

Abstract
The AAA+ molecular chaperone Hsp104 mediates the extraction of proteins from aggregates by unfolding and threading them through its axial channel in an ATP-driven process. An Hsp104-binding peptide selected from solid phase arrays enhanced the refolding of a firefly luciferase-peptide fusion protein. Analysis of peptide binding using tryptophan fluorescence revealed two distinct binding sites, one in each AAA+ module of Hsp104. As a further indication of the relevance of peptide binding to the Hsp104 mechanism, we found that it competes with the binding of a model unfolded protein, reduced carboxymethylated α-lactalbumin. Inactivation of the pore loops in either AAA+ module prevented stable peptide and protein binding. However, when the loop in the first AAA+ was inactivated, stimulation of ATPase turnover in the second AAA+ module of this mutant was abolished. Drawing on these data, we propose a detailed mechanistic model of protein unfolding by Hsp104 in which an initial unstable interaction involving the loop in the first AAA+ module simultaneously promotes penetration of the substrate into the second axial channel binding site and activates ATP turnover in the second AAA+ module.
Hsp104 is a AAA+ protein disaggregase that functions in yeast in the resolubilization and reactivation of thermally denatured and aggregated proteins (1, 2). In unstressed cells, Hsp104 is critical to the mitotic stability of the yeast prions [PSI+], [PIN+], and [URE3] (3–5). Hsp104 and its bacterial orthologue ClpB are members of the Hsp100/Clp family of proteins (6). Other Hsp100s, such as ClpA, ClpX, and ClpY (HslU), unfold and unidirectionally translocate polypeptides through a central axial channel (7–11). Crystal structures of HslU (12, 13) and cryoelectron microscopic reconstructions of ClpB (14) reveal that the diameter of the axial channel is regulated by flexible loops whose conformation is regulated by the nucleotide status of the nucleotide binding domain of each AAA+ module. Modification of these loops impairs protein translocation and/or degradation implying that these loops play critical roles in translocation (15–18). Likewise, mutation of the flexible loops of Hsp104 and ClpB results in refolding defects suggesting that all Hsp100s employ a similar unfolding/threading mechanism to process substrates whether they are ultimately degraded or refolded (16, 19, 20). Despite the growing body of knowledge regarding the unfolding and translocation mechanism of Hsp104, the determinants of the initial stage of the unfolding process, substrate recognition and binding, remain unclear.
In other Hsp100s, recognition of specific peptide sequences initiates unfolding and translocation. Protein substrates of ClpXP generally contain recognition signals of roughly 10–15 residues that can be located either at the N or C termini (21). The SsrA tag, an 11-amino acid peptide (AANDENYALAA) that is appended to the C terminus of polypeptides by the action of transfer-messenger RNA on stalled ribosomes (22), is a particularly well studied example of an Hsp100-targeting peptide. The SsrA tag physically interacts with both ClpA and ClpX, targeting the polypeptides for degradation by ClpAP and ClpXP (23). The N-terminal 15-aa3 peptide of RepA (MNQSFISDILYADIE) is another example of a peptide that, when fused either to the N or C termini of GFP, is sufficient to target the fusion protein for recognition and degradation by ClpAP (24).
Refolding of proteins trapped in aggregates requires not only Hsp104/ClpB but also a cognate Hsp70/40 chaperone system (2, 25). Evidence suggests that the Hsp70 system acts prior to the Hsp100, initially to produce lower order aggregates that still lack the ability to refold to the native state (26). A ClpB mutant containing a substitution in the coiled-coil domain is defective in processing aggregates that are dependent on the DnaK co-chaperone system but has no defect in the processing of unfolded proteins, suggesting a role for the coiled-coil domain in mediating a transfer of substrates from DnaK to ClpB (27). Although it is possible that the Hsp70/40 may act as adaptor proteins that present refolding substrates to Hsp104/ClpB, it is not an obligatory pathway. In the absence of Hsp70, Hsp104 alone remodels yeast prion fibers formed by Sup35 and Ure2 (28). Furthermore, Hsp104 in the presence of mixtures of ATP and slowly hydrolysable ATP analogues or a mutant of Hsp104 with reduced hydrolytic activity in the second AAA+ module can refold aggregated GFP and activate RepA for DNA binding without additional chaperones (29). These observations suggest that Hsp104 can directly and independently recognize and process substrates, although the molecular basis of this interaction is unclear.
We hypothesize that, like other Hsp100s, Hsp104 may initially engage substrates by binding to peptides displayed on the surface of aggregated proteins. Using peptide arrays, we found that potent Hsp104-binding peptides were enriched in charged and hydrophobic amino acids. Fusion of an Hsp104-binding peptide to the C terminus of firefly luciferase (FFL) enhanced its refolding in an Hsp104-dependent refolding reaction. Peptide binding to Hsp104, analyzed by fluorescence quenching using Hsp104 mutants containing tryptophan probes in the first (D1) and second (D2) AAA+ modules, revealed the existence of at least two unique peptide binding sites. Soluble peptide also competed for binding with a model unfolded protein, reduced carboxymethylated α-lactalbumin (RCMLa). We also analyzed protein and peptide binding of Hsp104 variants with amino acid substitutions in the axial channel and determined that inactivation of either pore loop abolished peptide and protein binding. However, stimulation of the ATPase activity in D2 was dependent on an intact loop in D1. These data demonstrate the potential for multiple modes of interaction between Hsp104 and segments of unfolded proteins as they are translocated through the axial channel and provide a hypothetical framework for allosteric triggering of ATP hydrolysis in protein unfolding.
EXPERIMENTAL PROCEDURES
Hsp104 Mutagenesis—All oligonucleotide sequences used are listed in Table 1. The E285A and Y257W mutants were constructed by following the QuikChange (Stratagene) method using pBSKII+ containing the EagI/SacI segment of Hsp104 or from Hsp104Y257A (19) and primers E285A(A) and E285A(B), and primers Y257W(A) and Y257W(B), respectively. The E687A mutant similarly was constructed using pBSKII+ containing the SalI/SpeI segment of Hsp104 using primers E687A(A) and E687A(B). Products were digested with DpnI, transformed into competent Escherichia coli cells, and colonies were screened for the destruction of a StyI-sensitive site for Y257W, the destruction of an NspI-sensitive restriction site for E285A, and the presence of a BtsI-sensitive restriction site for E687A. The expected sequences were confirmed by DNA sequencing. The altered segments were subcloned in place of the corresponding wild-type EagI/SacI segment or SalI/SpeI segment of pPROEX-HT-b 104. The Hsp104Y257A, Hsp104Y662A, and Hsp104Y662W mutants were previously constructed (19). The Hsp104Y257A/Y662W mutant was constructed by subcloning a BamHI/SalI fragment from pLA28SX104Y257A into pPROEX-HT-b 104Y662W. Hsp104ΔN and Hsp104ΔNtrap were constructed by amplification of a segment of DNA using pPROEX-HT-b 104 with primers ΔN(FWD) and ΔN(REV). The PCR product was digested with BamHI/EagI and ligated into pPROEX-HT-b 104 or pPROEX-HT-b 104trap digested with the same enzymes.
TABLE 1.
Primers used for plasmid construction The underlined residues indicate restriction endonuclease sites.
| Primer | Sequence |
|---|---|
| E285A(A) | GTGTTATTCATTGATGCAATTCATATGTTAATGGGTAATGga |
| E285A(B) | CCATTACCCATTAACATATGAATTGCATCAATGAATAACACa |
| E687A(A) | CTCCGTTTTGTTATTCGATGCAGTGGAAAAGGCACATCCTGb |
| E687A(B) | CAGGATGTCGCTTTTCCACTGCATCGAATAACAAAACGGAGb |
| Y257W(A) | GCATTAACCGCAGGTGCTAAGTGGAAAGGTGATTTCGAAGc |
| Y257W(B) | CTTCGAAATCACCTTTCCACTTAGCACCTGCGGTTAATGCc |
| ΔN(FWD) | ACGTGGATCCGGGATGACTAGAATTGACTCTCG |
| ΔN(REV) | ATCATCCGCGGTCTTGCGACGGCGACACC |
| LUC(FWD) | ATGCCATGGAAGACGCCAAAAACATAAAGAAAGGC |
| LUC(REV) | ACTGGATCCCAATTTGGACTTTCCGCCCTTCTTGGC |
| p370(A) | GATCCAAATTGTCTTTCGACGATGTTTTTGAAAGAGAGTATGCTTGAC |
| p370(B) | TCGAGTCAAGCATACTCTCTTTCAAAAACATCGTCGAAAGACAATTTG |
| p530(A) | GATCCAATGATTTTCAGGAGCAACAAGAACAAGCAGCTCCAGAATGAC |
| p530(B) | TCGAGTCATTCTGGAGCTGCTTGTTCTTGTTGCTCCTGAAAATCATTG |
| pSGG(A) | GATCCTCCGGAGGTTCAGGCGGTAGCGGTGGCAGTGGCGGGTCTTGAC |
| pSGG(B) | TCGAGTCAAGACCCGCCACTGCCACCGCTACCGCCTGAACCTCCGGAG |
Bold indicates codon 285
Bold indicates codon 687
Bold indicates codon 257
Construction of Firefly Luciferase-peptide Fusion Constructs—A segment of DNA was amplified from pPROEX-LUC, containing the WT FFL open reading frame and lacking a functional peroxisomal targeting signal, using the LUC(FWD) and LUC(REV) primers and digested with NcoI. The overhanging sequence was filled in using the large subunit of DNA polymerase I (Klenow) followed by BamHI digestion and ligation to BamHI-digested pBSKII+. The sequence was confirmed by DNA sequencing. The NcoI/BamHI fragment was then subcloned into p416Gal1 (p416Gal1-LUC) for expression in yeast. Cartridge-purified oligonucleotide pairs encoding 14-mer peptides (p370(A), p370(B), p530(A), p530(B), pSGG(A), and pSGG(B)) at a concentration of 5 nm in 10 mm Tris-HCl, pH 8, 50 mm NaCl, 1 mm EDTA, pH 8, were phosphorylated using polynucleotide kinase, annealed by heating to 95 °C, and slowly cooling to 25 °C (Δ0.1 °C/5 s), digested with BamHI/XhoI, and inserted into p416Gal1 LUC digested with the same enzymes. Correct insertion was confirmed by sequencing. For recombinant production of FFL fusion proteins, PacI/XhoI segments from p416Gal1-LUC series constructs were subcloned into pPROEX-LUC.
Protein Purification—All Hsp104 variants were expressed and purified as described elsewhere (19). Ydj1 was purified as described previously (30). For purification of recombinant Ssa1, a Saccharomyces cerevisiae strain (SSA1, ssa2, ssa3, ssa4, and pCAUHSEM-SSA1) was grown at 30 °C to mid-log phase in YP containing 2% glucose. The culture was then supplemented with 0.1 volume of 10× YP (1% (w/v) yeast extract, 2% (w/v) peptone), 2% glucose, and 100 μm CuSO4, and the cells were allowed to induce overnight. Ssa1 was then purified essentially as described elsewhere (30).
For expression and purification of FFL and mutant variants, plasmids were transformed into BL21Codon plus cells, and expression of N-terminal poly-histidine-tagged FFL was induced in mid-log phase with 100 μm isopropyl 1-thio-β-d-galactopyranoside at 18 °C overnight. Harvested cells were resuspended in 20 mm Tris, pH 8, 400 mm NaCl, 10 mm imidazole, and 1.4 mm β-mercaptoethanol and lysed by French press. Poly-histidine-tagged FFL was isolated by chromatography on nickel-nitrilotriacetic acid (Qiagen). Pooled peak fractions were diluted to 2 mg/ml, dialyzed twice against 20 mm Tris, pH 8, 50 mm NaCl, 1.4 mm β-mercaptoethanol, and 10% glycerol, and applied to anion exchange chromatography. Peak fractions were dialyzed twice against 50 mm Tris, pH 8, 150 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol, 0.8 m ammonium sulfate, and 2% glycerol, and frozen at -80 °C. Protein concentrations were determined using the Bio-Rad Assay Reagent with bovine serum albumin as a standard.
Peptide Synthesis—Peptides arrays were produced by spot synthesis on cellulose membranes according to the manufacturer's directions (Intavis, Germany). Soluble peptides were synthesized at the Advanced Protein Technology Center (Hospital for Sick Children, Toronto, Canada). Stock peptide solutions were made freshly by resuspending to 1 mm in sterile water. Concentrations were determined by measuring absorbance at 280 nm or using the Bio-Rad Assay Reagent with bovine serum albumin as a standard.
Hsp104 Binding to Peptide Arrays—Arrays were blocked in 1× Blocking Solution (Sigma-Aldrich) diluted in binding buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 10 mm MgCl2, 1 mm dithiothreitol), rinsed three times in binding buffer, and overlaid with 35 nm Hsp104trap in the presence of 2 mm ATP for 1 h at room temperature. Unbound Hsp104 was removed by extensive washing in binding buffer containing ATP. Bound protein was then transferred to polyvinylidene difluoride using a semidry blotter, and Hsp104 was detected with a rabbit polyclonal antibody. Immunoreactive spots were detected by enhanced chemiluminescence (Amersham Biosciences) and recorded on a Versadoc imaging system (Bio-Rad). Spot density was determined using IP Lab Gel 2.0. The frequency of amino acid occurrence was calculated as follows.
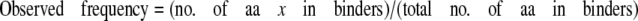 |
(Eq. 1) |
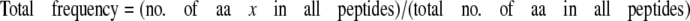 |
(Eq. 2) |
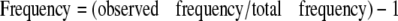 |
(Eq. 3) |
A poly-l-lysine spot on each array was used as an internal positive control for Hsp104 binding and as a standard to compare spot intensities between blots.
Fluorescein Labeling of Reduced α-Lactalbumin—Reduced carboxymethylated α-lactalbumin (RCMLa, Sigma) labeling with fluorescein isothiocyanate (Invitrogen) was performed according to the manufacturer's directions. The labeled protein was purified on a Sephadex G-25 column (Amersham Biosciences) equilibrated with 20 mm sodium phosphate, pH 7.5. Peak fractions were pooled, filtered, and stored at 4 °C in the dark until use.
Fluorescence Spectroscopy—Nucleotide binding measured by changes in Trp fluorescence was performed as previously described (19). All solutions were filtered (0.22 μm) or centrifuged (16,000 × g for 10 min) to remove particulate matter. To measure peptide binding, fluorescence of 0.6 μm Hsp104 containing 2 mm nucleotide was measured at 352 nm at 25 °C using a Spex Fluorolog-3 (Jobin-Yvon), with an excitation wavelength of 295 nm and a 5 nm bandpass. Peptides were titrated from a 100 μm stock solution. Each sample was stirred for 5 min before reading. Data were fitted to a single-site saturation equation for binding using MacCurveFit.
Fluorescence anisotropy was measured as previously described (31) in reaction buffer (20 mm HEPES KOH, pH 7.5, 150 mm NaCl, 10 mm MgCl2, and 1.4 mm β-mercaptoethanol) with several exceptions. 0.6 μm Hsp104trap was incubated with or without 2 mm nucleotide at 25 °C for 5 min. For inhibition of fluorescein-labeled RCMLa (fRCMLa) binding to Hsp104, competitors were added to a solution containing Hsp104 and ATP and incubated for 10 min, and reactions were initiated by the addition of fRCMLa to 0.06 μm. The fraction of fRCMLa bound to Hsp104 was calculated using Equation 4,
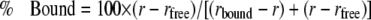 |
(Eq. 4) |
where r represents anisotropy. For competition of fRCMLa binding post-Hsp104-fRCMLa complex formation, fRCMLa was added to initiate the binding reaction, and upon completion of the reaction, competitors were added to 9 μm.
Refolding of Denatured Aggregated Luciferase—In vivo and in vitro refolding of FFL was performed as described elsewhere (32). In vitro refolding reactions were supplemented with 100 μm soluble peptides.
Luciferase Aggregation Assay—Experiments were performed as described elsewhere (33) with several modifications. FFL was thermally aggregated at 0.2 μm in a polystyrene 96-well flat-bottom plate (Sarstedt, Germany) at 42 °C in reaction buffer supplemented with 5 mm ATP in the presence or absence of 0.8 μm Ssa1 and 1.6 μm Ydj1. Rates of FFL aggregation were determined by monitoring increases in light scattering using a SpectraMax 340PC384 microplate reader (Molecular Devices) at 370 nm.
ATPase Activity—A coupled enzymatic spectrophotometric assay in combination with an ATP-regenerating system (34) was used to monitor ATP hydrolysis by Hsp104. All reagents were purchased from Sigma-Aldrich unless otherwise indicated. Reactions were carried out in reaction buffer containing 3 mm phosphoenolpyruvate, 0.23 mm NADH (Bioshop, Canada), 70 units/ml pyruvate kinase, and 100 units/ml l-lactate dehydrogenase (both obtained from rabbit muscle), 2 mm ATP, and 0.2 μm Hsp104. Assays were performed in a polystyrene 96-well flat-bottom plate using a SpectraMax 340PC384 microplate reader (Molecular Devices) at 30 °C monitoring NADH oxidation at 340 nm. The ATPase rate was calculated from the slope dA340 nm/dt using a molar extinction coefficient for NADH of ε340 nm = 6200 m-1cm-1. Data were fitted to either a line or a rectangular hyperbola.
RESULTS
Screen for Hsp104-interacting Peptides—We initiated our search for Hsp104-interacting peptides by screening solid-phase arrays of peptides corresponding to overlapping 13-mer segments of a variety of proteins. Array membranes were incubated with an Hsp104 “trap” mutant (E285A/E687A, Hsp104trap; see Fig. 1A for a schematic guide to Hsp104 domains and residues relevant to this work) that binds but does not hydrolyze ATP (35). After electrophoretic transfer of bound proteins, Hsp104 was detected with a polyclonal antibody. Strong Hsp104-binding peptides were defined as peptides in the 95th percentile by normalized spot intensity (156 of 3908 peptides). The relative fractional occurrence of each amino acid in the strongest binders against the natural occurrence of all 20 amino acids in all peptides was determined (Fig. 1B). We found that Hsp104-binding peptides were enriched in aromatic residues (phenylalanine and tyrosine) and charged residues, specifically lysine, asparagine, and aspartic acid. Serine, glycine, proline, and tryptophan were under-represented in these peptides. The abundances of cysteine and methionine residues on the arrays were too low to be considered statistically significant.
FIGURE 1.

Hsp104 binding to peptide arrays. A, the primary sequence elements of Hsp104. NTD, N-terminal domain; D1, AAA1+ module; CCD, coiled-coil domain; D2, AAA2+ module; CTD, C-terminal domain; A, Walker A; B, Walker B. B, frequency of amino acid occurrence in strong Hsp104-binding peptides. C, raw luminescence data from a 13-mer peptide array derived from the S. cerevisiae Sup35 GTPase domain. Amino acid position of the starting peptide in each row is indicated on the left. *, the end of the Sup35 sequence. D, ribbon diagram of homology model of the GTPase domain of S. cerevisiae Sup35 created by Swiss-Model (61) and based on the crystal structure of S. pombe Sup35 (1R5B) (36). Hsp104-binding peptides are colored by accessibility on a linear gradient (yellow = accessible, blue = buried) using Swiss-Pdb viewer (62) and are space-filled. The numbers correspond to amino acid number in Fig. 1C. The dagger indicates that the structure has been rotated 180° about the vertical axis.
Molecular chaperones are thought to be able to discriminate between folded and unfolded proteins by the high degree of exposure of hydrophobic residues on the surface of misfolded proteins compared with their native conformers. To provide insight into the location of Hsp104-binding peptides within a natively folded protein, we used binding data from a peptide array corresponding to the primary sequence of the globular domain of Saccharomyces cerevisiae Sup35 (Fig. 1C) and mapped them onto a model based on the crystal structure of the Schizosaccharomyces pombe protein (36). Analysis of the solvent accessibility of these peptides indicated that they were generally buried in the interior of the folded protein (Fig. 1C) consistent with their generally high content of hydrophobic amino acid residues. However, because further studies on peptide binding to Hsp104 in solution would be dependent on the solubility of peptides over a broad range of concentrations, we focused on those array peptides containing hydrophobic amino acids intermixed with charged or polar residues.
Peptides Can Enhance Refolding of Aggregated Protein—Other Hsp100s apparently initiate unfolding by binding to specific peptide sequences. For example, the SsrA tag appended onto the C terminus of GFP is sufficient to direct the degradation of GFP by the ClpXP protease (37). However, peptides selected for their ClpX binding properties from arrays conferred ClpX binding to a GFP peptide fusion protein but failed to promote GFP degradation in the presence of ClpP (38). This result could represent the manifestation of the formal possibility that some peptides on arrays could interact with the probe protein in an adventitious manner. For example, peptides could bind to the outer surfaces of the chaperone as opposed to within the axial channel where substrate processing most likely occurs.
We therefore adopted a functional approach to test whether candidate peptides could enhance the refolding of aggregated FFL, a robust model refolding substrate for Hsp104 in vivo (32, 39) and in vitro (2). In preliminary experiments candidate peptides were fused to FFL as C-terminal extensions and expressed in yeast. None of the peptides, that failed to bind Hsp104 on solid phase arrays and were incorporated into these experiments as negative controls, influenced FFL-peptide fusion protein refolding following thermal denaturation. However, some but not all peptides that were judged to be strong Hsp104-binders on solid phase arrays enhanced the recovery of thermally denatured FFL in vivo (data not shown).
To more rigorously determine the influence of peptide extensions on FFL refolding, two peptides that both bound Hsp104 on arrays and enhanced in vivo refolding of FFL, p370 (KLSFDDVFEREYA) and p530 (NDFQEQQEQAAPE), as well as a non-binding control peptide pSGG (SGGSGGSGGSGGS), were further tested in in vitro refolding reactions using Hsp104 along with the Hsp70/40 chaperones Ssa1 and Ydj1 (2). FFL-pSGG was refolded with the same efficiency as FFL lacking a peptide extension (Fig. 2A). Fusion of p530 to FFL modestly enhanced the refolding yield, whereas FFL-p370 was refolded completely. These results are consistent with the notion that Hsp104-binding peptides confer an additional element that enhances the recognition or processing of FFL that is not present in FFL lacking a peptide extension.
FIGURE 2.

Hsp104-dependent refolding and interaction with aggregated recombinant FFL-peptide fusion proteins. A, urea-denatured and aggregated FFL variants were incubated with Hsp104, Ssa1, and Ydj1 at 30 °C, and refolding was monitored. Error bars indicate the standard deviation of three independent experiments.B, FFL variants were thermally aggregated at 42 °C in the absence (black, -) or presence (gray, +) of Ssa1 and Ydj1. Turbidity at 370 nm was monitored.
However, it is possible that enhanced refolding of FFL-peptide fusions could be attributable to differences in the aggregation characteristics or in the ability of fusion proteins to interact with Hsp70/Hsp40 chaperones. To test this, FFL and the extended variants were heat-denatured under conditions where aggregation, measured by light scattering, was partially suppressed by the Hsp70/Hsp40 in the presence of ATP (33). The aggregation of FFL and FFL-p370 in the absence of chaperones and the degree of aggregation suppression in the presence of Hsp70/40 were not different (Fig. 2B). Addition of p530 and pSGG as C-terminal extensions on FFL modestly improved the Hsp70/40-dependent suppression of aggregation. However, because these differences did not correlate with enhanced refolding from the aggregated state, we conclude that peptide-mediated enhancement of refolding by peptide extension is primarily Hsp104-dependent.
Distinct Peptide Binding Sites in the First and Second AAA+ Modules—The axial channel of Hsp100s (12, 14) features flexible loops that govern the aperture of the pore. The position of these loops within the axial is controlled by nucleotide binding, and previously we exploited this property to measure nucleotide binding to D2 in a mutant Hsp104 containing a unique Trp substitution for a conserved Tyr residue on the 661GYVG664 D2 loop (19). In this work, we extended these measurements using Hsp104Y257W containing an analogous Trp residue on the 256KYKG259 D1 loop. Fluorescence of the Y257W probe was quenched in response to nucleotide binding (Fig. 3A). Dissociation constants for ATP and ADP binding to D1 (Table 2) were significantly higher than those previously reported for D2 indicating that indeed Hsp104Y257W and Hsp104Y662W independently measure nucleotide binding to each module. The lower affinity for nucleotide binding at D1 is consistent with the relatively high Km observed for ATP hydrolysis by D1 (40).
FIGURE 3.

Peptide binding to NBD1 and NBD2. A, fluorescence of single Trp mutant Hsp104Y257W titrated with increasing concentrations of ADP (left) or ATP (right). Each curve is derived from the combined data from three independent titrations. Error bars indicate the standard deviation at each point. Peptide binding to Hsp104Y257W (B) and Hsp104Y662W (C) was measured with 2 mm AMP-PNP (left) or ADP (right), and increasing concentrations of p370 (filled circles) or pSGG (open circles). Excitation and emission monochromators were set to 295 nm and 352 nm, respectively. Each data point is the mean of three independent experiments, and error bars indicate the standard deviation. Data were fitted to an equation for single-site saturated binding.
TABLE 2.
Summary of nucleotide-dependent fluorescence changes in single Trp-Hsp104 All data represent the mean of three independent trials ± S.D.
The flexible loops in the Hsp100 axial channel appear to play a role in binding substrates and in their subsequent processing by translocation through the axial channel (15–18). We hypothesized that peptide binding may also influence the conformation of residues in the axial channel of Hsp104 and therefore applied the site-specific probes to investigate peptide binding to Hsp104. The fluorescence of Hsp104Y257W in the D1 in the presence of AMP-PNP or ADP was quenched upon titration with p370 (Fig. 3B). Titration of the non-binding control peptide pSGG did not significantly alter the fluorescence of Hsp104Y257W. Calculated dissociation constants (Table 3) indicated that p370 binds with roughly the same affinity to D1 irrespective of the nucleotide bound.
TABLE 3.
Summary of fluorescence changes upon titration of p370 in single Trp-Hsp104 All data represent the mean of three independent trials ± S.D.
| Y257W | Y662W | E645K/Y662W | |
|---|---|---|---|
| AMP-PNP (2 mm) | |||
| Kd (nm) | 84 ± 1 | 300 ± 100 | 190 ± 30 |
| ΔF (1 – F/Fo)a | 0.106 ± 0.003 | 0.116 ± 0.006 | 0.133 ± 0.005 |
| ADP (2 mm) | |||
| Kd (nm) | 96 ± 3 | 160 ± 20 | not detected |
| ΔF (1 – F/Fo)a | 0.137 ± 0.007 | 0.060 ± 0.002 |
Percent change in fluorescence from peptide-free (Fo) to peptide-saturated protein
Parallel experiments with Hsp104Y662W indicated that titration of p370 into AMP-PNP or ADP-bound Hsp104 also quenched Trp fluorescence when the probe is incorporated into the D2 loop (Fig. 3C). No change in fluorescence was observed when Hsp104Y662W was titrated with pSGG in either nucleotide-bound state. The binding affinity of p370 to D2 was higher in the ADP-bound state when compared with the AMP-PNP-bound state. The distinct binding affinities for p370 to D1 compared with D2 suggest the existence of at least two peptide binding sites.
Surprisingly, even though p530 binds to Hsp104 on arrays and enhances refolding of FFL in vivo and in vitro, titration of p530 into solutions containing either Hsp104Y257W or Hsp104Y662W with AMP-PNP or ADP did not significantly change the fluorescence intensity of the probes (data not shown). This finding does not exclude the possibility that p530 is capable of binding to Hsp104 at a site that does not influence the fluorescence properties of the probes used in this analysis and thereby enhances the refolding of a FFL-p530 fusion protein.
Peptide Competition for Unfolded Protein Binding to Hsp104—We hypothesized that, if p370 binding represents a mechanistically relevant interaction with Hsp104, it should also compete for one or more binding sites used by unfolded proteins. To test this idea, soluble p370, p530, and pSGG were used as competitors for Hsp104 binding to RCMLa, an unfolded and soluble monomeric protein that has been used as a model for chaperone binding (30, 41–43), including Hsp104 where RCMLa binding is ATP-dependent (31). Preincubation of Hsp104trap and ATP with 2 μm p370 resulted in the inhibition of approximately half the binding of Hsp104 to fRCMLa (Fig. 4A). Preincubation with p530 at the same concentration had no inhibitory binding effect. An addition of 5 μm p370 completely abolished fRCMLa, whereas at the same concentration the control peptide, pSGG, had no effect. Preincubation of Hsp104trap-ATP with varying concentrations of p370 and fitting the resulting curve with an equation describing a single-site competition reported an IC50 of 2.1 μm (Fig. 4B).
FIGURE 4.

p370 competition with fRCMLa for binding to Hsp104. A, Hsp104trap was preincubated with ATP for 10 min. Subsequently, peptides at various concentrations were added and incubated for 5 min. fRCMLa binding was initiated by the addition of fRCMLa. Fluorescence anisotropy was measured with monochromators set to 494 nm (excitation) and 515 nm (emission). Experiments were performed in triplicate, and one representative data set is shown. B, the experiment was performed as described in A. p370 inhibited Hsp104trap from binding to fRCMLa with an IC50 of 2.1 ± 0.3 μm. Error bars indicate the standard deviation of three measurements. C, unlabeled RCMLa (gray circles), pSGG (empty diamonds), or p370 (filled diamonds) were added after Hsp104trap-fRCMLa-ATP complex formation, and the change in anisotropy was monitored. Data were fitted to an equation describing a three-component exponential decay process. D, refolding of denatured aggregated firefly luciferase by purified recombinant Hsp104, Ssa1, and Ydj1 in the absence or presence of peptide. Results were normalized to the refolding yield obtained in a refolding reaction in the absence of soluble peptide. Error bars indicate the standard deviation of three independent measurements.
Although it was previously reported that binding of fRCMLa to ATP-bound Hsp104trap was irreversible (31), we found that under our conditions, incubation of unlabeled RCMLa with preformed complexes resulted in exchange over time (Fig. 4C). Addition of p370 could also disrupt preformed fRCMLa-Hsp104trap complexes with a time course that nearly overlaps with that of RCMLa. Fitting these curves to equations describing three-component exponential decay could distinguish a rapid exchange component with a half-life (t½) of 1.2 min, a slower one with a t½ of 12 min, and a very slow component with a t½ of 20 h for RCMLa competition, and somewhat faster exchange by p370 with calculated t½ values of 1.3 min, 8.3 min, and 12 h.
Together these results indicate that at least one peptide, p370, binds to Hsp104 and competes with fRCMLa for binding. We next asked if p370 could inhibit a refolding reaction where ATP is constantly being hydrolyzed and exchanged. Addition of p370 or pSGG to high concentrations, even as high as 100 μm, did not reduce the yield of refolded FFL (Fig. 4D), suggesting that p370 may be readily displaced under conditions that permit turnover of ATP. Alternatively, p370 may be strongly outcompeted for binding during the processing of protein extracted from aggregates.
The N-terminal Domain Is Dispensable for Protein/Peptide Binding to Hsp104—We next set out to determine where in the Hsp104 molecule competition between protein and peptide binding occurs. To investigate the role of the Hsp104 N-terminal domain (NTD) we constructed an Hsp104 mutant lacking amino acids 1–152 (Hsp104ΔN). This truncation mutant was fully functional in vivo and in vitro in refolding FFL (Fig. 5, A and B). Although the NTD of Hsp104 is dispensable for thermotolerance and prion maintenance in yeast (44), some evidence suggests that the NTD of ClpB modulates its interaction with protein aggregates (45–47). To examine the role of the NTD in peptide and protein binding, we constructed a mutant that combined the N-terminal deletion with the Walker B E285A/E687A substitutions (Hsp104ΔNtrap). Hsp104ΔNtrap in the ATP-bound form was able to bind fRCMLa, and this binding closely approximated that of the full-length protein (Fig. 5C). p370 inhibited fRCMLa binding to Hsp104ΔNtrap with an IC50 of 1.3 μm, whereas excess pSGG had no inhibitory effect (Fig. 5D). Because the salient features of protein binding and peptide competition are preserved in the absence of the Hsp104 NTD, this indicates that this domain is dispensable for the formation of stable substrate-chaperone complexes.
FIGURE 5.

The N-terminal domain of Hsp104 is dispensable for protein and peptide binding. A, in vivo refolding of aggregated FFL. Cells were cultured in galactose to induce the expression of Hsp104 and FFL. Log phase cells were heated to 44 °C for 20 min to inactivate FFL. The recovery of FFL activity was normalized to the activity measured in each culture immediately before heat shock. One representative data set is shown. B, in vitro refolding of denatured aggregated firefly luciferase by purified recombinant Hsp104 without (-) and with (+) purified Ssa1 and Ydj1. Results were normalized to the refolding yield obtained in a complete refolding reaction containing wild-type Hsp104. Error bars indicate the standard deviation of three independent measurements. C, Hsp104trap (squares) and Hsp104ΔNtrap (diamonds) were incubated with fRCMLa, and the reaction was initiated by the addition of ATP (filled) or ADP (empty). Fluorescence anisotropy was measured as described in Fig. 4A. Experiments were performed in triplicate, and one representative data set is shown. D, inhibition of fRCMLa binding to Hsp104ΔNtrap by preincubation with peptides. The IC50 for p370 inhibition was 1.3 ± 0.05 μm.
Peptide Stimulation of ATP Hydrolysis—Recently, work by others has demonstrated that RCMLa strongly stimulates Hsp104 ATPase activity specifically in D2 (48). Given this observation, we next investigated whether p370 could substitute for unfolded protein in this stimulation. We constructed Hsp104 variants with Walker B substitutions that prevent ATP hydrolysis in Hsp104Trap (E285A or E687A) but separately to establish the ATPase activity of D1 and D2 independently. Consistent with previous observations with RCMLa, only the Walker B mutation in D2 abolished the stimulation of ATPase activity by p370 (Fig. 6A) indicating that p370 stimulates ATP hydrolysis in D2. As expected, addition of p530 or the non-binder pSGG to either mutant did not stimulate ATP hydrolysis (data not shown).
FIGURE 6.

Stimulation of ATP hydrolysis by peptide and protein. ATPase activity was measured at 30 °C in a reaction containing Hsp104, ATP, and an ATP-regeneration system in the presence of p370 or RCMLa. ATPase -fold stimulation was normalized to the rate of ATP hydrolysis in the absence of peptide or protein. Each data point is the mean of three independent experiments, and error bars represent standard deviations. Data were fitted linearly or to a rectangular hyperbola. Stimulation of ATP hydrolysis of Hsp104E285A (filled) and Hsp104E687A (open) by p370 (A), and of Hsp104WT (squares), Hsp104Y257A (filled circles), and Hsp104Y662A (open circles) by RCMLa (B) and p370 (C).
Because, in this work, we infer the existence of p370 binding sites in both D1 and D2 (Fig. 3, B and C), we next asked if an intact D2 loop is required to trigger the ATPase activity of D2. Surprisingly, the stimulation of Hsp104Y662A by p370 was not significantly different from that of the wild-type protein, whereas Hsp104Y257A was barely responsive to the addition of peptide (Fig. 6B). The profound defect in Hsp104Y257A stimulation was also observed in the presence of RCMLa (Fig. 6C). Taken together, these data suggest that an intact D1 loop in Hsp104 is required to activate ATP hydrolysis in D2 and that the D2 loop has no significant role in this stimulation.
Perturbation of the Axial Channel Alters Protein and Peptide Binding—Because an intact D1 loop is required for protein and peptide stimulation of Hsp104 ATPase activity while the D2 loop is dispensable, we predicted that only the D1 loop is critical for protein and peptide binding. Surprisingly, binding of fRCMLa to Hsp104Y257A or Hsp104Y662A in the presence of ATPγS was not detected suggesting that stable fRCMLa binding requires both loops (Fig. 7A).
FIGURE 7.

Perturbation of the axial channel alters protein and peptide binding. A, binding of fRCMLa to Hsp104WT, Hsp104Y257A, and Hsp104Y662A. Hsp104 was incubated with fRCMLa, and binding was initiated by the addition of ATPγS. Experiments were performed in triplicate, and one representative data set is shown. B, fluorescence quenching of Hsp104Y257A/Y662W (left), and Hsp104Y257W/Y662A (right), in response to p370 titration was monitored in the presence of AMP-PNP (closed circles) or ADP (open circles). Each data point is the mean of three independent experiments, and error bars indicate standard deviations. C, refolding of denatured aggregated firefly luciferase by purified recombinant Hsp104 as described in Fig. 5B.
Because we inferred the existence of two p370 binding sites in D1 and D2, we speculated that, in contrast to protein binding, peptide binding to D1 and D2 might occur independently. To test this we constructed a mutant of Hsp104 with the D1 loop inactivated by the Y257A substitution and incorporating the Trp probe into the D2 loop (Hsp104Y257A/Y662W). We also constructed the inverse mutant with the D2 loop inactivated and the Trp probe incorporated into the D1 loop (Hsp104Y257W/Y662A). No changes in fluorescence of Hsp104Y257A/Y662W or Hsp104Y257W/Y662A with either AMP-PNP or ADP were observed upon titration with p370 (Fig. 7B). Together, these observations suggest that stable protein and peptide binding requires intact D1 and D2 loops and that these loop function in an interdependent manner.
We previously observed that a non-conservative substitution in the D2 loop (Y662A) abolished Hsp104-mediated thermotolerance, whereas the analogous substitution in the D1 loop (Y257A) exhibited an intermediate loss of function (19). Given our observation that the D1 loop is critical for stable protein and peptide binding, we re-tested the activity of Hsp104Y662A in an in vitro refolding assay. Consistent with the protein and peptide binding data, we found that the refolding activity of Hsp104Y257A is severely impaired in vitro and only slightly more active than Hsp104Y662A (Fig. 7C).
DISCUSSION
Binding of Hsp104 to solid phase peptides supports the hypothesis that Hsp104 distinguishes misfolded proteins from their correctly folded conformers based on the exposure of hydrophobic amino acid side chains. First, the composition of Hsp104-binding peptides is enriched in certain hydrophobic residues, including Phe, Tyr, and Leu. Second, when the positions of Hsp104-interacting peptides from the globular domain of Sup35 are mapped onto a three-dimensional model of the domain, the peptides that display Hsp104 binding correspond to polypeptide segments that are only solvent-exposed at their ends in the folded protein. Although the exposure of these polypeptide segments in denatured conformers may be important for the ability of Hsp104 to discriminate between native and non-native protein complexes, for practical reasons the poor solubility of hydrophobic peptides limits their utility for exploration of the peptide-binding properties of Hsp104. In preliminary trials, hydrophobic peptides solubilized by polyionic tags (49) also strongly stimulate the ATPase activity of Hsp104.4
Nonetheless, soluble peptides that include hydrophobic as well as charged and polar amino acids appear to be appropriate substrate mimics in most respects. The enhanced refolding of the FFL-p370 fusion protein suggests that the p370 moiety provides an additional determinant that is not present in FFL lacking the extension and which promotes FFL extraction from aggregates and unfolding by Hsp104. Furthermore, p370 as a soluble peptide recapitulates the properties of an unfolded protein in that it competes for binding of the model unfolded protein RCMLa and displays a similar ability to stimulate the D2 ATPase activity of Hsp104. Neither unfolded protein binding nor the ability of peptide to compete is dependent on the N-terminal domain of Hsp104, suggesting that these interactions occur primarily in the axial channel formed by the AAA+ modules of Hsp104.
A common feature of chaperones is the cycling between high and low affinity states for substrate binding based on conformational changes driven by ATP hydrolysis. In other Hsp100s, including ClpA (50), ClpX (51), and ClpB (14, 35), the ATP-bound chaperone undergoes stable substrate binding. This is consistent with the formation of a stable RCMLa-Hsp104 complex with ATP or an ATP analogue bound but not ADP (this work and Ref. 31). Based on these observations we anticipated that, in parallel to the ATP-dependent formation of a stable unfolded protein-Hsp104 complex, peptide binding in D1 or D2 or both would exhibit a high affinity state with ATP bound and that in the ADP-bound state the affinity of peptide binding sites would be either greatly diminished or eliminated. In contrast we saw either no change peptide binding affinity in D1 or even an increase in affinity in the D2 binding site between the ATP and ADP states. We do not know at the present time whether this anomaly is a specific characteristic of p370 or a general feature of peptide binding that is distinct from protein binding.
A Model of the Hsp104 Reaction Cycle—Based on our own observations and those of others, we propose a model for protein unfolding and translocation by Hsp104 consisting of four distinct states (Fig. 8): the idling state, in which Hsp104 is poised to interact with incoming substrate; a primed state, in which ATPase activity is stimulated by an initial unstable interaction with a polypeptide at D1; a processing state, in which both D1 and D2 participate in binding and translocation; and a prerelease state, in which the polypeptide has traversed the axial channel at D1.
FIGURE 8.

A model of Hsp104-mediated unfolding and translocation. The substrate unfolding and translocation mechanism of Hsp104 consists of four distinct stages. In the idling state ATP is slowly turned over in D1 and hydrolytic activity at D2 is essentially quiescent. Upon polypeptide interaction with D1 in the primed complex, ATP hydrolysis at D2 is allosterically enhanced. Conversion of ATP to ADP at D2 in turn stimulates ATP hydrolysis at D1. The reversibility of this interaction indicates that it is unstable. Slowing of hydrolysis at D1 by the inclusion of slowly hydrolysable ATP analogue may enhance the formation of the primed complex. If a segment of polypeptide is sufficiently long to span the distance separating the D1 and D2 loops, the substrate becomes stably associated in the processing complex. The partial remodeling of aggregated proteins by Hsp70/40 chaperones may be required to generate extended polypeptide segments capable of efficiently forming the processing complex. In the prerelease complex the translocating polypeptide is released from D1 returning D2, and in turn, D1 to a less active state similar to the idling state but with the last segment of the polypeptide associated with D2. The polypeptide is either spontaneously released or is ejected from Hsp104 by the formation of a new primed complex. See “Discussion” for additional detail.
The Idling State—We define an Hsp104 molecule not engaged by polypeptide and hydrolyzing ATP at a basal rate to be in an idling state. In the absence of ligand, ATP hydrolysis at D1 is relatively slow at ∼20 min-1 (40) while hydrolysis at D2 is barely detectable. The low affinity of D1 for ADP (Fig. 3A) suggests that this domain is predominantly ATP-bound in the idling state. This characteristic may support the initial interaction with substrate and is consistent with the observation that RCMLa binding is not observed when Hsp104 is in the ADP-bound state (31, 48).
The Primed State—In other Hsp100s, substrates are translocated along the axial channel and extruded into the chamber of an associated protease for degradation (7, 9, 11, 16, 24, 37). Indeed, an Hsp104 mutant that interacts with ClpP is capable of translocating substrates into ClpP suggesting a directional mechanism for substrate binding and processing along the channel from D1 to D2 (52). An initial interaction with the D1 loop is consistent with experiments in which a ClpB-binding peptide can be cross-linked to the D1 loop of ClpB (53). In our experiments, stable protein and peptide binding required both D1 and D2 loops, whereas the activation of ATP hydrolysis at D2 required only an intact D1 loop. In our model, we call this initial D1 loop-dependent interaction the “primed” state. Previous work has suggested that ADP binding to D2 activates hydrolysis at D1 (40), and it is reasonable to propose that in the primed state, rapid conversion of ATP to ADP at D2 will result in simultaneous activation of ATP hydrolysis at D1.
Under standard conditions for Hsp104-dependent refolding, it is possible that the Hsp70/40 chaperones act at rate-limiting step. It has been recently suggested that although the action of Hsp70/40 on aggregates may not efficiently release free polypeptides, it can displace polypeptide segments from the surface of aggregates (26), and these may act at the formation of the primed state by presenting polypeptide segments in partially disaggregated proteins. When Hsp104-dependent refolding occurs under conditions that do not require Hsp70/40 (29), we propose that diminishing the hydrolysis of ATP at some NBDs using mixtures of ATP and ATPγS or slowing of ATP hydrolysis at D2 by mutation, may promote the formation of the primed state by prolonging a transient state in the idling complex, whichpotentiatessubstrateinteraction.
The Processing State—Activation of ATP hydrolysis in the primed state serves to capture a substrate at D1 driving it deeper into the axial. Because stable binding of RCMLa was abolished in the D2 loop mutant Hsp104Y662A, we propose that only when a substrate encounters the D2 loop, does it become stably associated with Hsp104 and that the interdependent action of D1 and D2 are required for full translocation. The slow formation of a stable RCMLa-Hsp104 complex (∼10 min) under conditions that prevent ATP hydrolysis may reflect the time required for a segment of RCMLa to reach the peptide binding site(s) present at D2 through spontaneous oscillation in the channel rather than a process facilitated by ATP hydrolysis-driven motion of the D1 loop. Using the T. thermophilus ClpB crystal structure (54) as a model we estimate the distance between the D1 and D2 loops to be ∼45 Å. Hsp70/40, in addition to promoting the primed state, could, by the same mechanism of partial unfolding of aggregates to expose polypeptide loops or termini, facilitate the formation of the processing state as well and may explain in part why binding of aggregates but not monomeric unfolded proteins to ATP-bound ClpB requires DnaK, DnaJ, and GrpE (27).
As long as there is contact between a substrate and the binding site(s) in D1, the reciprocal allosteric stimulation of ATP hydrolysis in both D1 and D2 will be maintained thus committing the processing complex to rapid unfolding and translocation of the substrate. The ability of Hsp104 to load substrate into ClpP suggests that at least some substrates are fully translocated (52). However, recent evidence obtained with ClpB demonstrated efficient refolding of protein fusions of misfolded and native domains without the unfolding of the folded domain, indicating that full translocation is not obligatory (55). Furthermore, ClpB hexamers are dynamic complexes and exchange subunits on a rapid timescale suggesting that hexamer disassembly may facilitate dissociation of ClpB from very stable aggregates after partial translocation thereby rescuing ClpB from substrate traps (55, 56).
The Prerelease State—Prior to the final release of substrate from the Hsp104 axial channel, the last segment of translocating polypeptide will be associated only with D2 in a complex that we define as the prerelease state. None of our experiments directly addressed how substrates might be released from the prerelease complex. Because a stable complex likely requires simultaneous interaction with both D1 and D2, it is also likely that a polypeptide, interacting with only D2, is released spontaneously. However, our model predicts that the formation of a hybrid state in which D1 interacts with an incoming substrate polypeptide will result in the restimulation of ATP turnover at D2 and thereby trigger efficient ejection of the previous substrate from D2.
Although proteins can be fully threaded through the axial channel of Hsp104, model substrates that are unable to completely traverse the axial channel, because they are fused to a stably folded domain that cannot be unfolded by ClpB, are nonetheless, released, and refolded (55). Subunit exchange experiments indicate that ClpB disassembles and reassembles under processing conditions suggesting an alternative mode of substrate release.
Structural Models of Hsp104—The crystal structure of the Hsp104 hexamer has yet to be determined. However, the structure of the bacterial ortholog ClpB (monomeric) has been solved and used to reconstruct a model of the native hexamer. The reconstructed hexamer describes ClpB as two-tiered, in which the two AAA+ modules in each monomer sit on top of one another. Additionally, the coiled-coil domains emerge from D1 and are displayed on the exterior of the hexamer (54). These features are corroborated by reconstructions of cryoelectron microscopy images of ClpB (14). Notably, a narrow channel penetrates the central axis of the ClpB hexamer. This channel is a common feature of all Hsp100s for which crystal structures are available (12, 13, 57–59). While this work was in progress, a cryoelectron microscopy study of ATPγS-bound Hsp104 (60) revealed a strikingly different picture of Hsp104 structure. In this model, Hsp104 forms a large central cavity up to 78 Å in diameter capped by the Hsp104 N-domains and with the coiled-coil arms intercalating between adjacent subunits where they form part of the walls of the central cavity and disrupt the domain interactions that are typical of all other AAA+ proteins. As this model lacks the narrow axial channel that is present in other Hsp100s, it is challenging to interpret our data in terms of the role of axial loop residues in protein or peptide binding. Additional structural and biochemical data are required to explore and corroborate the exceptional features of this model.
This work was supported in part by the Canadian Institutes for Health Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: aa, amino acid(s); GFP, green fluorescent protein; FFL, firefly luciferase; RCMLa, reduced carboxymethylated lactalbumin; fRCMLa, fluorescein-labeled RCMLa; NTD, N-terminal domain; ATPγS, adenosine 5′-O-(thiotriphosphate).
R. Lum and J. R. Glover, unpublished observation.
References
- 1.Parsell, D. A., Kowal, A. S., Singer, M. A., and Lindquist, S. (1994) Nature 372 475-478 [DOI] [PubMed] [Google Scholar]
- 2.Glover, J. R., and Lindquist, S. (1998) Cell 94 73-82 [DOI] [PubMed] [Google Scholar]
- 3.Chernoff, Y. O., Lindquist, S. L., Ono, B., Inge-Vechtomov, S. G., and Liebman, S. W. (1995) Science 268 880-884 [DOI] [PubMed] [Google Scholar]
- 4.Derkatch, I. L., Bradley, M. E., Zhou, P., Chernoff, Y. O., and Liebman, S. W. (1997) Genetics 147 507-519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moriyama, H., Edskes, H. K., and Wickner, R. B. (2000) Mol. Cell. Biol. 20 8916-8922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tkach, J., and Glover, J. (2006) in Chaperones, pp. 65-90, Springer Berlin, Heidelberg
- 7.Ortega, J., Singh, S. K., Ishikawa, T., Maurizi, M. R., and Steven, A. C. (2000) Mol. Cell 6 1515-1521 [DOI] [PubMed] [Google Scholar]
- 8.Weber-Ban, E. U., Reid, B. G., Miranker, A. D., and Horwich, A. L. (1999) Nature 401 90-93 [DOI] [PubMed] [Google Scholar]
- 9.Singh, S. K., Grimaud, R., Hoskins, J. R., Wickner, S., and Maurizi, M. R. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 8898-8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishikawa, T., Beuron, F., Kessel, M., Wickner, S., Maurizi, M. R., and Steven, A. C. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 4328-4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reid, B. G., Fenton, W. A., Horwich, A. L., and Weber-Ban, E. U. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 3768-3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang, J., Song, J. J., Franklin, M. C., Kamtekar, S., Im, Y. J., Rho, S. H., Seong, I. S., Lee, C. S., Chung, C. H., and Eom, S. H. (2001) Structure 9 177-184 [DOI] [PubMed] [Google Scholar]
- 13.Sousa, M. C., Trame, C. B., Tsuruta, H., Wilbanks, S. M., Reddy, V. S., and McKay, D. B. (2000) Cell 103 633-643 [DOI] [PubMed] [Google Scholar]
- 14.Lee, S., Choi, J. M., and Tsai, F. T. (2007) Mol. Cell 25 261-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinnerwisch, J., Fenton, W. A., Furtak, K. J., Farr, G. W., and Horwich, A. L. (2005) Cell 121 1029-1041 [DOI] [PubMed] [Google Scholar]
- 16.Weibezahn, J., Tessarz, P., Schlieker, C., Zahn, R., Maglica, Z., Lee, S., Zentgraf, H., Weber-Ban, E. U., Dougan, D. A., Tsai, F. T., Mogk, A., and Bukau, B. (2004) Cell 119 653-665 [DOI] [PubMed] [Google Scholar]
- 17.Siddiqui, S. M., Sauer, R. T., and Baker, T. A. (2004) Genes Dev. 18 369-374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park, E., Rho, Y. M., Koh, O. J., Ahn, S. W., Seong, I. S., Song, J. J., Bang, O., Seol, J. H., Wang, J., Eom, S. H., and Chung, C. H. (2005) J. Biol. Chem. 280 22892-22898 [DOI] [PubMed] [Google Scholar]
- 19.Lum, R., Tkach, J. M., Vierling, E., and Glover, J. R. (2004) J. Biol. Chem. 279 29139-29146 [DOI] [PubMed] [Google Scholar]
- 20.Kurahashi, H., and Nakamura, Y. (2007) Mol. Microbiol. 63 1669-1683 [DOI] [PubMed] [Google Scholar]
- 21.Flynn, J. M., Neher, S. B., Kim, Y. I., Sauer, R. T., and Baker, T. A. (2003) Mol. Cell 11 671-683 [DOI] [PubMed] [Google Scholar]
- 22.Gottesman, S., Roche, E., Zhou, Y., and Sauer, R. T. (1998) Genes Dev. 12 1338-1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haebel, P. W., Gutmann, S., and Ban, N. (2004) Curr. Opin. Struct. Biol. 14 58-65 [DOI] [PubMed] [Google Scholar]
- 24.Hoskins, J. R., Kim, S. Y., and Wickner, S. (2000) J. Biol. Chem. 275 35361-35367 [DOI] [PubMed] [Google Scholar]
- 25.Goloubinoff, P., Mogk, A., Zvi, A. P., Tomoyasu, T., and Bukau, B. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 13732-13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zietkiewicz, S., Lewandowska, A., Stocki, P., and Liberek, K. (2006) J. Biol. Chem. 281 7022-7029 [DOI] [PubMed] [Google Scholar]
- 27.Haslberger, T., Weibezahn, J., Zahn, R., Lee, S., Tsai, F. T., Bukau, B., and Mogk, A. (2007) Mol. Cell 25 247-260 [DOI] [PubMed] [Google Scholar]
- 28.Shorter, J., and Lindquist, S. (2006) Mol. Cell 23 425-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doyle, S. M., Shorter, J., Zolkiewski, M., Hoskins, J. R., Lindquist, S., and Wickner, S. (2007) Nat. Struct. Mol. Biol. 14 114-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cyr, D. M., Lu, X., and Douglas, M. G. (1992) J. Biol. Chem. 267 20927-20931 [PubMed] [Google Scholar]
- 31.Bosl, B., Grimminger, V., and Walter, S. (2005) J. Biol. Chem. 280 38170-38176 [DOI] [PubMed] [Google Scholar]
- 32.Tkach, J. M., and Glover, J. R. (2004) J. Biol. Chem. 279 35692-35701 [DOI] [PubMed] [Google Scholar]
- 33.Lu, Z., and Cyr, D. M. (1998) J. Biol. Chem. 273 27824-27830 [DOI] [PubMed] [Google Scholar]
- 34.Norby, J. G. (1988) Methods Enzymol. 156 116-119 [DOI] [PubMed] [Google Scholar]
- 35.Weibezahn, J., Schlieker, C., Bukau, B., and Mogk, A. (2003) J. Biol. Chem. 278 32608-32617 [DOI] [PubMed] [Google Scholar]
- 36.Kong, C., Ito, K., Walsh, M. A., Wada, M., Liu, Y., Kumar, S., Barford, D., Nakamura, Y., and Song, H. (2004) Molecular Cell 14 233-245 [DOI] [PubMed] [Google Scholar]
- 37.Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T., and Baker, T. A. (2000) Mol. Cell 5 639-648 [DOI] [PubMed] [Google Scholar]
- 38.Thibault, G., Yudin, J., Wong, P., Tsitrin, V., Sprangers, R., Zhao, R., and Houry, W. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 17724-17729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singer, M. A., and Lindquist, S. (1998) Mol. Cell 1 639-648 [DOI] [PubMed] [Google Scholar]
- 40.Hattendorf, D. A., and Lindquist, S. L. (2002) EMBO J. 21 12-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayer-Hartl, M. K., Ewbank, J. J., Creighton, T. E., and Hartl, F. U. (1994) EMBO J. 13 3192-3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kern, R. E., Malki, A., Holmgren, A., and Richarme, G. (2003) Biochem. J. 371 965-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scholz, C., Stoller, G., Zarnt, T., Fischer, G., and Schmid, F. X. (1997) EMBO J. 16 54-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hung, G. C., and Masison, D. C. (2006) Genetics 173 611-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka, N., Tani, Y., Hattori, H., Tada, T., and Kunugi, S. (2004) Protein Sci. 13 3214-3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu, Z., Tek, V., Akoev, V., and Zolkiewski, M. (2002) J. Mol. Biol. 321 111-120 [DOI] [PubMed] [Google Scholar]
- 47.Barnett, M. E., Nagy, M., Kedzierska, S., and Zolkiewski, M. (2005) J. Biol. Chem. 280 34940-34945 [DOI] [PubMed] [Google Scholar]
- 48.Schaupp, A., Marcinowski, M., Grimminger, V., Bosl, B., and Walter, S. (2007) J. Mol. Biol. 370 674-686 [DOI] [PubMed] [Google Scholar]
- 49.Cunningham, F., and Deber, C. M. (2007) Methods 41 370-380 [DOI] [PubMed] [Google Scholar]
- 50.Wickner, S., Gottesman, S., Skowyra, D., Hoskins, J., McKenney, K., and Maurizi, M. R. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 12218-12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wawrzynow, A., Wojtkowiak, D., Marszalek, J., Banecki, B., Jonsen, M., Graves, B., Georgopoulos, C., and Zylicz, M. (1995) EMBO J. 14 1867-1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tessarz, P., Mogk, A., and Bukau, B. (2008) Mol. Microbiol. 68 87-97 [DOI] [PubMed] [Google Scholar]
- 53.Schlieker, C., Weibezahn, J., Patzelt, H., Tessarz, P., Strub, C., Zeth, K., Erbse, A., Schneider-Mergener, J., Chin, J. W., Schultz, P. G., Bukau, B., and Mogk, A. (2004) Nat. Struct. Mol. Biol. 11 607-615 [DOI] [PubMed] [Google Scholar]
- 54.Lee, S., Sowa, M. E., Watanabe, Y. H., Sigler, P. B., Chiu, W., Yoshida, M., and Tsai, F. T. (2003) Cell 115 229-240 [DOI] [PubMed] [Google Scholar]
- 55.Haslberger, T., Zdanowicz, A., Brand, I., Kirstein, J., Turgay, K., Mogk, A., and Bukau, B. (2008) Nat. Struct. Mol. Biol. 15 641-650 [DOI] [PubMed] [Google Scholar]
- 56.Werbeck, N. D., Schlee, S., and Reinstein, J. (2008) J. Mol. Biol. 378 178-190 [DOI] [PubMed] [Google Scholar]
- 57.Guo, F., Maurizi, M. R., Esser, L., and Xia, D. (2002) J. Biol. Chem. 277 46743-46752 [DOI] [PubMed] [Google Scholar]
- 58.Bochtler, M., Hartmann, C., Song, H. K., Bourenkov, G. P., Bartunik, H. D., and Huber, R. (2000) Nature 403 800-805 [DOI] [PubMed] [Google Scholar]
- 59.Kim, D. Y., and Kim, K. K. (2003) J. Biol. Chem. 278 50664-50670 [DOI] [PubMed] [Google Scholar]
- 60.Wendler, P., Shorter, J., Plisson, C., Cashikar, A. G., Lindquist, S., and Saibil, H. R. (2007) Cell 131 1366-1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwede, T., Kopp, J., Guex, N., and Peitsch, M. C. (2003) Nucleic Acids Res. 31 3381-3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guex, N., and Peitsch, M. C. (1997) Electrophoresis 18 2714-2723 [DOI] [PubMed] [Google Scholar]