaction of about -1.4 kcal/mol.
DISCUSSION
The results presented here strongly indicate that specific disulfides in the IgG2 antibody class reshuffle while circulating in human serum. Changes to the disulfide class distribution in vivo can be reproduced in vitro using whole blood or PBS containing near physiological levels of low molecular weight free thiol compounds (6). Thus, a simple thiol exchange mechanism driven by serum small molecule thiol-containing compounds can account for the changes in vivo, and more complex mechanisms involving antibody recycling or enzymatic action do not need to be invoked to account for the changes observed.
As an IgG2 ages in the blood, the fraction of the B isoform increases. This change could lead to changes in activity of the antibody over time and provides a marker of the antibody's physiological age. Previous work has shown that serum polyclonal antibodies contain a mixture of A, B, and A/B isoforms (5). Such a mixture can be considered a large ensemble of antibodies differing in age, which results in the steady state disulfide isoform fractions obtained. Younger IgG2s would contain a higher percentage of the A isoform. Antibody-producing cells must then synthesize either pure A isoform or antibodies enriched in the A isoform over that obtained from serum.
Antibody disulfide bonds are formed in the ER of antibody-secreting cells. Because A to B conversion occurs in the serum, the ER lumen environment must differ somewhat from the extracellular environment. Either the ER conditions affect the thermodynamics of disulfide bond formation, resulting in a difference in the equilibrium distribution of forms, or the disulfides are temporarily kinetically trapped, or at least paused, in the ER. Because secretory proteins in the ER are both spatially and temporally separated from ones in the serum, either is possible.
This study was performed with a human monoclonal antibody containing a κ LC. Although all IgG2 antibodies studied have shown mixtures of the three disulfide isoforms, IgG2s with λ LCs contain lower levels of the A/B and B isoforms (5, 6). It is not known whether this distribution difference is due to differences in the kinetics or thermodynamics of the disulfide shuffling reaction.
The disulfide shuffling behavior of specific disulfide bonds in a circulating human IgG2 antibody contrasts with the classical view of disulfide function. Here, the structural forms, defined by different disulfide linkages, convert after leaving the secreting cell on a relatively slow time scale. Intermediate structural isoforms are not so much kinetically trapped as they are paused along the slow path to the most stable folded state.
Acknowledgments
We thank R. Remmele and D. Kelner for critical reading of the manuscript and R. Melara for help in obtaining critical reagents.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ER, endoplasmic reticulum; LC, light chain; HC, heavy chain; mAb, monoclonal antibody; PBS, phosphate-buffered saline; RP-HPLC, reversed-phase HPLC.
References
- 1.Ellgaard, L., and Helenius, A. (2003) Nat. Rev. 4 181-191 [DOI] [PubMed] [Google Scholar]
- 2.Tu, B. P., and Weissman, J. S. (2004) J. Cell Biol. 164 341-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thornton, J. M. (1981) J. Mol. Biol. 151 261-287 [DOI] [PubMed] [Google Scholar]
- 4.Hogg, P. J. (2003) Trends Biochem. Sci. 28 210-214 [DOI] [PubMed] [Google Scholar]
- 5.Wypych, J., Li, M., Guo, A., Zhang, Z., Martinez, T., Allen, M. J., Fodor, S., Kelner, D. N., Flynn, G. C., Liu, Y. D., Bondarenko, P. V., Ricci, M. S., Dillon, T. M., and Balland, A. (2008) J. Biol. Chem. 283 16194-16205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dillon, T. M., Ricci, M. S., Vezina, C., Flynn, G. C., Liu, Y. D., Rehder, D. S., Plant, M., Henkle, B., Li, Y., Deechongkit, S., Varnum, B., Wypych, J., Balland, A., and Bondarenko, P. V. (2008) J. Biol. Chem. 283 16206-16215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellgaard, L., and Ruddock, L. W. (2005) EMBO Rep. 6 28-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruber, C. W., Cemazar, M., Heras, B., Martin, J. L., and Craik, D. J. (2006) Trends Biochem. Sci. 31 455-464 [DOI] [PubMed] [Google Scholar]
- 9.Alarazi, P. N., Lascombe, M.-B., and Poljak, R. J. (1988) Annu. Rev. Immunol. 6 550-580 [DOI] [PubMed] [Google Scholar]
- 10.Frangione, B., and Milstein, C. (1968) J. Mol. Biol. 33 893-906 [DOI] [PubMed] [Google Scholar]
- 11.van der Neut Kolfschoten, M., Schuurman, J., Losen, M., Bleeker, W. K., Martinez-Martinez, P., Vermeulen, E., den Bleker, T. H., Wiegman, L., Vink, T., Aarden, L. A., De Baets, M. H., van de Winkel, J. G., Aalberse, R. C., and Parren, P. W. (2007) Science 317 1554-1557 [DOI] [PubMed] [Google Scholar]
- 12.Kosower, E. M., and Kosower, N. S. (1995) Methods Enzymol. 251 133-148 [DOI] [PubMed] [Google Scholar]
- 13.Newton, G. L., and Fahey, R. C. (1995) Methods Enzymol. 251 148-166 [DOI] [PubMed] [Google Scholar]
- 14.Chow, A. W. (2006) in Microchip Capillary Electrophoresis (Henry, C. S., ed) pp. 145-158, Humana Press Inc., Totowa, NJ
- 15.Chen, X., and Flynn, G. C. (2007) Anal. Biochem. 370 147-161 [DOI] [PubMed] [Google Scholar]
- 16.Dillon, T. M., Bondarenko, P. V., Rehder, D. S., Pipes, G. D., Kleemann, G. R., and Ricci, M. S. (2006) J. Chromatogr. 1120 112-120 [DOI] [PubMed] [Google Scholar]
- 17.Kornfeld, R., and Kornfeld, S. (1985) Annu. Rev. Biochem. 54 631-664 [DOI] [PubMed] [Google Scholar]
- 18.Di Giuseppe, D., Di Simplicio, P., Capecchi, P. L., Lazzerini, P. E., and Pasini, F. L. (2003) J. Lab. Clin. Med. 142 21-28 [DOI] [PubMed] [Google Scholar]
- 19.Andersson, A., Isaksson, A., Brattstrom, L., and Hultberg, B. (1993) Clin. Chem. 39 1590-1597 [PubMed] [Google Scholar]