

Abstract
The expression of the elongated fibrinogen γ chain, termed γ′, derives from alternative splicing of mRNA and causes an insertion sequence of 20 amino acids. This insertion domain interacts with the anion-binding exosite (ABE)-II of thrombin. This study investigated whether and how γ′ chain binding to ABE-II affects thrombin interaction with its platelet receptors, i.e. glycoprotein Ibα (GpIbα), protease-activated receptor (PAR) 1, and PAR4. Both synthetic γ′ peptide and fibrinogen fragment D*, containing the elongated γ′ chain, inhibited thrombin-induced platelet aggregation up to 70%, with IC50 values of 42 ± 3.5 and 0.47 ± 0.03 μm, respectively. Solid-phase binding and spectrofluorimetric assays showed that both fragment D* and the synthetic γ′ peptide specifically bind to thrombin ABE-II and competitively inhibit the thrombin binding to GpIbα with a mean Ki ≈ 0.5 and ≈35 μm, respectively. Both these γ′ chain-containing ligands allosterically inhibited thrombin cleavage of a synthetic PAR1 peptide, of native PAR1 molecules on intact platelets, and of the synthetic chromogenic peptide d-Phe-pipecolyl-Arg-p-nitroanilide. PAR4 cleavage was unaffected. In summary, fibrinogen γ′ chain binds with high affinity to thrombin and inhibits with combined mechanisms the platelet response to thrombin. Thus, its variations in vivo may affect the hemostatic balance in arterial circulation.
Fibrinogen is a key molecule in both primary and secondary hemostasis, because of its role in forming the platelet plug by connecting activated platelets and in forming plasma fibrin clot upon thrombin cleavage. Fibrinogen consists of two symmetric half-molecules, each containing a set of three different polypeptide chains termed Aα, Bβ, and γ. The latter contains several sites that interact with different ligands such as other fibrin(ogen) molecules, coagulation enzymes, growth factors, and integrins (1). The product of thrombin digestion of fibrinogen, i.e. fibrin, binds with a considerable specificity thrombin, so that in the early studies fibrin was termed antithrombin I (2). Thrombin has a divalent interaction with two classes of binding sites on fibrin, one of low affinity in the E domain and the other of high affinity in the D domain of fibrin(ogen) molecules (3). Binding of thrombin to fibrinogen involves sequences of both Aα and Bβ chain, which contain recognition sites in the fibrinogen E domain. These recognition sites are still able to interact with thrombin after cleavage of fibrinopeptide A and B and form the low affinity binding site for the enzyme. The D domains contain a γ chain variant, termed γ′, arising from an alternative mRNA splicing (4), resulting in an elongated chain composed of 427 instead of 411 residues. The inserted region at the C terminus is composed of 20 amino acids (408VRPEHPAETEYDSLYPEDDL427), rich of acidic side chains and two sulfate anions linked to Tyr418 and Tyr422 (5). The elongated γ chain, termed γ′, mainly heterodimerizes in the fibrinogen molecule with the more abundant γA chain, thus generating the γA/γ′ dimers (6). This fibrinogen, also called γA/γ′ fibrinogen, shows a high inter-individual variability in the ratio to the total fibrinogen γ chain (7). The different expression of γ′ chain has been variably associated with thrombotic disorders both in venous and arterial circulation.
Previous genetic studies showed, for instance, that the fibrinogen γ-H2 haplotype is characterized by a reduced fibrinogen γ′ levels and reduced fibrinogen γ′ to total fibrinogen ratio. This haplotype is associated with a significantly increased risk for venous thrombosis (6). Biochemical studies showed that γ′ chains bind to α-thrombin with high affinity (8) and that the 408–427 region of γ′ chain binds to the anion-binding exosite (ABE)2-II of thrombin (1, 9). Moreover, fibrinopeptide B cleavage by thrombin from γ′/γA fibrinogen is slower than in γA/γA fibrinogen (10). This effect was also associated with a reduced lateral aggregation of fibrin fibrils. All these findings may contribute to explain the reported enhanced risk for venous thromboembolism associated with a reduced expression of γ′ chain. However, at variance with these findings, other studies showed that fibrin fibers containing γ′ chains are more resistant than γ chains to proteolysis by fibrinolytic enzymes (11), so that fibrin clots containing a more abundant amount of γ′ chains could be associated with higher thrombotic risk. Notwithstanding the decreased sensitivity to fibrinolytic enzymes, the influence of a reduced expression of γ′/γA on enhanced risk for venous thromboembolism was prevalently demonstrated in clinical studies, although the detailed mechanism was only partially unraveled.
At variance with venous thromboembolism, the significance of altered expression of γ′ chain on arterial thrombosis remains largely elusive (6, 7, 12, 13). Platelets are major players of arterial thrombus formation, as also demonstrated by the clinical efficacy of anti-platelet agents in cardiovascular prevention. The fibrinogen γ′ chain, through its ability to bind to thrombin, might enhance the amount of clot-bound thrombin, known to be active in the presence of the heparin-antithrombin complex, and thus scarcely inactivated by traditional anticoagulants (heparins, indirect Factor Xa inhibitors) (3). Thus, clot-bound, active thrombin may represent a storage pool of the enzyme, facilitating arterial thrombus formation and growth.
In this study, we investigated the effect of the fibrinogen γ′ and also of its 20-amino acid-insertion peptide on the thrombin interaction with the platelet receptors glycoprotein (Gp) Ibα and protease-activated receptors 1 and 4 (PAR1 and PAR4), responsible for the thrombin-induced platelet activation. Fragment D was used as the best surrogate to selectively study the high affinity binding site for thrombin in γ chain in a conformation similar to that present in the native fibrinogen molecule and suitable for thrombin binding studies. This experimental approach was aimed at assessing whether γ′ chain can affect platelet activation by inhibiting competitively the interaction between the enzyme and GpIbα and by acting as an allosteric effector on PAR hydrolysis by thrombin. The obtained results may shed light on the possible role of fibrinogen γ′ chain on the thrombin-induced platelet activation and thus on possible implications on both anti-thrombotic and pro-thrombotic properties of fibrinogen in arterial circulation, where platelets play a central role in thrombo-hemorrhagic syndromes.
MATERIALS AND METHODS
Synthesis of Fibrinogen γ′ Peptide—The fibrinogen γ′ 408–427 peptide (408VRPEHPAETEYDSLYPEDDL427) and its scrambled sequence peptide (PTAHDYVDEERPYLPEELSD) as a control were synthesized by the peptide synthesis facility of the Brain Research Center at the University of British Columbia (Vancouver, Canada). The tyrosine residues 418 and 422 were phosphorylated, as these residues were sulfated in natural γ chains (5). The RP-HPLC analysis showed that these peptides were 95% pure, with a molecular mass of 2580.3 ± 0.2 atomic mass units, as determined by mass spectrometry.
Purification of Fibrinogen γA/γA and γA/γ′ D Fragments—Both γA/γA (D) and γA/γ′ (D*) fragments of fibrinogen were purified by a modified procedure, as reported previously (14). Human fibrinogen, free of plasminogen was purchased from Calbiochem. This preparation was chromatographed on a DEAE-Sepharose fast flow XK column connected to a fast protein liquid chromatography apparatus (GE Healthcare) to separate the fibrinogen fraction rich in γ′ chains. The column was equilibrated with 5 mm sodium phosphate, 40 mm Tris, pH 8.50, at a flow rate of 1 ml/min. One gram of fibrinogen was adsorbed on the column. After the elimination of nonadsorbed proteins, fibrinogen fractions were eluted using a stepwise gradient and three different eluting buffer solutions as follows: 1) 30 mm sodium phosphate, 60 mm Tris, pH 7.60; 2) 50 mm sodium phosphate, 80 mm Tris, pH 6.80; 3) 500 mm sodium phosphate, 0.5 m Tris, pH 4.40. All these buffers contained 1 mg/ml aprotinin as protease inhibitor. Three major peaks were obtained, and the fibrinogen fraction containing one γA and one γ′ chain was eluted with the third buffer solution, whereas the fraction containing two γA chains was obtained with the first buffer. D fragments were prepared from plasmin digests of the first and the third peak obtained by DEAE chromatography and gel-filtered on DG-10 columns (Bio-Rad) equilibrated with 50 mm Tris-HCl, 0.15 m NaCl, 10 mm CaCl2, pH 8.50. Human plasmin (specific activity, 5 units/mg; Calbiochem) was added at a final concentration of 0.05 unit/ml (0.01 mg/mg fibrinogen) and incubated with the pooled and concentrated fibrinogen peaks 1 and 3 of the DEAE chromatography in the above buffer at 25 °C for 120 min. The fibrinogen was pretreated for 15 min with 5 mm iodoacetamide to inhibit any minimal trace of contaminating Factor XIII before the addition of plasmin. The reaction was stopped by the addition of aprotinin (10 mg/ml final concentration). Fragment D (containing γA chains only) and D* (containing γ′ chain only) were purified from peaks 1 and 3, respectively, using a second DEAE column (Supelco, Sigma), 4.6 × 25 mm, and a two-pump HPLC apparatus (Jasco Easton, MD), equipped with a spectrophotometric device (model 2075), and a spectrofluorometric detector (FP-2020, Jasco). The spectrophotometric detection of the eluted peaks was accomplished at 280 nm, whereas the fluorescence of the proteins was monitored by using λex = 280 nm and λem = 340 nm. The developed gradient was 0–0.5 m NaCl in 20 mm Tris-HCl, pH 8.0, in 60 min. The flow rate was 1 ml/min. Fragment D was eluted at about 0.2 m NaCl, whereas fragment D* was obtained at ∼0.45 m NaCl. The concentration of fragment D and D* was calculated spectrophotometrically at 280 nm using an extinction coefficient of E0.1% = 2.0 cm2·mg-1, using the primary sequence of fragment D and the spectrophotometric method by Pace et al. (15). The fractions containing the fragment D and D* were pooled and concentrated, and their purity was checked by SDS-PAGE using 4–12% gradient gels under both not reducing and reducing conditions. The identity of the γ′ chain was checked by immunoblotting of the bands obtained in SDS-PAGE of reduced fragment D*, using a mouse anti-human monoclonal antibody (clone 2.G2.H9) from Millipore S.p.A. (Milano, Italy), a secondary anti-mouse horseradish peroxidase-conjugated antibody, and an ECL™ Western blotting detection system (GE Healthcare). The γA chains obtained from reduced fragment D did not react with the monoclonal antibody 2.G2.H9 (data not shown).
Thrombin-Fragment D* Interaction—Human α-thrombin was purified and characterized as reported previously (16). Binding of thrombin to purified fibrinogen fragment D* was studied by a solid-phase binding assay, immobilizing fragment D* (5 μg/ml) on microtiter plates (96-well; Nunc-Immuno Maxisorp Nunc), overnight at 4 °C in 50 mm bicarbonate buffer, pH 9.60. The plate surface was blocked at 37 °C for 4 h with 250 μl/well of a buffer solution containing 1 mg/ml bovine serum albumin, 50 mm Tris-HCl, pH 7.5. After aspiration of the blocking solution, plates were dried at room temperature and stored over desiccant at 4 °C. Use of the anti γ′ chain monoclonal antibody 2.G2.H9 conjugated to Alexa Fluor 488 (Invitrogen) allowed us to obtain a quantitative estimate of the amount of immobilized fragment D*. Under the above conditions, the amount of immobilized fragment D* was equal to about 10 ng/well (about 0.12 pmol/well). This estimate was based on the use of serial dilutions of a reference solution of the 2.G2.H9 monoclonal antibody, whose Alexa 488 fluorescence was measured using λex = 494 nm and λem = 520 nm. Thus, at maximum saturation using 100 μl of the buffer solution, about 1 nm thrombin could be bound by immobilized fragment D*. Control experiments were also performed using fragment D instead of fragment D* at the same concentration.
Thrombin (78 nm to 5 μm) was incubated for 30 min in the absence and presence of the C-terminal domain 45–57 of hemadin, a specific ligand for ABE-II of thrombin (17). The C-terminal peptide of hemadin had the sequence NH2-SEFEEFEIDEEEK-OH and was synthesized by the solid-phase Fmoc (N-(9-fluorenyl)methoxycarbonyl) method (18) on a p-alkoxybenzyl ester polystyrene resin, using a method detailed previously (19) The chemical identity of the purified material was established by high resolution mass spectrometry in positive ion mode on a Mariner electrospray ionization-time-of-flight instrument from PerSeptive Biosystems (Foster City, CA), which gave mass values in agreement with the expected amino acid composition within 10 ppm mass accuracy. The concentration of the peptide was determined by UV absorption at 257 nm on either a double-beam l-2 (PerkinElmer Life Sciences) or a Varian Cary 2200 spectrophotometer (Assoc. Inc., Sunnyvale, CA), using a molar absorption coefficient of 400 m-1·cm-1. The hemadin peptide was used at a fixed concentration spanning from 2 to 16 μm. The binding buffer was 10 mm Tris-HCl, 0.15 m NaCl, 0.1% PEG 6000, pH 7.50, at 25 °C (TBSP). In a different experimental setup, the C-terminal hemadin peptide was substituted by the γ′ peptide, used over a 22.5–180 μm concentration range, to test whether or not the latter behaves as a pure competitive inhibitor. After incubation at 25 °C for 30 min, and aspiration with three washing cycles with TBSP, a sheep anti-thrombin polyclonal antibody (∼10 mg/ml, from US Biological, Milan, Italy) was added at an optimal dilution of 1:500 in TBSP and incubated for 120 min. After aspiration of the solutions and three washing cycles, 100 μl of rabbit anti-sheep horseradish peroxidase-conjugated polyclonal antibody (∼2 mg/ml, dilution 1:250) from US Biological (Milan, Italy) was added and incubated for 60 min at 25 °C. After aspiration and three washing cycles, 100 μl of 5 mm 3,5,3′,5′-tetramethylbenzidine in the presence of 5 mm H2O2 was added, and the reaction was stopped after 15 min using 1 m H2SO4. This end point was chosen based on preliminary experiments showing a linear increase of the absorbance (15 points, R > 0.95) over that time interval even at the highest concentration of thrombin. This finding ruled out that the absorbance measured after 15 min of incubation did not reflect the real amount of thrombin bound to fragment D* and was not because of substrate depletion. An entire data set of thrombin binding to fragment D* (35 points) was simultaneously fitted to the Equation 1,
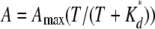 |
(Eq. 1) |
where A is the value of the absorbance measured at 450 nm; Amax is the asymptotic value of the absorbance; T is the thrombin concentration; and Kd* is the apparent equilibrium dissociation constant of thrombin binding to fragment D*, equal to K 0d (I/Ki), with K 0d as the real equilibrium binding constant; I is the concentration of either fragment D* or γ′ peptide, and Ki is the equilibrium dissociation constant of binding of these ligands to thrombin.
Binding of Fibrinogen γ′ Peptide to Thrombin Studied by Tryptophan Fluorescence—Binding of γ′ peptide to thrombin was studied by recording the increase in tryptophan fluorescence of thrombin at λmax (i.e. 334 nm) as a function of fibrinogen γ′ peptide. The interaction of the latter with thrombin was monitored by adding, under gentle magnetic stirring, to a solution of thrombin (1.4 ml, 50 nm) in 5 mm Tris-HCl buffer, pH 7.5, 0.1% PEG, in the presence of 0.15 m NaCl, aliquots (2–5 μl) of γ′ peptide (2.33 mm). Fluorescence spectra were recorded on a Jasco (Tokyo, Japan) model FP-6500 spectrofluorometer, equipped with a Peltier model ETC-273T temperature control system from Jasco. Excitation and emission wavelengths were 295 and 334 nm, respectively, using an excitation/emission slit of 10 nm. For all measurements, the long time measurement software (Jasco) was used. Control experiments were also performed to ruled out not specific effects, using the γ′ peptide scrambled peptide at a concentration of 100 μm. Under these conditions, at the end of the titration, a Trp photobleaching lower than 2% was observed. The absorbance of the solution at both 295 and 334 nm was always lower than 0.05 unit, and therefore no inner filter effect occurred during titration experiments. Fluorescence intensities were corrected for dilution (2–3% at the end of the titration) and subtracted for the contribution of the ligand at the indicated concentration. The fluorescence values, measured in duplicate, were analyzed as a function of the γ′ peptide concentration by a hyperbole equation to obtain the value of the Fmax (corresponding to the fluorescence at γ′ peptide concentration = ∞). This parameter was used to calculate ΔFmax = Fmax - Fo (where Fo is the fluorescence value in the absence of the peptide). The fluorescence changes expressed as (Fobs - Fo)/ΔFmax were analyzed as a function of the total γ′ peptide concentration according to a single site binding isotherm. Nonlinear least squares fitting was performed using the program Origin 7.5 (MicroCal Inc.), which allowed us to obtain the best fitting parameter values along with their standard errors.
Effect of γ′ Peptide and Fragment D* on Thrombin-GpIbα Interaction—Solid phase binding experiments to evaluate the effect of fibrinogen γ′ peptide on thrombin-GpIbα-(1–282) interaction were performed as detailed above by immobilizing purified GpIbα-(1–282) fragment (10 μg/ml) on polystyrene plates. Purification of platelet GpIbα-(1–282) fragment was performed as detailed previously (20). Thrombin (20 nm to 1.28 μm) was incubated in the presence of both 408–427 γ′ peptide and fragment D* at fixed concentrations spanning from 10 to 320 μm and from 0.2 and 3.2 μm, respectively. The binding buffer was TBSP. Both the experimental procedure of the binding assay and the analysis of the experimental data sets were the same as those used to study the thrombin-fragment D* interaction, detailed above. Control experiments, in which different concentrations of GpIbα(1–282) fragment from 0.31 to 10 μg/ml were immobilized on the microplate wells for binding to 10 nm thrombin, showed that in the time scale of the horseradish peroxidase reaction with 3,5,3′,5′-tetramethylbenzidine (15 min), the signal at 450 nm was always linear for all tested GpIb fragment concentrations. These results validated the assumption that in this solid-phase binding assay the absorbance measured at 450 nm after 15 min reflected the amount of thrombin bound to GpIb. Additional control experiments were also carried out with the synthetic peptide analog GpIbα-(268–282), as a competitive inhibitor of thrombin binding to the immobilized GpIb-(1–282) fragment. This peptide, encompassing the C-terminal tail 268–282 of GpIbα, was synthesized and characterized as detailed previously (19). The three sulfated tyrosines, present in the natural peptide sequence (residue 276, 278, and 279), were replaced by phosphotyrosine.
Effect of HD1 and HD22 Aptamers on Thrombin-Fragment D* Interaction—The ssDNA-aptamers 5′-GGTTGGTGTGGTTGG-3′ (HD1) and 5′-AGTCCGTGGTAGGGCAGGTTGGGGTGACT-3′ (HD22) were synthesized by Primm s.r.l. (Milano, Italy). HD1 and HD2 are ssDNA aptamers, which specifically bind to ABE-I and ABE-II, respectively (21). In these experiments 500 nm α-thrombin was added to fragment D*, immobilized on microplates as detailed in the previous paragraph, in the presence of different concentrations of HD22 (20–1280 nm) and HD1 (87.5–5600 nm) and incubated for 60 min. The detection of bound thrombin was performed by an immunoassay, as described previously.
Hydrolysis of Chromogenic Substrate d-Phe-Pip-Arg-pNA by Thrombin in the Presence of γ′ Peptide and Fibrinogen Fragment D*—Steady state hydrolysis of the chromogenic substrate S-2238 was studied in the absence and presence of six different γ′ peptide concentrations ranging from 2.5 to 320 μm and fragment D* concentrations spanning from 0.2 to 3.2 μm. Thrombin was used at 1 nm in 10 mm Tris-HCl, 0.15 m NaCl, 0.1% PEG 6000, pH 7.50, at 25 °C.
Hydrolysis of PAR1-(38–60) and PAR4(44–66) Peptide by Thrombin—PAR1-(38–60) (38LDPRSFLLRNPNDKYEPFWEDEE60) and PAR4-(44–66) (44PAPRGYPGQVCANDSDTLELPDS66) peptides were synthesized by PRIMM (Milan, Italy). Cleavage of these peptides by 0.1–1 nm thrombin was monitored by RP-HPLC as detailed previously (22). The Michaelis-Menten parameters kcat and Km were calculated in the absence and presence of fixed concentrations of the γ′ peptide ranging from about 2.5 to 320 μm. The kcat/Km values of PAR1 peptide hydrolysis in the presence of fragment D* (from 0.2 to 6.4 μm) was calculated at a peptide concentration of 1 μm, which is a concentration lower than the Km value of the thrombin-PAR interaction. Under these conditions, the first order rate constant of the peptide hydrolysis was proportional to the kcat/Km value, as experimentally verified. The hydrolysis reaction was performed in 10 mm Tris-HCl, 0.15 m NaCl, 0.1% PEG 6000, pH 7.50, at 25 °C. The kcat/Km values were analyzed as a function of both γ′ peptide and fibrinogen fragment D* using the following linkage Equation 2 (23),
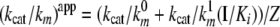 |
(Eq. 2) |
where Z = 1 + I/Ki; Ki is the equilibrium dissociation constant of either γ′ peptide or fragment D* binding to thrombin; I is the inhibitor concentration; and the superscript 0 and 1 refer to the kcat/Km value pertaining to free and γ′ peptide- or D*-bound thrombin form, respectively. Control experiments were also carried out using 320 μm scrambled γ′ peptide to exclude spurious effects generated by ionic strength phenomena.
Binding of [Fluorescein]-Hirudin54–65(PO3H2) to Human α-Thrombin in the Absence and Presence of γ′ Peptide—Fluorescein-conjugated and phosphorylated C-terminal hirudin-(54–65) peptide, [F]-hirudin54–65(PO3H2), having the sequence GDFEEIPEEY(PO3H2)LQ, was synthesized as described previously (24). Binding of this peptide to ABE-I of thrombin was studied by monitoring the decrease of the peptide fluorescence occurring upon interaction with thrombin, as reported previously (25). Fluorescence spectra were recorded on a Jasco (Tokyo, Japan) spectrofluorometer, as detailed above. Excitation and emission wavelengths were 492 and 516 nm, respectively, using an excitation/emission slit of 3/5 nm. During titration experiments, the decrease of fluorescence intensity at 516 nm was recorded as a function of thrombin concentration. For all measurements, the long time measurement software (Jasco) was used. Fluorescence intensities were corrected for dilution (i.e. 8–10%) at the end of the titration.
Data were analyzed by the following binding isotherm Equation 3 (26), using the program Origin 7.5 (MicroCal Inc.),
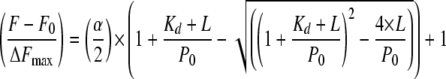 |
(Eq. 3) |
where α is the maximum fluorescence change; Kd is the dissociation constant; L is the total concentration of thrombin; and P0 is the concentration of [F]-hirudin54–65 (PO3H2).
Thrombin-induced Aggregation of Gel-filtered Platelets—Platelets from healthy volunteers were gel-filtered on Sepharose 2B columns (GE Healthcare) as reported previously (22). Born's aggregation of gel-filtered platelets, performed on a 4-channel PACKS-4 aggregometer (Helena Laboratories, Sunderland, UK) as detailed previously (22), was induced by 1 nm thrombin in the absence or presence of different concentrations of γ′ peptide and fibrinogen fragment D*. Control experiments were performed with both 50 μm PAR1- and 1 mm PAR4-activating peptides (PAR1-AP (SFLLRN-NH2) and PAR4-AP (AYPGKF-NH2), respectively, from PRIMM), 10 μm ADP, and 10 μg/ml collagen from Helena Laboratories. The specific effect of fragment D* was also evaluated by using fragment D at the same concentrations.
Monitoring of Full-length PAR1 Hydrolysis by Thrombin on Intact Platelets by Flow Cytometry—Gel-filtered platelets from healthy controls were mixed with 1 nm thrombin at 25 °C in the absence and presence of the γ′ peptide ranging from 27 to 310 μm and of fibrinogen fragment D* from 0.1 to 32 μm. After 120 s, the hydrolysis of PAR1 molecules on platelet membrane was stopped with 1 μm d-Phe-Pro-Arg-chloromethyl ketone, and the uncleaved PAR1 molecules were detected by flow cytometry, as described previously (22). Briefly, after cleavage reaction was stopped, platelets were labeled for 30 min at 4 °C with saturating amounts of phycoerythrin-conjugated antithrombin receptor monoclonal antibodies (SPAN-12 clone; Beckman Coulter, Milan, Italy), as detailed elsewhere (22). Isotype-matched, phycoerythrin-conjugated irrelevant antibodies were used to measure background fluorescence. Samples were run through a FACSCanto® flow cytometer (BD Biosciences) with standard equipment. Uncleaved PAR1 expression levels were reported in terms of mean fluorescence intensity (MFI) ratio of the SPAN-12+ platelet population.
RESULTS
Purification of Fragment D and D*—The purifications of both fibrinogen fragment D*, containing one γA and one γ′ chain, and of normal fragment D were successfully accomplished by DEAE chromatography. Fragment D in SDS-PAGE showed a molecular mass of about 85 kDa, whereas fragment D* had a slightly higher molecular mass as compared with fragment D, in agreement with the presence of the elongated γ′ chain (Fig. 1A). SDS-PAGE under reducing conditions and immunoblotting of the reduced sample with an anti-γ′ monoclonal antibody allowed us to identify the genuine presence of fibrinogen fragment D*, as shown in Fig. 1, B and C. Purified fragment D* was then used in the functional and solid-phase binding experiments, where the nominal concentration of the γ′ chain was assumed the same as that of the entire fragment D*.
FIGURE 1.

SDS-PAGE of purified fibrinogen fragment D. A, gel was 4–12% polyacrylamide under nonreducing conditions. Lane 1, purified fibrinogen fragment D; lane 2, total fibrinogen fraction eluted by 50 mm sodium phosphate, 80 mm Tris, pH 6.80, in the first chromatographic step using DEAE-Sepharose (see text); lane 3, fraction eluted by 0.5 m NaCl in 20 mm Tris-HCl, pH 8.0, in the second chromatographic step using DEAE-Sepharose (see text). The molecular mass markers are indicated on the right. B, gel was 4–12% polyacrylamide under reducing conditions. The sample was fragment D* obtained from the DEAE chromatography. The component with a molecular mass of ≈41,000 kDa is the elongatedγ chain fragment contained in fragment D*. The other bands pertain to theβ chain region of fragment D* (37.6 kDa) and theα chain fragment (12 kDa). The faint band below the Dγ* may be a minor fragment produced by plasmin digestion, possibly generated by cleavage at Ser86 of the γ chain (61). The molecular mass markers are indicated on the left. C, Western blot of the fragment D* sample shown on the left. Detection of the γ′ chain was obtained using the mouse monoclonal antibody 2.G2.H9, raised against the peptide sequence VRPEHPAETEYDSLYPEDDL of human fibrinogen elongated γ′ chain.
Characterization of the Fibrinogen Fragment D* Interaction with Thrombin—The interaction of purified fragment D* with thrombin was studied by a solid-phase binding assay that showed a specific interaction with a Kd value of 0.4 ± 0.03 μm (Fig. 2). The sequence of 20 amino acids of the γ′ peptide present in the fragment D* drives this interaction, as the purified γ′ peptide competitively inhibited with a Ki value of about 47 μm of the thrombin-fragment D* interaction, as shown by Fig. 2A. This interaction involved the ABE-II of thrombin, as its binding was competitively inhibited by specific ligands of this thrombin exosite. In fact, the C-terminal 45–57 peptide of hemadin, which binds to ABE-II (27), was able to competitively inhibit the thrombin-fragment D* interaction, with a Ki value of 4.2 ± 0.4 μm, as shown in Fig. 2B. No significant interaction was observed with fragment D (Fig. 2C). The involvement of the ABE-II of thrombin was also confirmed by the inhibition of the binding of 500 nm thrombin to immobilized fragment D* by the ssDNA aptamer HD22 (IC50 = 81 ± 6 nm; see Fig. 2C), whereas no effect was observed using the ssDNA aptamer HD1, which binds to ABE-I of the enzyme (data not shown).
FIGURE 2.

A, binding of thrombin to immobilized fragment D* in the presence of varying concentrations of purified γ′ peptide used at the following concentrations (○) 0, (•) 22.5 μm, (□) 45 μm, (▪) 90 μm, and 180 μm (Δ). The solid lines were drawn according to the best fit parameter values of a simultaneous fit to single site binding isotherm and a competitive inhibition scheme: Kd of thrombin binding to fragment D* = 0.41 ± 0.04 μm, and Ki of the γ′ peptide = 47.5 ± 6 μm. B, effect of the 45–57 C-terminal hemadin peptide on thrombin-fragment D* interaction. The solid lines were drawn according to the best fit parameter values of a single site binding isotherm and a competitive inhibition scheme: Kd of thrombin binding = 0.4 ± 0.03 μm, and Ki of the C-terminal hemadin peptide = 4.2 ± 0.4 μm. The concentrations of the hemadin peptide were (○) 0, (•) 2 μm, (□) 4 μm, (▪) 8 μm, and 16 μm (Δ). The experimental data set was analyzed by simultaneous fitting. C, control experiments showing the absence of interaction between thrombin and immobilized fragment D. D, binding of 500 nm thrombin to immobilized fibrinogen fragment D* as a function of the aptamer HD22 concentration. The solid line was drawn according to the best fit IC50 value equal to 81 ± 6 nm. In the inset, a similar experiment, carried out as a function of the aptamer HD1 concentration, is shown.
Binding of γ′ Peptide to Thrombin Monitored by Tryptophan Fluorescence—The binding of γ′ peptide to thrombin causes a significant increase of tryptophan fluorescence, without appreciable change in the λmax value. Hence, we exploited this change for estimating the affinity of the γ′ peptide for thrombin, as shown in Fig. 3. The corresponding Kd value for γ′ peptide binding was calculated as 30 ± 5 μm, in good agreement with the value determined by the solid-phase binding experiments reported above. The increase in the fluorescence quantum yield suggests that the chemical environment of Trp residues in thrombin becomes, on average, more rigid and apolar than in the ligand-free enzyme (28).
FIGURE 3.

The interaction of γ′ peptide with thrombin was monitored by adding, under gentle magnetic stirring, to a solution of thrombin (1. 4 ml, 50 nm) in 5 mm Tris-HCl buffer, pH 7.5, containing 0.1% PEG, and 0.15 m NaCl, aliquots (2–5 μl) of γ′ peptide (2.33 mm). The solid lines represent the least square fit with Kd value of 30 ± 5 μm.
Effect of γ′ Peptide and Fibrinogen Fragment D* on Platelet Aggregation—The fibrinogen γ′ peptide inhibited dose-dependently the thrombin-induced aggregation of gel-filtered platelets, up to about 70%, in a specific manner, as demonstrated by the lack of effect by the scrambled γ′ peptide (see Fig. 4, A and B). Likewise, purified fibrinogen fragment D* inhibited platelet aggregation up to about 70%, although it was impossible to reach full inhibition, even at higher fragment concentrations (Fig. 4B). At variance with these findings, no significant effect was observed with fragment D (Fig. 4B). The analysis of these data provided IC50 values of 42 ± 3.5 μm for the γ′ peptide and 0.47 ± 0.03 μm for the D* fragment. Aggregation induced by saturating concentrations of ADP (10 μm), collagen (10 μg/ml), PAR1-AP (50 μm), or PAR4-AP (1 mm) was not affected by the γ′ peptide or fragment D* (data not shown), indicating a specific interaction with thrombin.
FIGURE 4.

Aggregation of gel-filtered platelets by 1 nm thrombin in the absence (trace 1) and presence of 25 (trace 3), 50 (trace 4), 100 (trace 5), and 200 μm (trace 6) fibrinogen γ′ peptide. A, trace 2 was obtained in the presence of 200 μm scrambled γ′ peptide, whose sequence was generated by the RandSeq program, available the Expasy web site. B, inhibition analysis of thrombin-induced platelet aggregation in the presence of γ* peptide (•), fibrinogen fragment D* (○), and D (□). The solid lines were drawn according to the best fit IC50 values of 42 ± 3.5 μm for the γ′ peptide and 0.47 ± 0.03 μm for the D* fragment.
Effects of γ′ Peptide and Fibrinogen Fragment D* on Thrombin-GpIbα Interaction—Both γ′ peptide and fibrinogen fragment D* inhibited competitively the binding of thrombin to immobilized GpIbα-(1–282) with a Ki of about 40 and 0.5 μm, respectively (Fig. 5, A and B). Instead, no effect was observed with fragment D (data not shown). The competitive nature of the observed inhibition by both γ′ peptide and fragment D* was confirmed by control experiments performed with the synthetic peptide analog GpIbα-(268–282), which binds to thrombin with a Ki value of 9 μm (19). These findings can explain in part the inhibitory effect of the γ′ peptide and fragment D* on thrombin-induced platelet aggregation, because of the activating role of thrombin-GpIb interaction on platelet aggregation (22, 29).
FIGURE 5.

Binding of purified human α-thrombin to immobilized platelet GpIbα-(1–282) fragment. A, binding of thrombin in the presence of different concentrations of γ′ peptide. The solid lines were drawn according to the best fit parameter values of a single site binding isotherm: Kd of thrombin binding = 86 ± 7 nm, and Ki of γ′ peptide inhibition = 40 ± 6 μm. The concentrations of γ′ peptide were as follows: (○) 0, (•) 10 μm, (□) 20 μm, (▪) 40 μm, (Δ) 80 μm, (▴) 160 μm, and (▿) 320 μm. B, binding of thrombin in the presence of varying concentrations of fragment D* was as follows: (○) 0, (•) 0.2 μm, (□) 0.4 μm, (▪) 0.8 μm, (Δ) 1.6 μm, (▴) 3.2 μm. The solid lines were drawn according to the best fit parameter values of a single site binding isotherm: Kd of thrombin binding = 115 ± 7 nm, and Ki of fragment D* = 0.48 ± 0.37 μm. Each point represents the mean of two different measurements. Each experimental data set was analyzed by simultaneous fitting.
Effect of γ′ Peptide and Fibrinogen Fragment D* on Thrombin-catalyzed PAR1 and PAR4 Cleavage—Fibrinogen γ′ peptide inhibited the cleavage of the PAR1 substrate, as shown in Fig. 6A. The inhibitory effect was allosteric in nature, as PAR1-(38–60) substrate interacts with the ABE-I and the active site of thrombin and not with ABE-II (30), where γ′ peptide binds (9). Moreover, the inhibitory effect concerned mostly the kcat value, as shown in Table 1. When the kcat/Km values were analyzed by a linkage equation (Equation 2) as a function of γ′ peptide concentration, a best-fit Ki value of about 40 μm was obtained, in good agreement with the value derived from the GpIb solid-phase binding and fluorescence titration experiments (Fig. 6B). No significant effect was observed using 320 μm scrambled γ′ peptide, thus ruling out spurious ionic strength effects. Likewise, the kcat/Km values of PAR1 hydrolysis as a function of fragment D* concentration decreased, reaching an asymptotic value (Fig. 6C). In this case, the value of the equilibrium dissociation constant was about 0.5 μm, about 80-fold lower than that measured for γ′ peptide, in analogy to the results obtained in solid-phase binding experiments with GpIbα. No significant effect was instead observed using the fragment D (Fig. 6C).
FIGURE 6.

A, cleavage of 1 μm PAR1-(38–60) by 0.1 nm thrombin in the absence (○) and presence (•) of saturating concentrations (320 μm) of the γ′ peptide. The concentrations of the cleaved PAR1 peptide over time, [cleaved PAR1]t, were fitted to the first order equation as follows: [cleaved PAR1]t = 1 μm × (1 - exp(-kt)). Under these pseudo-first order conditions, k = e0 (kcat/Km) where e0 is the thrombin concentration. The solid lines were drawn according to the best fit values of the kcat/Km values equal to 3.83 ± 0.1 × 107 m-1 s-1 and 1.76 ± 0.1 × 107 m-1 s-1 in the absence and presence of γ′ peptide, respectively. B, values of kcat/Km of PAR1-(38–60) hydrolysis by thrombin as a function of γ′ peptide. The solid line was drawn according to Equation 2 with the best fit values as follows: (kcat/Km)0 = 4 ± 0.1 × 107 m-1 s-1, (kcat/K)1 = 1.47 ± 0.1 × 107 m-1 s-1, Ki = 37.4 ± 6 μm. C, values of kcat/Km of PAR1-(38–60) hydrolysis by thrombin as a function of fragment D* (•) and fragment D (□) concentration. The solid line was drawn according to Equation 2 with the best fit values as follows: (kcat/Km)0 = 3.97 ± 0.13 × 107 m-1 s-1 (kcat/Km)1 = 1.60 ± 0.15 × 107 m-1 s-1, Ki = 0.54 ± 0.15 μm. The vertical bars are the standard deviations from two determinations.
TABLE 1.
Michaelis-Menten parameters of PAR-1(38–60) hydrolysis by human α-thrombin in the presence of different concentration of γ′ peptide
| kcata | Km | kcat/Km | ||
|---|---|---|---|---|
| s–1 | μm | ×107m–1 s–1 | ||
| γ′ peptide concentration (μm) | ||||
| 0 | 78 | 2 | 3.9 | |
| 2.5 | 79 | 2.02 | 3.9 | |
| 5 | 74 | 1.99 | 3.71 | |
| 10 | 72 | 2.1 | 3.43 | |
| 20 | 68 | 2.3 | 2.96 | |
| 40 | 59 | 2.13 | 2.77 | |
| 80 | 55 | 2.4 | 2.29 | |
| 160 | 48 | 2.5 | 1.92 | |
| 320 | 45 | 2.55 | 1.76 | |
| Scrambled γ′ peptide (μm) | ||||
| 320 | 3.7b | |||
The standard error percentage was equal to 7% of the parameter value
Data were determined at 1 μm PAR-1 concentration, following the pseudo-first order kinetics of its hydrolysis by 0.1 nm thrombin
At variance with PAR1, the hydrolysis of PAR4 was not affected by γ′ peptide, as the kcat/Km values measured as a function of the γ′ peptide concentration were scattered around a mean of about 4 × 105 m-1 s-1 (data not shown). In addition, experiments were carried out using the synthetic peptide substrate d-Phe-Pip-Arg-pNA (S-2238). Both γ′ peptide and fragment D* reduced the catalytic competence of thrombin toward S-2238, with this effect linked mostly to a reduction of the kcat values, as listed in Table 2.
TABLE 2.
Michaelis-Menten parameters of S-2238 hydrolysis by human α-thrombin in the presence of different concentrations of γ′ peptide and fibrinogen fragment D*
| kcata | Km | kcat/Km | ||
|---|---|---|---|---|
| s–1 | μm | ×107m–1 s–1 | ||
| γ′ peptide concentration (μm) | ||||
| 0 | 85.0 | 2.00 | 4.30 | |
| 2.5 | 84.5 | 2.20 | 3.84 | |
| 5 | 79.6 | 2.10 | 3.79 | |
| 10 | 79.8 | 2.30 | 3.47 | |
| 20 | 72.4 | 2.30 | 3.18 | |
| 40 | 64.7 | 2.40 | 2.70 | |
| 80 | 63.6 | 2.80 | 2.27 | |
| 160 | 56.6 | 2.90 | 1.95 | |
| 20 | 52.2 | 2.98 | 1.75 | |
| Fibrinogen fragment D* (μm) | ||||
| 0.2 | 74 | 2.10 | 3.50 | |
| 0.4 | 60 | 2.06 | 2.91 | |
| 0.8 | 55 | 1.95 | 2.82 | |
| 1.6 | 50 | 1.88 | 2.66 | |
| 3.2 | 47 | 1.94 | 2.42 | |
The standard error percentage was equal to 5% of the parameter value
A dose-dependent inhibition of the hydrolysis of full-length PAR1 molecules on intact platelets was also observed as a function of increasing concentrations of γ′ peptide, as shown in Fig. 7A. At high peptide concentrations the inhibition reached an asymptotic value, in agreement with the results obtained with the synthetic PAR1 peptide. Similar effects were observed with the fragment D* (Fig. 7B). Thus, γ′ peptide can exert its inhibitory effect on platelet activation by inhibiting competitively the interaction between the enzyme and GpIbα and by causing an allosteric inhibition of PAR1 hydrolysis.
FIGURE 7.

Effect of γ′ peptide (A) and fibrinogen fragment D* (B) on thrombin cleavage of PAR1 molecules on gel-filtered platelets. The MFI ratio was defined as MFI of test histograms/MFI of control histograms (background fluorescence without γ′ peptide). Data are representative of mean ± S.D. recorded in three independent experiments run in duplicate.
The effect of γ′ peptide on the interaction of thrombin with [F]-hirudin54–65(PO3H2) was also investigated to assess whether or not the negative influence of the γ′ peptide on PAR1 but not PAR4 hydrolysis arose from a conformational change induced in the ABE-I, where PAR1 but not PAR4 binds. These experiments showed that the Kd value of [F]-hirudin54–65 (PO3H2) was not significantly changed in the presence of a high concentration of the γ′ peptide (i.e. 74 μm), as shown in Fig. 8. The equilibrium dissociation constants of the hirudin peptide was in fact equal to 19.7 ± 1.2 and 14.3 ± 1.5 nm, in the absence and presence of the γ′ peptide, respectively. Altogether, these findings suggest that the inhibiting effect of the γ′ peptide on the cleavage of both PAR1 and the synthetic substrate S-2238 stems mainly from conformational changes induced in the catalytic site of thrombin.
FIGURE 8.

Plot of the normalized fluorescence fractional change (F - F0/ΔFmax) of [F]-hirudin54–65(PO3H2) (110 nm) as a function of human α-thrombin concentration in the absence (•) or in the presence (○) of 74 μm of γ′ peptide. The solid lines were drawn according to Equation 3, with best fit parameters Kd = 19.7 ± 1.2 nm (•) and 14.3 ± 1.5 nm (○).
DISCUSSION
This study showed for the first time that the fibrinogen sequence 408–427 in the elongated γ′ chain inhibits the thrombin-induced aggregation of platelets through a combined mechanism, impairing both GpIbα and PAR1 interactions. Because it has been shown previously that binding of thrombin to GpIbα could enhance the efficiency of thrombin cleavage of PAR1 (22), the double effect of γ′ peptide on both GpIbα interaction and PAR1 cleavage may cooperatively determine a strong inhibition on platelet activation/aggregation, as indeed observed. These results are unprecedented, as they show how the same ligand may hinder at the same time the thrombin interaction with the two thrombin-elicited receptors involved in platelet activation, i.e. GpIb and PAR1.
The interaction of fragment D* is energetically driven by the insertion of the γ′ sequence 408–427, which specifically binds to ABE-II, as demonstrated by various experimental strategies. In fact, both γ′ peptide and fragment D* are able to displace from ABE-II any ligand, which is known to interact with this site, such as GpIbα, the C-terminal hemadin peptide and the ssDNA aptamer HD22. However, the synthetic γ′ peptide and the fragment D* showed a different affinity for thrombin. The Kd value of fragment D* was about 80-fold lower than that of the synthetic peptide. These results parallel previous findings on the binding of fibrinogen γ chain to platelet GpIIb-IIIa, where a 70-fold difference in affinity between the synthetic peptide 400–411 of the fibrinogen γ chain and the native fibrinogen fragment D was found (31). Similar results were obtained for the binding of the N-terminal domain 1–282 of GpIbα to thrombin, where the affinity of the properly sulfated C-terminal peptide 268–282 is about 50 times lower than that of the full-length GpIbα-(1–282) fragment (19, 20). These observations can be reasonably explained by assuming that the protein core may orient the C-terminal tail of the γ chain in a conformation productive for binding or that the main body of the protein, beyond the C-terminal extension, enhances affinity by directly interacting with thrombin. The latter situation is documented by the crystal structure of the GpIbα-thrombin complex (32), where numerous hydrophobic and electrostatic interactions, not involving the C-terminal tail, further stabilize the complex. Although the isolated C-terminal segment of the γ chain displays some nascent secondary structure element, NMR data indicate that it is highly flexible and intrinsically disordered in solution (33). This conformational flexibility is also confirmed by the poor electron density observed for the C-terminal γ-segment in the crystallographic structures of fibrinogen D fragment (PDB codes 1LT9, 1FZC, and 1FIC) (34–36). On the other hand, the structure of a smaller γ chain fragment (PDB code 1FIC) reveals that the segment Leu392–Gly403 extends along the protein surface making numerous hydrogen bonds with the rest of the γ chain (36). Hence, these contacts may facilitate interaction with thrombin by orienting the elongated γ′-segment in a conformation productive for binding.
It is known that ∼10% of circulating fibrinogen molecules contain the elongated γ′ chain. If we refer to a normal plasma fibrinogen concentration (200–400 mg/dl corresponding to ≈6–12 μm), this would correspond to a concentration of elongated γ′ chain of about 0.6–1.2 μm, nicely overlapping the Kd value of the fragment D* interaction with the enzyme (Kd ∼ 0.5 μm). Thus, variations in the ratio between normal and elongated γ′ chain can significantly affect thrombin′s ligation in vivo, in keeping with the notion that the maximum change of the fractional saturation of a macromolecule as a function of its ligand concentration occurs when the latter is present at levels similar to the Kd value (37).
Moreover, it has to be outlined that in this study a surrogate for γ′ fibrin was used. The latter actually interacts with thrombin engaging both exosite 2 and 1 (3), although exosite 1 binds to fibrinogen fragment E with low affinity (3). Thus, we can speculate that γ′ fibrin can inhibit thrombin-induced platelet activation more extensively than fragment D*, as it competitively blocks both PAR1 cleavage, via engagement of exosite 1, and GpIb ligation, via binding to exosite 2, that allosterically down-regulates also PAR1 cleavage, as demonstrated in this study.
The allosteric effect linked to binding of the elongated γ′ chain to thrombin ABE-II resulted in a decrease of the catalytic specificity of the enzyme for good substrates such as PAR1 and the synthetic tripeptide S-2238, whereas no significant effect was observed with the PAR4 peptide-(44–66). Thus, platelet activation induced by PAR4 hydrolysis, which occurs mainly at high thrombin concentrations and signals independently from PAR1 (38), is not affected by either γ′ peptide or fragment D*. This may also contribute to explain the lack of complete inhibition of the thrombin-induced platelet aggregation observed even at high γ′ peptide and fragment D* concentrations (see Fig. 4B). The extracellular region of PAR1 interacts with thrombin active site through the sequence 38LDPR41 and with ABE-I using a hirudin-like sequence (24, 30). This is not the case for PAR4, which orients Pro44 and Pro46 of the sequence 44PAPR47 in the catalytic pocket but does not interact with ABE-I residues, using the C-terminal segment (39, 40). Recently, the crystal structure of murine thrombin in complex with the extracellular fragment of murine PAR4 confirmed this mode of binding (41). Perturbation of ABE-I by hirudin or PAR1 exodomain (residues 42–60) allosterically induces significant structural changes in the free catalytic pocket of thrombin, mainly at and around Ser195 (16, 30, 42), that result in altered reactivity of the enzyme toward synthetic and natural substrates (24–26, 43) and for binding of inhibitors (44–46). Notably, ligand binding to ABE-I can either enhance or inhibit the cleavage of small chromogenic substrates carrying an Arg residue at P1 position, according to their chemical structure at P2 and P3 positions (24, 26). Similar conclusions can be drawn for the perturbation of ABE-II, where binding of some ligands such as prothrombin fragment F2 or GpIbα causes negligible or even opposite effects on thrombin-mediated cleavage of chromogenic substrates (26, 47). For instance, hydrolysis of S-2238 is inhibited in the presence of F2, whereas cleavage of tosyl-Gly-Pro-Arg-pNA is enhanced at a similar extent (26). These findings confirm the extreme molecular plasticity of thrombin. Unfortunately, crystal structures of thrombin bound to several different ligands, including the prothrombin F2 fragment (48) (PDB code 2HPQ), heparin (49) (PDB code 1XMN), and GpIbα-(1–282) (32, 50) (PDB codes 1P8V and 1OOK), indicate that thrombin accommodates ABE-II ligands with little, if any, change in its folded structure and thus do not explain the observed variations in thrombin function upon ligand binding. These discrepancies likely arise from crystal packing effects (49) or from the presence of the d-Phe-Pro-Arg-chloromethyl ketone inhibitor (32, 48), which locks the active site and the specificity exosites of the enzyme into a fixed conformation, thus abrogating the structural changes that may be induced by ligand binding in solution. Very recently, the crystal structure of thrombin complex with the fibrinogen γ′ peptide-(408–427) has been solved at 2.4 Å resolution. No significant change in the structure of the enzyme-peptide complex could be detected when compared with that of free thrombin (9) (PDB code 2HWL). Conversely, solution studies involving hydrogen-deuterium exchange coupled with matrix-assisted laser desorption ionization time-of-flight mass spectrometry showed that the gamma′ peptide interacts at or near the thrombin ABE-II residues Arg93, Arg97, Arg173, and Arg175. Moreover, the binding of the γ′ peptide induces a conformational perturbation to the enzyme as a whole, by significantly protecting from deuterium exchange other regions of thrombin, such as the autolysis loop, the edge of the active site region, some portion of ABE-I, and the A chain (51). Most of Trp residues in thrombin (i.e. Trp96, Trp141, Trp148, Trp207, and Trp215) are embedded in segments whose hydrogen-deuterium exchange efficiency is reduced upon γ′ peptide binding, whereas other tryptophans (i.e. Trp29 and Trp237) are in direct contact with the perturbed segments (51). Hence, it is not surprising that the formation of γ′ peptide-thrombin complex results in a higher fluorescence intensity of the enzyme, as shown in Fig. 3, compatible with a conformational change of thrombin in which the chemical environment of Trp residues becomes more rigid and hydrophobic (28). Thus, it is conceivable that the structural perturbations caused by γ′ peptide binding propagates from the ABE-II residues toward the S2-S4 subsites of the catalytic cleft of the enzyme and that these changes are sensed differently by the various P3 residues present in S-2238, PAR1, and PAR4. In agreement with this allosteric hypothesis, the kcat/Km value of S-2288 (d-Ile-Pro-Arg-pNA) by thrombin was increased by 40% at high concentration of the synthetic γ′ peptide (160 μm, data not shown), demonstrating that even subtle changes in the side chain volume (Ile = 124 Å3; Phe = 135 Å3) and electronic properties at the P3 site can significantly change the allosteric linkage between binding to ABE-II and hydrolytic activity of thrombin.
In principle, the inhibition of PAR1 cleavage by thrombin might also arise from conformational transitions in ABE-I induced long range by γ′ peptide binding to ABE-II, leading to a lower affinity of the C-terminal PAR1 segment for ABE-I. The effect ofγ′ peptide on PAR4 cleavage would be negligible because this latter substrate does not bind to ABE-I. This working hypothesis is worthy of attention in the light of the proposed, but still debated, allosteric linkage existing between ABE-I and ABE-II (25, 26). To test this hypothesis, we investigated the effect of γ′ on the binding of the fluorescein-conjugated C-terminal 54–65 peptide of hirudin, [F]-hirudin54–65(PO3H2), a well known ligand for ABE-I (52). The fluorescence experiments showed that the binding of [F]-hirudin54–65(PO3H2) was not significantly affected by the γ′ peptide, as shown by Fig. 8. Thus, it is likely that binding of the elongated γ chain to thrombin induces a conformational change mainly occurring at the catalytic site of the enzyme.
The influence of γ′ chain on venous thrombosis has been largely recognized (6). In particular, a decrease of this chain was associated with a net increase of the risk factor for venous thromboembolism (6). In contrast, as anticipated above, the role of this fibrinogen chain for arterial thrombosis is still debated (6, 7, 13). Clinical studies were conducted in the attempt to demonstrate whether altered levels of γ′ chain are inversely or directly correlated with increased risk for arterial thrombosis. These studies showed that the association of γ′ chain expression with arterial thrombotic diseases is paradoxically different from that shown in venous thrombosis. In particular, the reduced γ′/γA ratio occurring in certain fibrinogen polymorphisms, such as FGG-H2, was not associated with either acute myocardial infarction (53) or ischemic stroke. Instead, increased γ′ levels were shown to be positively associated with an increased risk for both acute myocardial infection and ischemic stroke (54, 55). However, this association was shown to be strengthened by the presence of increased levels of plasma fibrinogen concentration (54, 55) and by FGG 9340T and FGA 2224G polymorphisms (54, 55). Other factors, such as total fibrinogenemia (56), the ability of the γ′ chain to protect thrombin by the heparin-antithrombin inhibition (3), and to confer to fibrin clots resistance to fibrinolytic degradation, may represent confounding factors in these studies. Thus, whether or not the thrombin interaction with the γ′ chain of fibrinogen plays any pathophysiological role in particular clinical settings remains controversial. In a recent study on ischemic stroke (55), both γ′ chain and total fibrinogen levels were elevated in the acute phase of the disease and subsequently decreased in the convalescent phase. The increased γ′ chain in the acute phase stems from the elevation of interleukin-6, which can promote the synthesis of the γA and γ′ chains (57, 58). Thus, further studies aimed at investigating the γ′/γA ratio rather than the absolute content of γ′ are needed, especially in clinical situations like acute thrombosis, where plasma fibrinogen is usually increased, and thus investigating the absolute γ′ content alone might be misleading.
Based on our data, indicating a net platelet inhibitory effect of fibrinogen γ′ chain upon thrombin stimulation, we can infer that elevation of γ′ chain level, as a possible result of acute phase response, might exert a beneficial effect on the acute phase of thrombotic syndromes. A recent study in a baboon thrombosis model showed indeed that the 410–427 γ′ peptide is able not only to inhibit fibrin-rich thrombus formation, because of the inhibition of the intrinsic coagulation pathway (related to inhibition of FVIII activation by thrombin), but also platelet-rich thrombus formation in the arterial circulation (59). These findings may be also relevant for clinical applications of ssDNA aptamers, like HD22, whose specific target is ABE-II of thrombin (60). On the contrary, the anti-thrombin and anti-platelet effect of the γ′ chain may be deleterious in hemorrhagic sequelae of vascular accidents, such as hemorrhagic stroke. In the latter condition, the expansion of the volume of the hematoma causes the post-stroke complications often responsible for the high mortality from the disease. The findings reported in this study predict that the presence of enhanced expression of the γ′ chain could exert deleterious effects on the thrombin-induced platelet activation and thus on either the arrest or onset of the hemorrhage. In conclusion, the role of different expressions of the γ′ chain in circulating fibrinogen may variably influence the thrombotic and hemorrhagic manifestations in different clinical settings or different phases of a vascular disease.
Acknowledgments
We thank the Blood Bank of the Catholic University School of Medicine of Rome for the generous gift of outdated platelet concentrates used to purify the GpIbα-(1–282) fragment.
This work was supported by Italian Ministry of University “PRIN-2005.” The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ABE-I/II, thrombin anion binding exosite I/II; MFI, mean fluorescence intensity; PAR, protease-activated receptor; AP, activating peptide; RP-HPLC, reversed-phase high performance liquid chromatography; S-2238, d-Phe-Pip-Arg-pNA; Pip, pipecolyl; pNA, p-nitroanilide; PEG, polyethylene glycol; Gp, glycoprotein; ssDNA, single-stranded DNA; PDB, Protein Data Bank.
References
- 1.Mosesson, M. W. (2003) J. Thromb. Haemost. 1 231-238 [DOI] [PubMed] [Google Scholar]
- 2.Seegers, W. H., Nieft, M., and Loomis, E. C. (1945) Science 101 520-521 [DOI] [PubMed] [Google Scholar]
- 3.Fredenburgh, J. C., Stafford, A. R., Leslie, B. A., and Weitz, J. I. (2008) J. Biol. Chem. 283 2470-2477 [DOI] [PubMed] [Google Scholar]
- 4.Chung, D. W., and Fujikawa, K. (2002) Biochemistry 41 11065-11070 [DOI] [PubMed] [Google Scholar]
- 5.Farrell, D. H., Mulvihill, E. R., Huang, S. M., Chung, D. W., and Davie, E. W. (1991) Biochemistry 30 9414-9420 [DOI] [PubMed] [Google Scholar]
- 6.Uitte de Willige, S., de Visser, M. C., Houwing-Duistermaat, J. J., Rosendaal, F. R., Vos, H. L., and Bertina, R. M. (2005) Blood 106 4176-4183 [DOI] [PubMed] [Google Scholar]
- 7.Lovely, R. S., Falls, L. A., Al-Mondhiry, H. A., Chambers, C. E., Sexton, G. J., Ni, H., and Farrell, D. H. (2002) Thromb. Haemostasis 88 26-31 [PubMed] [Google Scholar]
- 8.Meh, D. A., Siebenlist, K. R., and Mosesson, M. W. (1996) J. Biol. Chem. 271 23121-23125 [DOI] [PubMed] [Google Scholar]
- 9.Pineda, A. O., Chen, Z. W., Marino, F., Mathews, F. S., Mosesson, M. W., and Di Cera, E. (2007) Biophys. Chem. 125 556-559 [DOI] [PubMed] [Google Scholar]
- 10.Cooper, A. V., Standeven, K. F., and Ariens, R. A. (2003) Blood 102 535-540 [DOI] [PubMed] [Google Scholar]
- 11.Falls, L. A., and Farrell, D. H. (1997) J. Biol. Chem. 272 14251-14256 [DOI] [PubMed] [Google Scholar]
- 12.Mannila, M. N., Eriksson, P., Lundman, P., Samnegard, A., Boquist, S., Ericsson, C. G., Tornvall, P., Hamsten, A., and Silveira, A. (2005) Thromb. Haemostasis 93 570-577 [DOI] [PubMed] [Google Scholar]
- 13.Drouet, L., Paolucci, F., Pasqualini, N., Laprade, M., Ripoll, L., Mazoyer, E., Bal dit Sollier, C., and Vanhove, N. (1999) Blood Coagul. Fibrinolysis 10 S35-S39 [PubMed] [Google Scholar]
- 14.Kuyas, C., Haeberli, A., and Straub, P. W. (1982) J. Biol. Chem. 257 1107-1109 [PubMed] [Google Scholar]
- 15.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995) Protein Sci. 4 2411-2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Cristofaro, R., Rocca, B., Bizzi, B., and Landolfi, R. (1993) Biochem. J. 289 475-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richardson, J. L., Kroger, B., Hoeffken, W., Sadler, J. E., Pereira, P., Huber, R., Bode, W., and Fuentes-Prior, P. (2000) EMBO J. 19 5650-5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atherton, E., and Sheppard, R. C. (1989) Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, Oxford, UK
- 19.De Cristofaro, R., and De Filippis, V. (2003) Biochem. J. 373 593-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Cristofaro, R., De Candia, E., Rutella, S., and Weitz, J. I. (2000) J. Biol. Chem. 275 3887-3895 [DOI] [PubMed] [Google Scholar]
- 21.Tasset, D. M., Kubik, M. F., and Steiner, W. (1997) J. Mol. Biol. 272 688-698 [DOI] [PubMed] [Google Scholar]
- 22.De Candia, E., Hall, S. W., Rutella, S., Landolfi, R., Andrews, R. K., and De Cristofaro, R. (2001) J. Biol. Chem. 276 4692-4698 [DOI] [PubMed] [Google Scholar]
- 23.De Cristofaro, R., Peyvandi, F., Palla, R., Lavoretano, S., Lombardi, R., Merati, G., Romitelli, F., Di Stasio, E., and Mannucci, P. M. (2005) J. Biol. Chem. 280 23295-23302 [DOI] [PubMed] [Google Scholar]
- 24.Liu, L. W., Vu, T. K., Esmon, C. T., and Coughlin, S. R. (1991) J. Biol. Chem. 266 16977-16980 [PubMed] [Google Scholar]
- 25.Verhamme, I. M., Olson, S. T., Tollefsen, D. M., and Bock, P. E. (2002) J. Biol. Chem. 277 6788-6798 [DOI] [PubMed] [Google Scholar]
- 26.Fredenburgh, J. C., Stafford, A. R., and Weitz, J. I. (1997) J. Biol. Chem. 272 25493-25499 [DOI] [PubMed] [Google Scholar]
- 27.Richardson, J. L., Fuentes-Prior, P., Sadler, J. E., Huber, R., and Bode, W. (2002) Biochemistry 41 2535-2542 [DOI] [PubMed] [Google Scholar]
- 28.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishing Corp., New York
- 29.Soslau, G., Class, R., Morgan, D. A., Foster, C., Lord, S. T., Marchese, P., and Ruggeri, Z. M. (2001) J. Biol. Chem. 276 21173-21183 [DOI] [PubMed] [Google Scholar]
- 30.Mathews, I. I., Padmanabhan, K. P., Ganesh, V., Tulinsky, A., Ishii, M., Chen, J., Turck, C. W., Coughlin, S. R., and Fenton, J. W., II (1994) Biochemistry 33 3266-3279 [DOI] [PubMed] [Google Scholar]
- 31.Kirschbaum, N. E., Mosesson, M. W., and Amrani, D. L. (1992) Blood 79 2643-2648 [PubMed] [Google Scholar]
- 32.Dumas, J. J., Kumar, R., Seehra, J., Somers, W. S., and Mosyak, L. (2003) Science 301 222-226 [DOI] [PubMed] [Google Scholar]
- 33.Mayo, K. H., Burke, C., Lindon, J. N., and Kloczewiak, M. (1990) Biochemistry 29 3277-3286 [DOI] [PubMed] [Google Scholar]
- 34.Kostelansky, M. S., Betts, L., Gorkun, O. V., and Lord, S. T. (2002) Biochemistry 41 12124-12132 [DOI] [PubMed] [Google Scholar]
- 35.Spraggon, G., Everse, S. J., and Doolittle, R. F. (1997) Nature 389 455-462 [DOI] [PubMed] [Google Scholar]
- 36.Yee, V. C., Pratt, K. P., Cote, H. C., Trong, I. L., Chung, D. W., Davie, E. W., Stenkamp, R. E., and Teller, D. C. (1997) Structure (Lond.) 5 125-138 [DOI] [PubMed] [Google Scholar]
- 37.Wyman, J., and Gill, S. J. (1990) Binding and Linkage: Functional Chemistry of Biological Macromolecules, pp. 33-39 University Science Books, Mill Valley, CA
- 38.Faruqi, T. R., Weiss, E. J., Shapiro, M. J., Huang, W., and Coughlin, S. R. (2000) J. Biol. Chem. 275 19728-19734 [DOI] [PubMed] [Google Scholar]
- 39.Cleary, D. B., Trumbo, T. A., and Maurer, M. C. (2002) Arch. Biochem. Biophys. 403 179-188 [DOI] [PubMed] [Google Scholar]
- 40.Jacques, S. L., and Kuliopulos, A. (2003) Biochem. J. 376 733-740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bah, A., Chen, Z., Bush-Pelc, L. A., Mathews, F. S., and Di Cera, E. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 11603-11608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vijayalakshmi, J., Padmanabhan, K. P., Mann, K. G., and Tulinsky, A. (1994) Protein Sci. 3 2254-2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacques, S. L., LeMasurier, M., Sheridan, P. J., Seeley, S. K., and Kuliopulos, A. (2000) J. Biol. Chem. 275 40671-40678 [DOI] [PubMed] [Google Scholar]
- 44.Bock, P. E. (1992) J. Biol. Chem. 267 14974-14981 [PubMed] [Google Scholar]
- 45.Bock, P. E. (1992) J. Biol. Chem. 267 14963-14973 [PubMed] [Google Scholar]
- 46.Henry, B. L., Monien, B. H., Bock, P. E., and Desai, U. R. (2007) J. Biol. Chem. 282 31891-31899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jandrot-Perrus, M., Clemetson, K. J., Huisse, M. G., and Guillin, M. C. (1992) Blood 80 2781-2786 [PubMed] [Google Scholar]
- 48.Arni, R. K., Padmanabhan, K., Padmanabhan, K. P., Wu, T. P., and Tulinsky, A. (1993) Biochemistry 32 4727-4737 [DOI] [PubMed] [Google Scholar]
- 49.Carter, W. J., Cama, E., and Huntington, J. A. (2005) J. Biol. Chem. 280 2745-2749 [DOI] [PubMed] [Google Scholar]
- 50.Celikel, R., McClintock, R. A., Roberts, J. R., Mendolicchio, G. L., Ware, J., Varughese, K. I., and Ruggeri, Z. M. (2003) Science 301 218-221 [DOI] [PubMed] [Google Scholar]
- 51.Sabo, T. M., Farrell, D. H., and Maurer, M. C. (2006) Biochemistry 45 7434-7445 [DOI] [PubMed] [Google Scholar]
- 52.Rydel, T. J., Tulinsky, A., Bode, W., and Huber, R. (1991) J. Mol. Biol. 221 583-601 [DOI] [PubMed] [Google Scholar]
- 53.Furlan, M., Robles, R., and Lamie, B. (1996) Blood 87 4223-4234 [PubMed] [Google Scholar]
- 54.Mannila, M. N., Lovely, R. S., Kazmierczak, S. C., Eriksson, P., Samnegard, A., Farrell, D. H., Hamsten, A., and Silveira, A. (2007) J. Thromb. Haemost. 5 766-773 [DOI] [PubMed] [Google Scholar]
- 55.Cheung, E. Y., de Willige, S. U., Vos, H. L., Leebeek, F. W., Dippel, D. W., Bertina, R. M., and de Maat, M. P. (2008) Stroke 39 1033-1035 [DOI] [PubMed] [Google Scholar]
- 56.Thomas, D. P., and Roberts, H. R. (1997) Ann. Intern. Med. 126 638-644 [DOI] [PubMed] [Google Scholar]
- 57.Mannila, M. N., Eriksson, P., Leander, K., Wiman, B., de Faire, U., Hamsten, A., and Silveira, A. (2007) J. Intern. Med. 261 138-147 [DOI] [PubMed] [Google Scholar]
- 58.Duan, H. O., and Simpson-Haidaris, P. J. (2003) J. Biol. Chem. 278 41270-41281 [DOI] [PubMed] [Google Scholar]
- 59.Lovely, R. S., Boshkov, L. K., Marzec, U. M., Hanson, S. R., and Farrell, D. H. (2007) Br. J. Haematol. 139 494-503 [DOI] [PubMed] [Google Scholar]
- 60.Lancellotti, S., and De Cristofaro, R. (2008) Cardiovascular and Haematological Agents in Medicinal Chemistry, Bentham Sciences, in press
- 61.Everse, S. J., Pelletier, H., and Doolittle, R. F. (1995) Protein Sci. 4 1013-1016 [DOI] [PMC free article] [PubMed] [Google Scholar]