

Abstract
Reactivation of the androgen receptor (AR) signaling pathway represents a critical step in the growth and survival of androgen-independent (AI) prostate cancer (CaP). In this study we show the DU145 and PC3 AI human CaP cell lines respond to androgens and require AR expression for optimal proliferation in vitro. Interestingly, AR gene transcripts in DU145 and PC3 cells harbored a large number of single base pair nucleotide transitions that resulted in missense mutations in selected AR codons. The most notable lesion detected in AR gene transcripts included the oncogenic codon 877T→A gain-of-function mutation. Surprisingly, AR gene transcript nucleotide transitions were not genome-encoded substitutions, but instead the mutations co-localized to putative A-to-I, U-to-C, C-to-U, and G-to-A RNA editing sites, suggesting the lesions were mediated through RNA editing mechanisms. Higher levels of mRNA encoding the A-to-I RNA editing enzymes ADAR1 and ADARB1 were observed in DU145 and PC3 cells relative to the androgen-responsive LNCaP and 22Rv1 human CaP cell lines, which correlated with higher levels of AR gene transcript A-to-I editing detected in DU145 and PC3 cells. Our results suggest that AR gene transcripts are targeted by different RNA editing enzymes in DU145 and PC3 cells. Thus RNA editing of AR gene transcripts may contribute to the etiology of hormone-refractory phenotypes in advanced stage AI CaP.
Androgen-independent (AI)2 prostate cancer (CaP) typically develops from the selective outgrowth of tumor cells to castrate levels of testosterone in response to androgen-deprivation therapy (1). AI CaP cells have evolved different strategies for overriding the androgen-dependent (AD) growth and survival characteristics of early stage, organ-confined CaP or early stage metastatic CaP (2). Aberrant AR activation is the primary mechanism for the growth and survival of AI CaP in response to castrate levels of androgen (2). With the exception of prostatic small cell neuroendocrine carcinoma (3), the most parsimonious model of AI CaP arises from the inappropriate activation of AR-dependent cell growth and survival pathways. Unfortunately, this model is incomplete because examples of AR-independent CaP do exist. Most notably, the well established human CaP cell lines DU145 and PC3, which were derived from brain and bone metastases, respectively (4, 5), are supposedly devoid of AR mRNA and protein (6, 7) and thus represent bona fide models of AR-independent CaP. Interestingly, this classification was recently called into question as detectable levels of AR mRNA and protein were observed in both DU145 and PC3 cells (8). More importantly, several studies have demonstrated low AR activity in PC3 cells, suggesting these cells also utilize AR-dependent mechanisms for growth and survival similar to other established AD CaP cell lines (e.g. LNCaP, 22Rv1, and C4-2) (8–11).
AR displays variable immunoreactivity in individual tumor cells of AI CaP, with cells showing a strong, weak, or undetectable AR signal (12). These results illustrate the heterogeneity of AR expression in AI CaP and suggest that both AR-dependent and AR-independent mechanisms of growth and survival are present in AI CaP. Practically speaking, determining whether AI CaP utilizes AR-dependent or AR-independent mechanisms for growth and survival is a controversial and arduous endeavor that has profound clinical ramifications on how to treat advanced stage, AI CaP. Here we have investigated whether DU145 and PC3 cells are representative models of AR-independent CaP by testing if they respond to androgens or require AR expression for cell growth in vitro. We show that DU145 and PC3 cells are androgen-responsive and require AR for optimal growth in vitro, thus demonstrating each cell line is a bona fide model of androgen-responsive, AR-dependent CaP. We show AR gene transcripts in DU145 and PC3 cells harbor nucleotide transitions suggesting the AR pre-mRNA is the target of multiple RNA editing enzymes. We propose that RNA editing enzymes are modulators of AR activity through the introduction of loss-of-function or gain-of-function mutations into AR gene transcripts in advanced stage AI CaP.
EXPERIMENTAL PROCEDURES
Reagents
The following reagents were purchased: AR agonist R1881 (methyltrienolone) (PerkinElmer Life Sciences); 17β-estradiol, progesterone, cyproterone acetate (CPA), hydroxyflutamide (HF) (Sigma); double-stranded siRNAs (Dharmacon Research, Lafayette, CO); Oligofectamine reagent, 4–12% SDS-polyacrylamide gels, and the TOPO TA cloning kit (Invitrogen); prestained Precision Plus protein standards and goat anti-mouse horseradish peroxidase-conjugated secondary (Bio-Rad); mouse monoclonal AR antibody (AR441) (Santa Cruz Biotechnology (Santa Cruz, CA); MEGAscript® high yield transcription kit (Ambion, Austin, TX); BCA protein assay kit (Pierce); ECL reagents kit and Hyperfilm ECL film (GE Healthcare); RNeasy midi kit, Oligotex Midi kit, and DNA oligonucleotides (Qiagen, Valencia, CA); proteinase K solution, DNase-free RNase, transcriptor reverse transcriptase enzyme, and FastStart TaqDNA polymerase (Roche Applied Science); fetal bovine serum and charcoal-stripped fetal bovine serum (Hyclone Laboratories, Logan, UT); fluorescent short tandem repeat (STR) oligonucleotides (Applied Biosystems, Foster City, CA); GeneRuler 1-kb DNA ladder (MBI Fermentas, Hanover, MD); rat genomic DNA (ATCC, Manassas, VA); and qPCR primers (SuperArray, Frederick, MD).
Cell Lines
LNCaP, 22Rv1, DU145, and PC3 cells were from American Tissue Type Culture Collection. LNCaP and 22Rv1 cells were grown in phenol red-deficient RPMI 1640 medium (Invitrogen) containing 10% FBS (Hyclone Laboratories Inc., Logan, UT). DU145, PC3, and 293HEK cells were grown in phenol red-deficient DMEM high glucose media containing 10% FBS. STR analysis was used to authenticate the genotype of all human CaP cell lines (supplemental Figs. 2 and 3).
Western Blot Analysis
LNCaP, 22Rv1, PC3, and DU145 cells were seeded into Falcon (BD Biosciences) 6-well tissue culture dishes (10,000 cells/cm2) for 48 h. Cells were subsequently solubilized in 0.3 ml of buffer A (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, pH 7.4, 1% SDS) and boiled for 5 min. Total protein lysates were quantified using the Pierce BCA protein assay kit. 8 μg of total protein lysates were subjected to SDS-PAGE (4–12% gradient precast gels; Invitrogen). The proteins were transferred onto a polyvinylidene difluoride membrane, incubated in Tris-buffered saline containing 0.1% Tween 20 (TBST), and blocked with 5% nonfat milk (w/v) for 1 h. Membranes were incubated in TBST containing 5% bovine serum and a 1:250 dilution of the AR441 mouse monoclonal antibody (Santa Cruz Biotechnology) overnight at 4 °C. The blots were washed three times for 5 min in TBST and incubated with a goat anti-mouse horseradish peroxidase secondary at a 1:10,000 dilution in TBST for 1 h at room temperature. The blots were washed three times for 5 min in TBST, and immunoreactive bands were developed and visualized using the ECL reagent kit (GE Healthcare). The blots were exposed to Hyperfilm ECL film (GE Healthcare) for <1 min. The same protocol was used to probe whole cell lysates of siRNA-transfected PC3 and DU145 cells with a mouse monoclonal antibody to β-tubulin (Sigma). For silver staining, SDS-polyacrylamide gels of DU145 and PC3 cells transfected with control and AR siRNAs were processed for silver stain visualization as detailed previously (13).
siRNAs
The control, AR1 (5′-AAGCCCATCGTAGAGGCCCCA-3′), or AR siGENOME SMARTpool siRNAs (Dharmacon) were transfected into DU145 and PC3 cells. DU145 and PC3 cells seeded at 2000 cells/cm2 into medium A (phenol red-deficient DMEM high glucose medium supplemented with 10% FBS lacking antibiotics) for 24 h were transfected with the control, AR1, or the SMARTpool siRNA at a final concentration of 50 nm using the Oligofectamine reagent (Invitrogen) according to the manufacturer's guidelines.
Cell Growth Assays
Androgen Treatment—DU145 and PC3 cells were seeded at 2000 cells/cm2 into AD (phenol-red deficient DMEM high glucose medium supplemented with 2% charcoal-stripped FBS plus antibiotics) or androgen-supplemented (AS) medium containing 1.0 or 100 nm R1881 (PerkinElmer Life Sciences) for 240 h. LNCaP cells were subjected to the same experimental conditions except cells were grown in RPMI 1640 growth medium. Growth medium was replenished every 72 h, and cell growth was determined by trypan blue dye exclusion (240 h). Data represent the mean ± S.D. Student's t test compared AD with AS cells (asterisk denotes p values <0.05).
siRNA Transfection—DU145 and PC3 cells were seeded at 2000 cells/cm2 into medium A for 24 h. Cells were transfected with control, AR1, or SMARTpool siRNAs at a final concentration of 50 nm for 120 h. Cells were fed medium A for 144 h, and cell density was determined by trypan blue dye exclusion. Data represent the mean ± S.D. Student's t test compared control with AR1-transfected cells; and control and SMARTpool siRNA-transfected cells (asterisk denotes p value <0.05).
AR Cloning
For RNA purification, total RNA (RNeasy midi kit, Qiagen) isolated from LNCaP, 22Rv1, PC3, and DU145 CaP cells (passage number <10) was used to clone AR gene transcripts (amino acids 487–919) using standard RT-PCR cloning methods. LNCaP and 22Rv1 total RNA and DU145 and PC3 mRNA (Oligotex Midi kit, Qiagen) were used to clone AR gene transcripts, respectively.
RT-PCR, cDNA Cloning, and DNA Sequencing
RT-PCR—Three independent RT reactions were used to clone AR gene transcripts in LNCaP, 22Rv1, PC3, and DU145 cells. The individual RT reactions included 200 ng of total RNA (LNCaP and 22Rv1 cells) or 1 μg of mRNA (DU145 and PC3 cells). The gene-specific AR primer was 5′-TCACTGGGTGTGGAAATAGATGGGCTTGACTTTCCCAGAAAGG-3′. A 13-μl reaction volume contained 1 μl (25 μm) of the gene-specific AR primer 5′-TCACTGGGTGTGGAAATAGATGGGCTTGACTTTCCCAGAAAGG-3′. Purified RNA and RNase-free water were combined and incubated at 75 °C for 10 min in an RNase-free microcentrifuge tube. 4 μl (five times) of Transcriptor RT Reaction Buffer, 0.5 μl of Protector RNase (40 units/μl), 2 μl (10 mm) of dNTP mix, and 0.5 μl of Transcriptor Reverse Transcriptase (Roche Applied Science) were added to the ice-cooled tube at a final reaction volume of 20 μl. The tube was mixed and incubated at 60 °C for 45 min. 2 μl of the RT reactions were used in the following PCRs.
PCR—The 50-μl PCR contained 39.6 μl of sterile double-distilled water, 5 μl of PCR buffer (10×, 20 mm MgCl2), 1 μl of dNTP mixture (25 mm), 1 μl of the 5′ DNA primer, 5′-CAGGGGCTGGCGGGCCAGGAAAGCGACTTCACCGCACCTGATGTGTGGTACCCTGGCGGCATGGTGAGCA-3′ (25 μm), 1 μl of the 3′ DNA primer, 5′-TCACTGGGTGTGGAAATAGATGGGCTTGACTTTCCCAGAAAGG-3′ (25 μm), 0.4 μl of FastStart TaqDNA polymerase (250 units/μl), and 2 μl of RT product. The PCR cycle conditions consisted of a 4-min 95 °C incubation cycle followed by 40 cycles of a 30-s 95 °C denaturation step, a 60 °C annealing step, and a 3-min 72 °C elongation step. This 40 cycle reaction was followed by a 72 °C 7-min extension that terminated the PCR. Separate RT-PCRs were resolved onto separate 0.8% agarose gels containing ethidium bromide, and the cDNAs were gel-purified onto DEAE-cellulose membranes. Membranes were high salt (1.5 m NaCl, 25 mm Tris, pH 8.0) extracted, and eluted cDNAs were ethanol-precipitated and quantified with a UV spectrophotometer.
cDNA Cloning—30 ng of gel-purified RT-PCR product was ligated to 10 ng to the pCR 4-TOPO vector using the TOPO TA cloning kit (Invitrogen) and transformed into bacteria according to the manufacturer's protocol. Transformant plasmids containing DNA inserts were submitted for automated DNA sequencing (DNA Sequencing Facility, Davis, CA). Plasmid inserts were sequenced with the following primers to the pCR 4-TOPO vector and AR: M13 reverse 5′-CAGGAAACAGCTATGAC-3′, M13-21 5′-GTAAAACGACGGCCAGT-3′, T7 5′-TAATACGACTCACTATAGGG-3′, 5′-CAGCCATCTGGTCGTCCACGTGTAAGTTGCG-3′, 5′-ATGACTCTGGGAGCCCGGAAGCTGAAGAAACTTG-3′, 5′-TAATGCTGAAGAGTAGCAGTGCTTTCATGCACAG-3′, and 5′-ACACATTGAAGGCTATGAATGTCAG-3′. DNA sequences were translated into C-terminal AR polypeptides (amino acids 507–919) and aligned with the ClustalW algorithm (14).
DNA Sequencing
Recombinant AR cDNAs were sequenced using the following: for DNA primers, M13 reverse 5′-CAGGAAACAGCTATGAC-3′, M13-21 5′-GTAAAACGACGGCCAGT-3′, T7 5′-TAATACGACTCACTATAGGG-3′; for AR primers, 5′-CAGCCATCTGGTCGTCCACGTGTAAGTTGCG-3′, 5′-ATGACTCTGGGAGCCCGGAAGCTGAAGAAACTTG-3′, 5′-TAATGCTGAAGAGTAGCAGTGCTTTCATGCACAG-3′, 5′-ACACATTGAAGGCTATGAATGTCAG-3′. DNA sequences were translated into polypeptides (507–919) and aligned with ClustalW.
Genomic DNA Subcloning and Sequencing
Genomic DNA in LNCaP, 22Rv1, DU145, and PC3 cells was extracted and processed as detailed under “STR Analysis.” Genomic DNA was used to clone fragments of DNA spanning AR amino acids 665–717, 725–768, and 844–919 in LNCaP, 22Rv1, DU145, and PC3 cells with the following DNA primer pairs: 665–717, 5′-GAAGGCTATGAATGTCAGCCCATC-3′ and 5′-CTTGACCACGTGTACAAGCTGTCTC-3′; 725–768, 5′-CGCAACTTACACGTGGACGACCAGATGGCTG-3′ and 5′-CCAGATCAGGGGCGAAGTAGAGCATCC-3′; and 844–919, 5′-GAGGCCACCTCCTTGTCAACCCTG-3′ and 5′-GGAACATGTTCATGACAGACTGTACATC-3′. The same PCR amplification, subcloning, and DNA sequencing methods detailed under “cDNA Cloning” were used with the following modifications: PCR extension reaction, 30 s; agarose DNA gel purification, 2.5% agarose gel; cloning reaction, 5 ng of DNA ligated to the pCR 4-TOPO vector; DNA sequencing, T7 and M13-21 primer reactions only. The DNA chromatograms were inspected for nucleotide transitions against the wild-type AR sequence, and the results are presented in Table 2.
TABLE 2.
Genome-encoded AR mutations The total number of sequenced clones with missense mutations at codons 695, 757, 874, and 877 in LNCaP, 22Rv1, DU145, and PC3 cells are shown.

CTAP-AR Mutant Expression Vectors
The CTAP-AR vector was used to construct the CTAP-AR-D695G/V757A/D819G and CTAP-AR-T877A expression vectors using the QuikChange multisite-directed mutagenesis kit (Stratagene) as detailed in the manufacturer's protocol. The AR-D695G/V757A/D819G and AR-T877A missense mutations were introduced into the CTAP-AR expression vector using the following mutagenic DNA primers: D695G mutation primer, 5′-GACACGACAACAACCAGCCCGGCTCCTTTGCAGCCTTGCTCTC-3′; V757A mutation primer, 5′-CTGGCGATCCTTCACCAATGCCAACTCCAGGATGCTCTACTTC-3′; D819G mutation primer, 5′-CTCTTCAGCATTATTCCAGTGGGTGGGCTGAAAAATCAAAAATTC-3′; and T877A mutation primer, 5′-GAGAGAGCTGCATCAGTTCGCTTTTGACCTGCTAATCAAGTCAC-3′. Two sequential site-directed mutagenesis experiments were performed to construct the CTAP-AR-D695G/V757A/D819G vector. The first mutagenesis experiment used the mutagenic D695G and V757A primers to construct the CTAP-AR-D695G/V757A expression vector. The second mutagenesis experiment used the mutagenic D819G primer to construct the CTAP-AR-D695G/V757A/D819G expression vector. The CTAP-AR-T877A expression vector was constructed with the mutagenic T877A primer. DNA sequence analysis verified the mutations, and the authenticity of the constructs was determined by Western blot confirmation of AR expression in transiently transfected 293HEK cells.
pGL4.10-Luc2-Probasin Vector
A 751-bp DNA fragment (KpnI/XhoI) containing the proximal promoter of the rat probasin gene (base positions -750 to +1) was cloned into the promoterless pGL4.10 luciferase expression vector (Promega) using standard DNA cloning protocols. The Pfu polymerase enzyme was used to amplify rat genomic DNA (ATCC) using the 5′ end primer 5′-GATCGGTACCGTAATCATACATATTATGATTATCCAATAAGCTTTCTGG-3′ and the 3′ end primer 5′-GATCCTCGAGCGTGTGTGAGCTCTGTAGGTATCTGGACCTCACTGACAAGGTGC-3′ according to the manufacturer's protocol. The amplified rat probasin DNA promoter was agarose gel-purified, KpnI- and XhoI-digested, and subcloned into the pGL4.10-Luc2 vector to produce the pGL4.10-Luc2-Probasin vector.
qPCR Experiments
RNA Extraction—RNA purification methods detailed above were used to isolated mRNA from LNCaP, 22Rv1, DU145, and PC3 cells (passage <15) for qPCR analyses. For siRNA knockdown experiments, cells were transfected with the siRNA duplexes as detailed above, and total RNA was isolated from cells 96 h post-transfection and processed for qPCR analyses as detailed below.
RT and qPCR Reactions—RT experiments were executed using
purified mRNA as a template, except random primers were included in the cDNA
synthesis reactions. ADAR1, ADARB1, and GAPDH qPCR primers (SuperArray,
Frederick, MD) were used for the qPCRs with the RT2 SYBR Green/ROX
qPCR Master Mix (SuperArray, Frederick, MD) according to the manufacturer's
protocol. 200 ng of cDNA template was used in each qPCR. GAPDH served as the
control gene for the qPCRs. Results were analyzed according to the
manufacturer's protocol using the ΔΔCt method.
ΔCt is the difference between the target gene (ADAR1 or ADARB1)
and the reference gene (GAPDH). ΔΔCt is the difference in
ΔCt between the target gene and the control gene. The fold
change was calculated as 2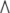 (-ΔΔCt).
(-ΔΔCt).
STR Analysis
DNA Extraction—Genomic DNA was isolated from LNCaP, 22Rv1, PC3, and DU145 cells (passage number <10) using the following steps: confluent Falcon T-175-cm2 tissue culture dishes (BD Biosciences) were washed in phosphate-buffered saline, and cells were solubilized in 0.6 ml of buffer D (10 mm Tris-HCl, 1 mm EDTA, 0.1% SDS, pH 8.0) containing 10 μl of proteinase K solution (20 mg/ml) (Roche Applied Science) and incubated at 55 °C overnight. 5 μl of DNase-free RNase (Roche Applied Science) was added to the samples and incubated at 37 °C for 1 h. 0.6 ml of potassium acetate solution was added, and samples were spun at 13,000 rpm for 5 min at 4 °C. Supernatants were mixed with an equal volume of isopropyl alcohol, and DNA was recovered by spinning the sample at 13,000 rpm for 1 min. DNA samples were washed with 70% ethanol, and DNA was dissolved in nuclease-free water.
STR Profiling—LNCaP, 22Rv1, DU145, and PC3 genomic DNA was used to identify short tandem repeats. 250 ng of genomic DNA was used in each TaqPCR. The cycling conditions included 32 cycles of a 95 °C denaturation step, a 55 °C annealing step, and a 72 °C elongation step. The nine markers and fluorescent DNA primer pairs included in the DNA profiling experiments were as follows: 1) Amelogenin (6FAM-5′-AGGCTTGAGGCCAACCATCAG-3′ and 5′-ACCTCATCCTGGGCACCCTGG-3′); 2) CSF1PO (NED-5′-AACCTGAGTCTGCCAAGGACTAGC-3′ and 5′-TTCCACACACCACTGGCCATCTTC-3′); 3) D13S317 (PET-5′-GGCAGCCCAAAAAGACAGA-3′ and 5′-ACAGAAGTCTGGGATGTGGAGGA-3′); 4) D16S539 (VIC-5′-GGGGGTCTAAGAGCTTGTAAAAAG and 5′-GTTTGTGTGTGCATCTGTAAGCAT-3′); 5) D5S818 (6FAM-5′-AGCCACAGTTTACAACATTTGTATCT-3′ and 5′-GGTGATTTTCCTCTTTGGTATCC-3′); 6) D7S820 (NED-5′-ATGTTGGTCAGGCTGACTATG-3′ and 5′-GATTCCACATTTATCCTCATTGAC-3′); 7) TH01(PET-5′-GTGGGCTGAAAAGCTCCCGATTAT-3′ and 5′-ATTCAAAGGGTATCTGGGCTCTGG-3′); 8) TPOX (VIC-5′-GGAGGAACTGGGAACCACACAGGTTA-3′ and 5′-ACTGGCACAGAACAGGCACTTAGG-3′); and 9) vWA (6FAM-5′-GGACAGATGATAAATACATAGGATGGATGG-3′ and 5′-GCCCTAGTGGATGATAAGAATAATCAGTATGTG-3′). The nine individual STR reactions representing LNCaP, 22Rv1, DU145, or PC3 cells were run separately on an ABI 3730 DNA sequencer using a loose 500-bp internal ladder. The results were processed and annotated by the Veterinary Genetics Laboratory (University of California, Davis).
Cloning of T7 Synthesized AR mRNA
RNA encoding full-length AR (amino acids 1–919) was synthesized in vitro with the MEGAscript kit according to the manufacturer's protocol. Briefly, a microcentrifuge tube containing buffer, ribonucleotides, linearized pSG5-AR expression vector, and T7 RNA polymerase was incubated at 37 °C for 4 h. After the 15-min incubation with TURBO DNase, the transcribed RNA was purified by a phenol/chloroform extraction and precipitated with isopropyl alcohol as described in the manufacturer's protocol. The RNA was quantified with a UV spectrophotometer, and 100 ng of RNA was subjected to RT-PCRs as detailed in the cDNA subcloning and sequencing sections. In parallel, no PCR product was detected in the following negative control experiments. 1) RNA was pretreated with RNase (500 ng, DNase-free) for 1 h at 37°C prior to the RT-PCR; 2) transcriptor reverse transcriptase was excluded from the RT reaction. The results are presented in supplemental Table 3 and supplemental Fig. 6.
Src Cloning and Sequencing
The same methods used to clone AR gene transcripts with DU145 mRNA were used to clone Src gene transcripts. This experiment used the following reagents: 3′ SRC RT primer, 5′-CTAGAGGTTCTCCCCGGGCTGGTACTGGGGCTCGGTGGACGTG-3′; PCR amplification primers, 5′-CAGCAGCTGGTGGCCTACTACTCCAAACAC-3′ and 5′-CTAGAGGTTCTCCCCGGGCTGGTACTGGGGCTCGGTGGACGTG-3′. The 933-bp PCR-amplified cDNA fragment encoding Src amino acids 227–536 was subcloned, and the DNA sequencing results are depicted in supplemental Table 2 and supplemental Fig. 5.
Luciferase Assay
293HEK cells seeded at a density of 30,000 cells/cm2 into Falcon (BD Biosciences) 24-well tissue culture dishes for 24 h in phenol red-deficient DMEM high glucose media supplemented with 2% CS-FBS and antibiotics (medium B) were transfected by calcium phosphate co-precipitation (15) in triplicate with 400 ng of total plasmid DNA/well consisting of the pGL4.10-Luc2-Probasin (10 ng), pRLSV40 Renilla (Promega) (25 ng), pcDNA3 (165 ng), and (200 ng) of the CTAP-AR, CTAP-AR-D695G/V757A/D819G, or CTAP-AR-T877A vectors. Vehicle (ethanol), synthetic androgen (R1881), 17β-estradiol, progesterone, CPA, and HF were added to the cells 24 h post-transfection. Cell lysates were measured for dual luciferase activity 48 h later according to the manufacturer's protocol. All firefly and Renilla luciferase measurements were performed on a Veritas microplate luminometer (Turner BioSystems, Inc., Sunnyvale, CA). The mean ± S.D. were determined for all firefly luciferase values.
RESULTS
A recent study showed the AI CaP cell lines DU145 and PC3 express very low levels of AR (8), which prompted us to explore if either cell line responded to androgens in vitro. As reported previously (8), low AR expression was detected in DU145 and PC3 cells relative to the androgen-responsive LNCaP and androgen-refractory 22Rv1 human CaP cell lines by Western blot (Fig. 1A). Androgens are known to influence the proliferative capacity of AD CaP cell lines in vitro (6), with low levels of androgen (10 pm to1 nm) stimulating proliferation, and high levels of androgen (10–100 nm) inducing growth arrest and differentiation (16, 17). Thus we decided to test if physiologic (1 nm) or supraphysiologic doses (100 nm) of androgen had any influence on the proliferation of DU145 and PC3 cells. Cell growth was measured after a 10-day incubation in androgen-depleted (2% charcoal-stripped FBS) or androgen-supplemented (2% charcoal-stripped FBS + R1881, androgen analogue) growth medium (Fig. 1, B and C). Crystal violet staining showed cultures containing 100 nm R1881 were noticeably reduced relative to cells grown in androgen-depleted growth medium (supplemental Fig. 1). Quantitatively, the growth of DU145 cells was reduced by ∼20 and ∼ 30% in response to 1 and 100 nm R1881, respectively (Fig. 1B), whereas PC3 cell growth was reduced by ∼40% in response to 100 nm R1881 (Fig. 1C). We included the androgen-responsive LNCaP cell line in these studies and found that 1 nm R1881 increased growth greater than 60% (Fig. 1D), whereas 100 nm R1881 reduced growth more than 40% (Fig. 1D). These results show DU145 and PC3 cells are androgen-responsive for growth in vitro, suggesting both cell lines express functional levels of AR.
FIGURE 1.

AR expression in human CaP cell lines. A, Western blot analysis of AR in LNCaP, 22Rv1, PC3, and DU145 cells. Arrows denote AR protein expression. B–D, DU145, PC3, and LNCaP cell growth in response to androgen (R1881). B, 10 days of PC3 cell growth in androgen-depleted (2% CS-FBS) or androgen-supplemented (2% CS-FBS + 1.0 or 100 nm R1881). Graph depicts total number of viable cells. Mean ± S.D. Asterisk = Student's t test (p value <0.05). C, DU145 cells, exact conditions as detailed for B. D, LNCaP cells, exact conditions as detailed for B except cells were grown in RPMI 1640 growth medium. Asterisk = Student's t test (p value <0.05). E, AR expression in AR knockdown DU145 and PC3 cells. Upper panel, Western blot analysis of AR (96 h) (control siRNA, 50 nm; AR1 siRNA, 50 nm); middle panel, silver-stained SDS-PAGE of extracts in upper panel to demonstrate equivalent protein loading; lower panel, loading control Western blot of β-tubulin. F, photograph of crystal violet-stained control or AR1 siRNA-transfected DU145 and PC3 cells 11 days post-transfection. (i), PC3 cells transfected with control siRNA; (ii), PC3 cells transfected with AR1 siRNA; (iii), DU145 cells transfected with control siRNA; (iv), DU145 cells transfected with AR1 siRNA. G, AR knockdown on DU145 and PC3 cell growth. 120 h post-transfection, cells were grown in siRNA-deficient growth medium for 144 h. Graph depicts total number of viable cells 264 h post-transfection. The total number of AR1 (50 nm) and AR SMARTpool (50 nm) siRNA transfected cells (x axis) is plotted relative to the total number of control siRNA (50 nm) transfected cells (y axis). Mean and ± S.D. Asterisk = Student's t test (p value < 0.05). Data are representative of at least two independent experiments.
To show the androgen-responsive cell growth phenotypes of DU145 and PC3 cells were not mediated by cell line contamination (18), a parallel passage (<10) of cells was subjected to short tandem repeat (STR) analysis to authenticate the genotype of each cell line. LNCaP and 22Rv1 cells were included as positive controls for these analyses. These experiments verified the genotype of all four human CaP cell lines (supplemental Figs. 2 and 3), thus showing the results were not compromised by cell line contamination.
The growth-inhibited phenotype of DU145 and PC3 cells to exogenous androgens prompted us to test if either cell line required AR expression for cell growth in vitro (Fig. 1, B and C). Both cell lines were transfected with an siRNA duplex directed to AR called AR1, which was previously shown to validate the AR-dependent cell growth phenotype of LNCaP cells through AR knockdown (11). Western blot analysis showed the AR1 siRNA effectively reduced AR levels in transfected DU145 and PC3 cells relative to the control siRNA (Fig. 1E, lanes 1–4). Crystal violet staining showed the growth of cells transfected with the AR1 siRNA was reduced relative to the control siRNA (Fig. 1F). In fact, the AR1 siRNA reduced DU145 and PC3 cell growth by more than 60% relative to the control siRNA (Fig. 1G). To exclude the possibility the AR1 siRNA had a nonspecific effect of DU145 and PC3 cell growth, each cell line was transfected with an optimized pool of AR siRNA duplexes (4-plex) (Dharmacon siGENOME SMARTpool). Similar to the AR1 siRNA, the SMARTpool siRNAs strongly inhibited the growth of DU145 and PC3 cells relative to the control siRNA (Fig. 1G). These results show that DU145 and PC3 cells require AR expression for optimal cell growth in vitro and demonstrate an AR-dependent cell growth phenotype in each cell line.
DU145 and PC3 cell lines are the representative prostate cancer models for studying mechanisms of AR-independent CaP growth and survival (6). However, our results showed that both cell lines were androgen-responsive and required AR expression for optimal proliferation in vitro (Fig. 1). These findings prompted the search for AR mutations (e.g. constitutively activated AR) that could explain the androgen-responsive, AR-dependent phenotype in DU145 and PC3 cells. Thus, AR gene transcript mutations in DU145 and PC3 cells were probed by cloning the C-terminal AR mRNA (amino acids 487–919) encoding the DNA (exons 2 and 3) and ligand-binding domains (exons 4–8) of AR, because lesions tend to cluster to these regions in CaP (19). AR gene transcripts in LNCaP and 22Rv1 cells were cloned in parallel to verify the genome-encoded T877A (ACT→GCT) and H874Y (TAT→CAT) missense mutations in LNCaP and 22Rv1 cells respectively. The analysis would also validate the AR exon 3 duplication in 22Rv1 cells (20, 21). Thus, a single translated AR cDNA clone (amino acids 509–919) representative of each CaP cell line was ClustalW aligned to the wild-type AR C-terminal polypeptide sequence (supplemental Fig. 4A). This analysis verified the missense mutations in T877A and H874Y in LNCaP and 22Rv1 cells, and it confirmed the exon 3 duplication in 22Rv1 cells. This lesion increases AR length by 39 amino acids and causes it to migrate as a slower, immunoreactive species in 22Rv1 cells (Fig. 1A) (14, 20, 21). Remarkably, the DU145 and PC3 clones contained the same exact missense mutations at codons 695 (Asp → Gly), 757 (Val → Ala), and 819 (Asp → Gly), which were all caused by single base pair nucleotide transitions at the selected codons (codon 695, GAC→ GGC; codon 757, GTC→ GCC; codon 819, GAC→ GGC) (supplemental Fig. 4A). Interestingly, the missense mutation at codon 695 (Asp → Val) was previously detected in a patient afflicted by androgen-insensitivity syndrome, which is associated with a decrease in AR transactivation (22, 23). In contrast, the functional consequences of the V757A mutation are unknown, but this lesion was originally identified in a lymph node CaP metastasis (24). Finally, no mutation has been reported for codon 819 to date, suggesting the putative D819G lesion is a novel AR missense mutation.
These findings prompted us to verify AR mutations as authentic AR gene transcript lesions. Thus, independent RT-PCR cloning experiments (n = 3) were performed to isolate multiple AR cDNA clones in LNCaP, 22Rv1, DU145, and PC3 cells, to determine the frequency and distribution of nucleotide transitions in AR gene transcripts. All sequenced clones were ClustalW aligned at the nucleotide and polypeptide level to determine the abundance of nonsense, missense, and silent mutations in all cDNA clones (Fig. 2 and supplemental Fig. 4, B–E, supplemental Table 1). As anticipated, the genome-encoded T877A lesion was detected in >90% of LNCaP clones (Fig. 2 and Table 1), whereas >70% of 22Rv1 clones contained the H874Y and exon 3 duplication (Fig. 2 and Table 1). Surprisingly, more than 60% of DU145 and PC3 clones contained the D695G, V757A, and D819G mutations (Fig. 2 and Table 1), whereas another ∼20% of the clones also harbored the Y551C and T877A mutations (Fig. 2 and Table 1). The D695G/V757A/D819G mutant was a prominent lesion detected in >30% of DU145 and PC3 clones (Fig. 2 and Table 1). Of note, exons 2 and 3 were missing in several LNCaP clones, whereas more than 10% of the 22Rv1 clones harboring the H874Y lesion, also lacked the exon 3 duplication (Table 1). This finding demonstrated uncoupling of the exon 3 duplication and H874Y mutation in 22Rv1 cells, suggesting the exon 3 duplication may arise through a nongenomic mechanism (e.g. RNA splicing) (20, 21). Interestingly, many of the missense mutations represented AR lesions in CaP samples (Table 1) (23). These findings show AR gene transcripts harbor both silent and missense mutations (supplemental Table 1), and the high frequency of D695G, V757A, and D819G lesions in DU145 and PC3 clones strongly suggested they were bona fide AR mutations in DU145 and PC3 cells. However, the larger number (>3-fold) of nucleotide transitions observed in DU145 and PC3 clones relative to the LNCaP and 22Rv1 clones could also support the notion that the lesions were artifacts if the cloning process was inherently more mutagenic in DU145 and PC3 cells (supplemental Table 1).
FIGURE 2.

Frequency of missense mutations in cloned AR gene transcripts (amino acids 507–919) in human CaP cell lines. AR missense mutations are listed on the x axis. The total number of clones observed in LNCaP, 22Rv1, DU145, and PC3 cells is represented on the y axis. Missense mutations observed in two or more independent clones are denoted in the graph. Number of clones analyzed as follows: LNCaP (44 clones), 22Rv1 (52 clones), DU145 (48 clones), and PC3 (47 clones).
TABLE 1.
Androgen receptor gene transcript missense mutations The table shows the number and distribution of missense mutations/deletions/insertions of cloned AR gene-transcripts (amino acids 507–919) in LNCaP, 22Rv1, DU145, and PC3 cells.

CaP indicates somatic
PAIS indicates partial AIS, constitutional
CAIS indicates complete AIS, constitutional
MAIS indicates mild AIS, constitutional
Missense mutation with different amino acid is indicated
The above sequencing results raised several important observations regarding the putative AR mutations in DU145 and PC3 cells. First, if the nucleotide transitions were authentic AR gene transcript mutations, these lesions should be present at the AR locus in DU145 and PC3 cells. Second, if the RT-PCR cloning process was highly mutagenic (e.g. incorporation of nucleotide transitions) in DU145 and PC3 cells, nucleotide transitions should be present at a high frequency in a cloned gene transcript unrelated to AR.
First, we tested if the D695G, V757A, and T877A lesions were by-products of genome-encoded nucleotide transitions at the AR locus in DU145 and PC3 cells (25, 26). Individual PCR-amplified genomic DNA segments encompassing AR codons 695, 757, or 877 were cloned and sequenced to validate the predicted adenosine to guanosine transition at codon 695 (GAC→ GGC), the predicted thymidine to cytosine transition at codon 757 (GTC→ GCC), and the predicted adenosine to guanosine transition in codon 877 (ACT→GCT) in DU145 and PC3 cells. In parallel, the genomic DNA segments spanning AR codons 874–877 in LNCaP and 22Rv1 cells were also cloned to verify the cytosine to thymidine transition at codon 874 (CAT→TAT) in 22Rv1 cells, and the adenosine to guanosine transition at codon 877 (ACT→GCT) in LNCaP cells (Table 2) (27, 28). All LNCaP clones contained the T877A lesion, whereas the H874Y lesion was detected in all 22Rv1 clones (Table 2). Surprisingly, wild-type AR sequences at codons 695 (GAC), 757 (GTC), and 874 (ACT) were present in all DU145 and PC3 clones (Table 2). These findings showed that nucleotide transitions responsible for the AR gene transcript lesions (e.g. D695G, V757A, and T877A) in DU145 and PC3 cells were not genome-encoded AR lesions.
These findings prompted us to test if the mRNA used to clone AR was chemically modified so that nucleotide transitions were indiscriminately incorporated into gene transcripts at a high frequency during the cloning process as stated above. Thus, a C-terminal portion of the human nonreceptor tyrosine kinase Src (P12931) gene transcript (amino acids 227–536) was cloned with the same mRNA used to clone AR in DU145 cells. Thirty two independent cDNA clones were translated into C-terminal Src polypeptides and ClustalW aligned (supplemental Fig. 5). The total population of clones contained four nucleotide transitions, and three of the lesions resulted in missense mutations (supplemental Fig. 5 and supplemental Table 2). These findings validated the integrity of mRNA used to clone AR gene transcripts in DU145 cells, and showed mutagenic nucleotide transitions were incorporated into cloned gene transcripts at a low frequency in vitro.
Next, we wanted to rule out the remote possibility that the AR mRNA adopted secondary structures that selectively incorporated nucleotide transitions into specific codons (e.g. codons 695, 757, and 819) during the cloning process. Thus, a T7 RNA polymerase synthesized AR mRNA transcript was cloned in vitro (AR exon 2–8) to determine the frequency and distribution of nucleotide transitions of cloned products in vitro (supplemental Table 3). A total of 18 nucleotide transitions were detected in 32 independent cDNA clones (supplemental Table 3). This included a total of 6 silent and 12 missense mutations (supplemental Fig. 6 and supplemental Table 3). More importantly, each missense mutation was unique with no more than one missense mutation present in a single clone (supplemental Table 3). The results showed that nucleotide transitions were randomly incorporated into cloned AR gene transcripts at a low frequency in vitro. Overall, the findings supported the notion that the D695G, V757A, D819G, and T877A lesions were authentic AR gene transcript mutations in DU145 and PC3 cells.
These results prompted us to explore if the missense mutations could be incorporated into the AR gene transcript by a nongenomic mechanism. The post-transcriptional process of RNA editing is an extra-genomic process capable of introducing nucleotide transitions into pre-mRNA transcripts, which result in codon changes that modify the activity of a target protein (29). For example, developmentally regulated RNA editing of the mammalian glutamate receptor-2 (GluR2) at the Gln/Arg (Q/R) site in the GluR2 subunit channel-pore-loop domain makes the channel impermeable to Ca2+ ions (30). Thus, RNA editing could explain the AR gene transcript missense mutations if the nucleotide transitions correlated to the substrate specificities of RNA editing enzymes. The substrate specificities of RNA editing enzymes include A-to-I nucleotide transitions, which are mediated by adenosine deaminase RNA-specific enzymes (ADAR1, ADARB1), C-to-U transitions, which are mediated by the APOBEC 1 cytidine deaminase, and U-to-C and G-to-A transitions, which are mediated by yet-to-be identified enzymes (29). Interestingly, the single base pair nucleotide transitions at the D695G, V757A, D819G, Y874H, and T877A lesions were positioned at putative A-to-I, U-to-C, or G-to-A RNA editing sites (Table 3). Thus, A-to-I editing could mediate the nucleotide transitions at codons 695 (GAC→ GGC), 819 (GAT→ GGT), and 877 (ACT→GCT), whereas U-to-C editing could mediate the nucleotide transitions at codons 757 (GTC→ GCC) and 874 (TAT→CAT). G-to-A editing could mediate the A877T lesion (GCT→ACT), which is an RNA editing event originally detected in human immunodeficiency virus transcripts (31). Therefore, the mechanism of RNA editing could account for the AR gene transcript missense mutations in CaP cells.
TABLE 3.
Putative edited sites The table shows putative A-to-I, U-to-C, and G-to-A RNA editing sites in AR codons 551, 695, 757, 819, 874, and 877. Bold italic = edited nucleotide.

Secondary RNA structures, which include RNA bulges and hairpin loops, facilitate RNA editing through the intramolecular folding of complementary exon and intron sequences (32). The AR pre-mRNA is more than 180 kb in length, thus making it impractical to use RNA folding algorithms to calculate secondary RNA structures to a molecule of this size (33). However, if a smaller 17.9-kb AR pre-mRNA transcript is partitioned into smaller ∼6-kb segments, the pre-mRNA can be analyzed for secondary RNA structures using the MFOLD RNA folding algorithm. The pre-mRNA spans a portion of intron 3 (∼3.1 kb) and extends through to the 3′-untranslated region (∼1.85 kb), which includes codons 695, 757, 819, 874, and 877. The pre-mRNA segments that span this region contain many RNA bulges and hairpin loops (supplemental Fig. 7) (33). These highly speculative double-stranded (ds) foldback structures provide in silico evidence that the AR pre-mRNA can adopt complex ds foldback structures that are preferred targets of RNA editing enzymes. More evidence to suggest the AR pre-mRNA is a target of RNA editing would be the presence of repetitive elements that could promote the formation of complex ds foldback structures through the complementary base pairing of inversely spaced repeat elements (32, 34).
These repetitive elements include both short interspersed elements and long interspersed elements that typically localize to introns and untranslated regions (34). We used the repeat data base program CENSOR to identify repetitive elements in the complex ds foldback structures in the 17.9-kb AR pre-mRNA (35). Multiple repetitive elements were identified along the 17.9-kb pre-mRNA AR transcript (Table 4), which included the simple repeat elements (AC)n, and (GAAAA)n, and the more complex short interspersed elements and long interspersed elements (Table 4). Most importantly, inversely oriented repeat elements were present in the pre-mRNA structure and thus provided the opportunity to form foldback structures through the complementary annealing of sense and antisense elements (Table 4). These in silico analyses suggest repetitive elements may prompt complex ds foldback structures in the 17.9-kb pre-mRNA AR transcript.
TABLE 4.
Repetitive elements (intron 3 to exon 8) Repetitive elements: segment A, 1A = L1; 2A = (AC)n; 3A = (AG)n; 4A = AluJo; 5A, 7A, 8A, 9A = MIRb; 6A = MIR3. Segment B, 1B, 7B, 12B = MIRb; 2B = MIR3; 3B = L2B; 4B = THER1; 5B, 8B, 11B = MIR; 6B = LIP_MA2; 9B = AluSg; 10B = (GAAA)n. Segment C, 1C, 3C, 6C = (AC)n; 2C = (CAAA)n; 4C, 5C = (AG)n; 7C = (GAAAAA)n; 8C = (CAAAAA)n; 9C = (TAVA)n.

These analyses prompted us to examine the relationship, if any, between RNA editing enzyme expression levels and the quantity of AR gene transcript editing in CaP cells. Focusing on the ADAR1 and ADARB1 enzymes, which mediate A-to-I RNA editing, qPCR showed ADAR1 expression was 3–4-fold higher in DU145 and PC3 cells relative to LNCaP and 22Rv1 cells (Fig. 3A), whereas ADARB1 levels were 1.5–2-fold higher in PC3 cells relative to LNCaP and 22Rv1 cells (Fig. 3A). Repeated experiments to directly test if ADAR1 or ADARB1 knockdown (e.g. transient or stable short hairpin RNA) could reduce A-to-I editing of AR gene transcripts in DU145 and PC3 cells were unsuccessful (data not shown). Although a transient ∼80 and ∼50% reduction (e.g. 96 h post-transfection) in ADAR1 and ADARB1 mRNA expression was achieved by siRNA knockdown in DU145 cells (Fig. 3B), a statistically significant reduction in A-to-I editing of AR gene transcripts was not observed (data not shown). We also reduced ADAR1 and ADARB1 mRNA levels by ∼92 and ∼80%, respectively, in PC3 cells by siRNA-mediated knockdown (data not shown). Again, a statistically significant reduction in AR gene transcript A-to-I editing was not observed (data not shown). Plasmid-based microRNAs directed against ADAR1 and ADARB1 also failed to reduce AR gene transcript A-to-I editing in PC3 cells (data not shown). Multiple scenarios could explain why effective ADAR1 and ADARB1 mRNA knockdown (e.g. > 90% reduction) did not bring about a statistically significant decrease in AR gene transcript A-to-I editing. First, despite causing a significant reduction in ADAR1 and ADARB1 mRNA levels using siRNA or miRNA methods, sufficient levels of these enzymes may still be present under these experimental conditions to effectively catalyze AR gene transcript A-to-I editing in PC3 and DU145 cells. Second, there is the possibility that an A-to-I RNA editing activity, not encoded by ADAR1 or ADARB1 (e.g. ADAR3), could also mediate AR gene transcript in PC3 and DU145 cells. Importantly, neither scenario is mutually exclusive, as both processes could be at play to facilitate AR gene transcript A-to-I editing in DU145 and PC3 cells. Finally, high ADAR1 levels have been shown to decrease the efficacy of siRNA knockdown in mammalian cells and also change miRNA target specificity in cells (36, 37). These findings could help to explain why a statistically significant reduction in AR gene transcript editing in DU145 and PC3 cells was not observed when both ADAR1 and ADARB1 were targeted for knockdown using siRNA or miRNA reagents.
FIGURE 3.

ADAR1 and ADARB1 RNA expression in human CaP cell lines. A, qPCR analysis of ADAR1 and ADARB1 expression in LNCaP, 22Rv1, DU145, and PC3 cells. Expression values were normalized to GAPDH and determined by the comparative Ct method. The y axis shows normalized expression by percent to LNCaP cells. Mean and ± S.D. of values. B, qPCR analysis of ADAR1 and ADARB1 expression in DU145 cells 96 h post-transfection with the control siRNA or an ADAR1/ADARB1 siRNA mixture (50 nm siRNA per gene) (ON-TARGETplus SMART-pool siRNA). Results were analyzed as described above in A.
Finally, we tested if the AR-D695G/V757A/D819G mutant, which represented the most abundant AR gene transcript detected in DU145 and PC3 cell populations (Fig. 2), could influence AR-mediated transcription in mammalian cells. The androgen-responsive rat probasin-luciferase reporter was used to measure the transcriptional activity of AR-D695G/V757A/D819G in response to androgen (R1881), estrogen (17β-estradiol), progesterone, HF, and CPA in transfected 293HEK cells (Fig. 4, A–E) (38). These transcriptional reporter experiments clearly showed AR-D695G/V757A/D819G lacked transcriptional activity toward androgenic or anti-androgenic ligands (Fig. 4, A–E). This loss-of-function phenotype is concordant with the loss of AR transcriptional activity in androgen-insensitivity syndrome patients carrying the D695V lesion (22, 23). Future studies will determine whether the V757A and D819G lesions in isolation can perturb AR-mediated transcription.
FIGURE 4.

AR transcriptional activity in mammalian cells. A–E, transcriptional activity of mutant AR in 293HEK cells. 293HEK cells transfected with pGL4.10-Luc2-Probasin vector (10 ng), pRLSV40 Renilla (25 ng), CTAP-AR, CTAP-AR-D695G/V757A/D819G, or CTAP-AR-T877A vectors (200 ng/vector). Ethanol (A–E), androgen (R1881) (A), 17β-estradiol (B), progesterone (C), CPA (D), or HF (E) were added 24 h post-transfection and dual luciferase activity was measured (48 h). Results were expressed as the fold induction in luciferase activity relative to vehicle-treated cells as the ratio of Firefly and Renilla luciferase activities in each sample. The values are the means ± S.D. of at least two independent experiments. WT, wild type.
DISCUSSION
We demonstrate the established human DU145 and PC3 CaP cell lines are androgen-responsive, AR-dependent models of human CaP. We provide evidence that AR gene transcripts contain nucleotide transitions likely mediated through RNA editing enzymes. These findings are important because AR gene transcript RNA editing may represent a novel molecular mechanism for introducing and accumulating AR mutations that give rise to androgen-independent growth and survival phenotypes in advanced, hormone-refractory CaP. One of the unresolved questions in the prostate cancer field is determining if AR mutations promote prostate tumorigenesis through genomic (transcription-mediated) or nongenomic (nontranscription-mediated) pathways. For example, it was recently shown that AR-mediated transcription and androgen-mediated cell proliferation can be uncoupled in prostate cancer cells (39). The study showed that partial agonists of classical AR transcriptional responses are as efficacious as dihydrotesterone in stimulating proliferation in human prostate cancer cells. These findings are very important on several levels. First, AR mutations that uncouple AR-mediated transcription and androgen-mediated cell proliferation responses in human prostate cancer cells have yet to be determined. Thus we lack the knowledge to quantify the biological impact of AR mutations, either individually or collectively, have on AR-mediated transcription or androgen-mediated cell proliferation during prostate tumorigenesis. Second, human prostate cancer cell lines can be misclassified as “androgen-independent” based upon transcriptional responses to androgen (e.g. androgenic increases in prostate-specific antigen expression) (39). A transcriptional response to androgen lacks the sensitivity to accurately classify human prostate cancer cells as androgen-independent. For example, many studies have shown that high AR expression retards the growth of established human prostate cancer cell lines in vitro (16, 17, 40, 41). Under these circumstances lower levels of androgen (<1 nm) stimulate proliferation, whereas higher levels of androgen (>1 nm) retard cellular growth. Thus, low AR levels may be advantageous to the proliferative state of DU145 and PC3 cells in vitro. For example, low levels of AR might be necessary for critical cellular processes such as proliferation (e.g. DNA replication) (42), whereas high levels of AR could prompt growth-arrest through the expression of genes associated with cellular differentiation (e.g. expression of PSA). We envision that RNA editing may stimulate or abolish AR-mediated proliferation responses in low level AR expressing prostate cancer cells through the introduction of gain-of-function or loss-of-function AR mutations. This model is supported by our findings as both DU145 and PC3 cells are androgen-responsive and require endogenous low level AR expression for optimal cellular growth in vitro. More importantly, nucleotide transitions may result in loss- or gain-of-function AR mutations, thus providing a novel mechanism for introducing and propagating disease-associated AR lesions into human CaP. Overall, these findings provide a plausible explanation of the androgen-responsive, AR-dependent phenotype of DU145 and PC3 cells. For example, prominent AR-D695G/V757A/D819G expression would allow cells to appear phenotypically androgen-independent because this mutant lacks detectable transcriptional activity (e.g. undetectable change in PSA expression, low reporter activity). However, this same population of cells may also co-express AR gene transcripts harboring gain-of-function mutations (e.g. AR-Y551C/V757A/T887A mutant), albeit at lower levels, which expand the ligand specificity of AR and potentiate AR activity to suboptimal levels of androgen (43, 44). Under this scenario CaP cells would phenotypically appear androgen-independent but truly require AR activity. This is similar to the phenotypes of DU145 and PC3 cells described in this study. Alternatively, AR transcriptional activity may be dispensable for CaP growth and survival (39). Under this circumstance, even near undetectable levels of AR would be required to carry out essential, AR-mediated cellular functions (e.g. DNA replication) in CaP cells (42). Future studies to test individual components of this hypothesis are forthcoming. Although this study focused on nucleotide transitions in AR exons 2–8, exon 1 may also be targeted by RNA editing machinery and accumulate missense mutations through the formation of secondary hairpin loop RNA structures nucleated by the polyglycine or polyglutamic acid repeat sequences in this region. Direct biochemical evidence demonstrating the AR gene transcript is an RNA editing enzyme target in vitro awaits further investigation. Molecular characterization of RNA editing enzymes in CaP lines, mouse xenografts, and CaP samples is greatly anticipated because this family of enzymes may impact the etiology of AI CaP. Thus, RNA editing enzymes may represent a novel class of biomarkers of diagnostic, prognostic, or therapeutic value in patients afflicted by AI CaP.
Supplementary Material
Acknowledgments
We thank Drs. Cliff G. Tepper, David K. Han, and Peter S. Nelson for helpful discussions. M. E. W. also extends a personal acknowledgment to Dr. Richard W. Michelmore for the unwavering support.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–7 and Tables 1–3.
Footnotes
The abbreviations used are: AI, androgen-independent; AD, androgen-dependent; AR, androgen receptor; CaP, prostate cancer; ds, double-stranded foldback; siRNA, short interfering RNA; STR, short tandem repeat; qPCR, quantitative PCR; FBS, fetal bovine serum; CS, charcoal-stripped; DMEM, Dulbecco's modified Eagle's medium; HF, hydroxyflutamide; CPA, cyproterone acetate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; RT, reverse transcription.
References
- 1.Miyamoto, H., Messing, E. M., and Chang, C. (2004) Prostate 61 332-353 [DOI] [PubMed] [Google Scholar]
- 2.Richter, E., Srivastava, S., and Dobi, A. (2007) Prostate Cancer Prostatic Dis. 10 114-118 [DOI] [PubMed] [Google Scholar]
- 3.Clegg, N., Ferguson, C., True, L. D., Arnold, H., Moorman, A., Quinn, J. E., Vessella, R. L., and Nelson, P. S. (2003) Prostate 55 55-64 [DOI] [PubMed] [Google Scholar]
- 4.Kaighn, M. E., Narayan, K. S., Ohnuki, Y., Lechner, J. F., and Jones, L. W. (1979) Investig. Urol. 17 16-23 [PubMed] [Google Scholar]
- 5.Stone, K. R., Mickey, D. D., Wunderli, H., Mickey, G. H., and Paulson, D. F. (1978) Int. J. Cancer 21 274-281 [DOI] [PubMed] [Google Scholar]
- 6.Sobel, R. E., and Sadar, M. D. (2005) J. Urol. 173 342-359 [DOI] [PubMed] [Google Scholar]
- 7.Tilley, W. D., Wilson, C. M., Marcelli, M., and McPhaul, M. J. (1990) Cancer Res. 50 5382-5386 [PubMed] [Google Scholar]
- 8.Alimirah, F., Chen, J., Basrawala, Z., Xin, H., and Choubey, D. (2006) FEBS Lett. 580 2294-2300 [DOI] [PubMed] [Google Scholar]
- 9.Yuan, X., Li, T., Wang, H., Zhang, T., Barua, M., Borgesi, R. A., Bubley, G. J., Lu, M. L., and Balk, S. P. (2006) Am. J. Pathol. 169 682-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zegarra-Moro, O. L., Schmidt, L. J., Huang, H., and Tindall, D. J. (2002) Cancer Res. 62 1008-1013 [PubMed] [Google Scholar]
- 11.Wright, M. E., Tsai, M. J., and Aebersold, R. (2003) Mol. Endocrinol. 17 1726-1737 [DOI] [PubMed] [Google Scholar]
- 12.Shah, R. B., Mehra, R., Chinnaiyan, A. M., Shen, R., Ghosh, D., Zhou, M., MacVicar, G. R., Varambally, S., Harwood, J., Bismar, T. A., Kim, R., Rubin, M. A., and Pienta, K. J. (2004) Cancer Res. 64 9209-9216 [DOI] [PubMed] [Google Scholar]
- 13.Echan, L. A., and Speicher, D. W. (1996) in Current Protocols in Protein Science (Coligan, J. E., ed) pp. 10.5.1-10.5.18, Wiley Interscience, New York [DOI] [PMC free article] [PubMed]
- 14.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kingston, R. E., Chen, C. A., and Rose, J. K. (1987) in Current Protocols in Molecular Biology (Ausubel, F. M., ed) pp. 9.1.1-9.1.11, Greene Publishing Associates, New York
- 16.Berns, E. M., de Boer, W., and Mulder, E. (1986) Prostate 9 247-259 [DOI] [PubMed] [Google Scholar]
- 17.Langeler, E. G., van Uffelen, C. J., Blankenstein, M. A., van Steenbrugge, G. J., and Mulder, E. (1993) Prostate 23 213-223 [DOI] [PubMed] [Google Scholar]
- 18.Lacroix, M. (2008) Int. J. Cancer 122 1-4 [DOI] [PubMed] [Google Scholar]
- 19.Buchanan, G., Greenberg, N. M., Scher, H. I., Harris, J. M., Marshall, V. R., and Tilley, W. D. (2001) Clin. Cancer Res. 7 1273-1281 [PubMed] [Google Scholar]
- 20.Tepper, C. G., Boucher, D. L., Ryan, P. E., Ma, A. H., Xia, L., Lee, L. F., Pretlow, T. G., and Kung, H. J. (2002) Cancer Res. 62 6606-6614 [PubMed] [Google Scholar]
- 21.Chlenski, A., Nakashiro, K., Ketels, K. V., Korovaitseva, G. I., and Oyasu, R. (2001) Prostate 47 66-75 [DOI] [PubMed] [Google Scholar]
- 22.Hiort, O., Sinnecker, G. H., Holterhus, P. M., Nitsche, E. M., and Kruse, K. (1998) J. Pediatr. 132 939-943 [DOI] [PubMed] [Google Scholar]
- 23.Gottlieb, B., Lehvaslaiho, H., Beitel, L. K., Lumbroso, R., Pinsky, L., and Trifiro, M. (1998) Nucleic Acids Res. 26 234-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcelli, M., Ittmann, M., Mariani, S., Sutherland, R., Nigam, R., Murthy, L., Zhao, Y., DiConcini, D., Puxeddu, E., Esen, A., Eastham, J., Weigel, N. L., and Lamb, D. J. (2000) Cancer Res. 60 944-949 [PubMed] [Google Scholar]
- 25.Culig, Z., Klocker, H., Eberle, J., Kaspar, F., Hobisch, A., Cronauer, M. V., and Bartsch, G. (1993) Prostate 22 11-22 [DOI] [PubMed] [Google Scholar]
- 26.van Bokhoven, A., Varella-Garcia, M., Korch, C., Johannes, W. U., Smith, E. E., Miller, H. L., Nordeen, S. K., Miller, G. J., and Lucia, M. S. (2003) Prostate 57 205-225 [DOI] [PubMed] [Google Scholar]
- 27.Tan, J.-A., Sharief, Y., Hamil, K. G., Gregory, C. W., Zang, D.-Y., Sar, M., Gumerlock, P. H., DeVere White, R. W., Pretlow, T. G., Harris, S. E., Wilson, E. M., Mohler, J. L., and French, F. S. (1997) Mol. Endocrinol. 11 450-459 [DOI] [PubMed] [Google Scholar]
- 28.Veldscholte, J., Ris-Stalpers, C., Kuiper, G. G., Jenster, G., Berrevoets, C., Claassen, E., van Rooij, H. C., Trapman, J., Brinkmann, A. O., and Mulder, E. (1990) Biochem. Biophys. Res. Commun. 173 534-540 [DOI] [PubMed] [Google Scholar]
- 29.Nishikura, K. (2006) Nat. Rev. Mol. Cell Biol. 7 919-931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higuchi, M., Single, F. N., Kohler, M., Sommer, B., and Sprengel, R. (1993) Cell 75 1361. [DOI] [PubMed] [Google Scholar]
- 31.Bourara, K., Litvak, S., and Araya, A. (2000) Science 289 1564-1566 [DOI] [PubMed] [Google Scholar]
- 32.Bass, B. L. (2002) Annu. Rev. Biochem. 71 817-846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuker, M. (2003) Nucleic Acids Res. 31 3406-3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Athanasiadis, A., Rich, A., and Maas, S. (2004) PLoS Biol. 2 e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohany, O., Gentles, A. J., Hankus, L., and Jurka, J. (2006) BMC Bioinformatics 7 474-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang, W., Wang, Q., Howell, K. L., Lee, J. T., Cho, D. S., Murray, J. M., and Nishikura, K. (2005) J. Biol. Chem. 280 3946-3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawahara, Y., Zinshteyn, B., Sethupathy, P., Iizasa, H., Hatzigeorgiou, A. G., and Nishikura, K. (2007) Science 315 1137-1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jasavala, R., Martinez, H., Thumar, J., Andaya, A., Gingras, A. C., Eng, J. K., Aebersold, R., Han, D. K., and Wright, M. E. (2007) Mol. Cell. Proteomics 6 252-271 [DOI] [PubMed] [Google Scholar]
- 39.Sathya, G., Chang, C.-Y., Kazmin, D., Cook, C. E., and McDonnell, D. P. (2003) Cancer Res. 63 8029-8036 [PubMed] [Google Scholar]
- 40.Litvinov, I. V., Antony, L., Dalrymple, S. L., Becker, R., Cheng, L., and Isaacs, J. T. (2006) Prostate 66 1329-1338 [DOI] [PubMed] [Google Scholar]
- 41.Yuan, S., Trachtenberg, J., Mills, G. B., Brown, T. J., Xu, F., and Keating, A. (1993) Cancer Res. 53 1304-1311 [PubMed] [Google Scholar]
- 42.Litvinov, I. V., Vander Griend, D. J., Antony, L., Dalrymple, S., De Marzo, A. M., Drake, C. G., and Isaacs, J. T. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 15085-15090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun, C., Shi, Y., Xu, L. L., Nageswararao, C., Davis, L. D., Segawa, T., Dobi, A., McLeod, D. G., and Srivastava, S. (2006) Oncogene 25 3905-3913 [DOI] [PubMed] [Google Scholar]
- 44.Duff, J., and McEwan, I. J. (2005) Mol. Endocrinol. 19 2943-2954 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.