

Abstract
Caspase-3 is an attractive therapeutic target for treatment of diseases involving disregulated apoptosis. We report here the mechanism of caspase-3 inactivation by isoquinoline-1,3,4-trione derivatives. Kinetic analysis indicates the compounds can irreversibly inactivate caspase-3 in a 1,4-dithiothreitol (DTT)- and oxygen-dependent manner, implying that a redox cycle might take place in the inactivation process. Reactive oxygen species detection experiments using a chemical indicator, together with electron spin resonance measurement, suggest that ROS can be generated by reaction of isoquinoline-1,3,4-trione derivatives with DTT. Oxygen-free radical scavenger catalase and superoxide dismutase eliciting the inactivation of caspase-3 by the inhibitors confirm that ROS mediates the inactivation process. Crystal structures of caspase-3 in complexes with isoquinoline-1,3,4-trione derivatives show that the catalytic cysteine is oxidized to sulfonic acid (-SO3H) and isoquinoline-1,3,4-trione derivatives are bound at the dimer interface of caspase-3. Further mutagenesis study shows that the binding of the inhibitors with caspase-3 appears to be nonspecific. Isoquinoline-1,3,4-trione derivative-catalyzed caspase-3 inactivation could also be observed when DTT is substituted with dihydrolipoic acid, which exists widely in cells and might play an important role in the in vivo inactivation process in which the inhibitors inactivate caspase-3 in cells and then prevent the cells from apoptosis. These results provide valuable information for further development of small molecular inhibitors against caspase-3 or other oxidation-sensitive proteins.
Apoptosis (or programmed cell death) is a physiological mechanism that is crucial for normal development of organisms during embryogenesis, maintenance of tissue homeostasis in adults, and elimination of diseased or otherwise harmful cells during pathogenesis (1–4). Disregulated apoptosis has been implicated in many human diseases, including neurodegenerative diseases such as Alzheimer disease and Huntington disease, ischemic damage, autoimmune disorders, and several forms of cancer (5–7).
Genetic and biochemical studies have demonstrated that a proteolytic cascade controlled by the caspase (cysteine aspartyl-specific protease) family is central to the apoptotic process (8–11). Caspase-3 is a member of effector caspases and has been found to integrate upstream signals into final execution of cell death (12). This makes caspase-3 a potential therapeutic target for treatment of diseases involving disregulated apoptosis.
Ongoing endeavors of many pharmaceutical companies and academic institutions have led to the identification of a series of small-molecule inhibitors of caspase-3, including N-nitrosoanilines (13), dithiocarbamate (14), anilinoquinazolines (15), isatin sulfonamide analogues (16–18), 1,3-dioxo-2,3-dihydro-1H-pyrrolo[3,4-c]quinolines (19), 2-(2,4-dichlorophenoxy)-N-(2-mercaptoethyl)-acetamide, and 5-fluoro-1H-indole-2-carboxylic acid (2-mercaptoethyl)-amide (20). These inhibitors can inhibit the caspase-3 activity with different inhibition mechanisms: N-nitrosoanilines are general inhibitors of cysteine-dependent enzymes; dithiocarbamates are able to block the activation of the caspase-3 proenzyme (21); anilinoquinazolines and isatin sulfonamide analogues are competitive inhibitors of caspase-3 (16); 1,3-dioxo-2,3-dihydro-1H-pyrrolo[3,4-c]quinolines are reversible and noncompetitive inhibitors of caspase-3 (19); and 2-(2,4-dichlorophenoxy)-N-(2-mercaptoethyl)-acetamide and 5-fluoro-1H-indole-2-carboxylic acid (2-mercaptoethyl)-amide are able to bind at an allosteric site and can inhibit both caspase-3 and caspase-7. Several of these compounds could prevent apoptosis in cell-based models such as Jurkat cells, chondrocytes, and mouse bone marrow neutrophils (15–17, 19).
Previously we have identified isoquinoline-1,3,4-trione as a novel caspase-3 inhibitor through high-throughput screening of a small molecule library using recombinant caspase-3 (22, 23). A series of isoquinoline-1,3,4-trione derivatives developed through structural modification exhibit nanomolar potency against caspase-3 in vitro and are validated as selective, irreversible, slow-binding and pancaspase inhibitors. Furthermore, we have found that these inhibitors could attenuate apoptosis induced by β-amyloid (25–35) in PC12 cells and primary neuronal cells (23) and decrease in infarct volume in the transient middle cerebral artery occlusion stroke model (22). These caspase-3 inhibitors with novel structures have provided a new therapeutic strategy directed against diseases involving abnormally up-regulated apoptosis. With kinetic assay and structural analysis, we report here that a redox cycle is responsible for the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives.
EXPERIMENTAL PROCEDURES
Materials—1,4-Dithiothreitol (DTT),4 (±)-α-lipoic acid, dihydrolipoic acid, 2′,7′-dichlorofluorescein diacetate (H2DCFDA), 5,5-dimethyl-1-pyrolline-N-oxide, catalase from bovine liver (2000–5000 units/mg), and superoxide dismutase from horseradish (3000 units/mg) were purchased from Sigma. Caspase-3 peptide substrate Ac-DEVD-pNA was synthesized in this laboratory. Peptide inhibitor Ac-DEVD-CHO was purchased from Bachem Bioscience (King of Prussia, PA). H2DCFDA and peptide were dissolved in dimethyl sulfoxide (DMSO). The concentrated catalase and superoxide dismutase stock solutions were prepared in 50 mm K2HPO4, pH 7.4. Other reagents and solvents are of analytical grade.
Expression and Purification of Recombinant Caspase-3 and Its Mutants—The catalytic domain of human caspase-3 was expressed according to the procedure described previously (23). The cells that contained the His6-tagged caspase-3 were lysed on ice by sonication and the insoluble cell debris was removed by centrifugation at 15,000 × g at 4 °C.
The lysis supernatant was loaded onto HiTrap Chelating HP column (GE Healthcare) or nickel-nitrilotriacetic acid beads (Novagen, Darmstadt, Germany) equilibrated with buffer A (50 mm HEPES, pH 7.4) and the His6-tagged caspase-3 was eluted with an elution buffer (buffer A supplemented with 250 mm imidazole). The eluted fractions were dialyzed against buffer A for 4 h and then loaded onto a HiPrep 16/10 Q anion exchange column (GE Healthcare) and the target protein was eluted in fractions containing buffer A with 200–350 mm NaCl. The eluted fractions were pooled together and then loaded on a HiPrep 16/10 Phenyl column (GE Healthcare), which was preequilibrated with buffer B (buffer A supplemented with 500 mm NaCl) and the target protein existing in the flow-through fractions was frozen at -80 °C for further use. The protein sample was desalted into buffer C (buffer A supplemented with 50 mm NaCl and 10 mm DTT) with a HiPrep 26/10 desalting column (GE Healthcare) for crystallization and into buffer D (buffer A supplemented with 2 mm EDTA) prior to kinetic assay. Protein concentrations were determined by the Bradford method with bovine serum albumin as the standard. SDS-PAGE and dynamic light scattering analyses indicated that the protein sample had high purity and homogeneity.
The mutant caspase-3 were constructed using the QuikChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA) following the manual of the supplier with the pET32b-caspase-3 (23) as the template. All constructs were verified by DNA sequencing. The mutant caspase-3 were expressed and purified using the same method as the wild type caspase-3.
Caspase-3 Activity Assay and Kinetic Study—The activity assay of caspase-3 was carried out in a system of 100 μl containing 50 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, 100 μm Ac-DEVD-pNA, 20 nm caspase-3, and 2 mm of a reducing agent (as indicated in the text), in the presence or absence of 2 μl of inhibitor in DMSO. The rate of hydrolysis product, pNA, was monitored continuously by change of absorbance at 405 nm for 5 min, and the initial rate of hydrolysis was determined using the early linear region of the enzymatic reaction curve. Continuous kinetic monitoring was performed in clear 96-well plates (Corning, Lowell, MA) on Envision (PelkinElmer Life Sciences) controlled by Wallac EnVision Manager.
To obtain the observed inactivation rate (kobs) at a specific concentration of inhibitor, caspase-3 was preincubated with the compound for various times prior to initiation of the reaction with Ac-DEVD-pNA. Data from such an experiment can be fit to the following equation: vt = vi exp(-kobs t) (24), where vt is the measured steady state velocity after preincubation of time t, and vi is the steady state velocity at preincubation of time 0. By these methods we can obtain the kobs at varying concentrations of inhibitor, and then use the results to compute an apparent inactivation rate constant (kobs/I) to quantify the effectiveness of the inhibitor.
To create oxygen-free conditions, caspase-3 in the assay buffer (containing 2 mm DTT) was exchanged with nitrogen for three cycles in a rubber septum-sealed round-bottom flask. Then a concentrated inhibitor solution (30 times) was injected into the mixture under nitrogen. After incubation for a specified time, 90 μl of the mixture was taken out by syringe to measure the residual activity.
Analysis of Kinetic Data—The kinetic data were analyzed by GraphPad Prism 4.00 (GraphPad Software) and presented as the mean ± S.E. from at least three independent experiments unless otherwise specified. The IC50 value was calculated in GraphPad Prism using nonlinear regression analysis and a sigmoidal dose-response (variable slope) equation. The time-dependent reaction progress curve was approximated by a first-order equation (24). The slope of the linear response region, indexed as the apparent inactivation rate constant, was used to determine the potency of the inhibitors.
ROS Production Determination—ROS production was monitored by using H2DCFDA. This dye is a stable non-fluorescent compound until oxidized by hydrogen peroxide or low molecular weight peroxides (25, 26). The 100-μl assay system contained 10 μm H2DCFDA, 2 μl of inhibitor, and reducing agents (as specified in the text) in the caspase-3 assay buffer. Oxidation of the probe was detected in a clear 96-well plate by monitoring the increase of fluorescence with Envision, at 490 nm excitation and 525 nm emission (top readout). The fluorescence intensity after reaction for 90 min (except continuous kinetic monitoring) was used to quantify the ROS signal.
Crystallization and Diffraction Data Collection—Crystallization was performed at 4 °C using the hanging-drop vapor diffusion method. The inhibitors I–IV were dissolved in DMSO to a concentration of 25 mm. To prepare crystals of the binary complexes, the purified caspase-3 protein solution (∼0.2 mg/ml) was first mixed with the inhibitor solution in a molar ratio of 1:40 and then incubated at 4 °C overnight. The protein samples were concentrated to about 4 mg/ml before crystallization setup. Sheet-shaped crystals of the binary complexes were grown in drops containing equal volumes (2 μl) of the protein mixture solution and the reservoir solution (4–6% polyethylene glycol 6000, 0.1 m HEPES, pH 7.6, 20 mm l-cysteine, and 5% glycerol) to a maximum size of 0.3 × 0.3 × 0.02 mm3 in 1 week. Diffraction data were collected to 2.4–2.8-Å resolution from flash-cooled crystals at -176 °C at beamline 17A of Photon Factory, Japan, and processed with the HKL2000 suite (27). Crystals of these complexes belong to space group P212121 containing four caspase-3 molecules and one inhibitor molecule in the asymmetric unit with a solvent content of ∼50%. Summary of the diffraction data statistics are given in Table 3.
TABLE 3.
Summary of diffraction data and structure refinement statistics
|
Caspase-3
|
||||
|---|---|---|---|---|
| Compound I | Compound II | Compound III | Compound IV | |
| Statistics of diffraction data | ||||
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Resolution range (Å)a | 50.0–2.50 (2.59–2.50) | 50.0–2.80 (2.90–2.80) | 50.0–2.60 (2.69–2.60) | 50.0–2.40 (2.49–2.40) |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell parameters | ||||
| a (Å) | 65.1 | 65.7 | 65.4 | 65.2 |
| b (Å) | 96.9 | 96.5 | 96.7 | 96.3 |
| c (Å) | 180.4 | 180.5 | 180.4 | 180.2 |
| Observed reflections | 180,300 | 137,332 | 156,710 | 196,328 |
| Unique reflections | 36,312 | 27,196 | 35,800 | 41,447 |
| Average redundancy | 5.0 (4.7) | 5.0 (4.7) | 4.4 (3.0) | 4.7 (2.9) |
| Average I/σ(I) | 13.9 (2.0) | 15.0 (1.9) | 13.2 (1.3) | 17.1 (1.8) |
| Completeness (%) | 90.1 (70.8) | 94.3 (94.7) | 99.2 (94.1) | 91.5 (90.8) |
| Rmerge (%)b | 11.4 (43.5) | 12.8 (53.0) | 12.1 (48.2) | 9.1 (39.6) |
| Statistics of refinement | ||||
| Number of reflections | 36,280 | 27,133 | 35,739 | 41,413 |
| Working set | 34,487 | 25,767 | 33,954 | 39,360 |
| Free R set | 1,793 | 1,366 | 1,785 | 2,053 |
| R factor (%)c | 20.8 | 21.1 | 21.4 | 20.9 |
| Free R factor (%) | 26.8 | 29.1 | 27.4 | 25.3 |
| Number of residues | 930 | 933 | 930 | 936 |
| Number of water molecules | 181 | 74 | 124 | 241 |
| Average B factor of all atoms (Å2) | 59.9 | 72.4 | 61.7 | 56.3 |
| Caspase-3 | 60.0 | 72.6 | 61.9 | 56.4 |
| Inhibitor | 48.8 | 63.0 | 50.6 | 48.5 |
| Water molecule | 55.4 | 53.8 | 50.7 | 53.7 |
| r.m.s. deviationd bond lengths (Å) | 0.010 | 0.012 | 0.010 | 0.009 |
| r.m.s. deviation bond angles (°) | 1.2 | 1.4 | 1.2 | 1.2 |
| Ramachandran plot (%) | ||||
| Ramachandran favored | 96.3 | 95.3 | 96.5 | 97.2 |
| Ramachandran allowed | 99.7 | 99.6 | 99.6 | 99.8 |
| Ramachandran outliers | 0.3 | 0.4 | 0.4 | 0.2 |
Numbers in parentheses refer to the highest resolution shell
Rmerge = ΣhklΣi|Ii(hkl)i – 〈I(hkl)〉|/ΣhklΣiIi(hkl)
Rfactor = Σhkl||Fo| – |Fc||/Σhkl|Fo|
r.m.s. deviation, root mean square deviation
Structure Determination and Refinement—The structures of the caspase-3-inhibitor complexes were solved by molecular replacement with the program CNS (28) using the coordinates of caspase-3 in complex with an isatin sulfonamide inhibitor (PDB access code 1GFW) (16) as the search model. In the initial difference Fourier map there was evident electron density corresponding to the bound inhibitor in each complex.
Structure refinement was carried out with CNS using standard protocols (energy minimization, simulated annealing, and B factor refinement) and model building was facilitated using the program O (29). There are four caspase-3 molecules in the asymmetric unit; thus strict 4-fold non-crystallographic symmetry constraints were applied in the early stage of refinement, but released in the later stage of refinement. The final structure refinement was performed using the maximum likelihood algorithm implemented in the program REFMAC5 (30). A bulk solvent correction and a free R factor monitor (calculated with 5% of randomly chosen reflections) were applied throughout the refinement. The stereochemical quality of the structure models was evaluated with the program MOLPROBITY (31). The statistics of structure refinement are summarized in Table 3.
RESULTS
Time-dependent Irreversible Inactivation of Caspase-3 by Isoquinoline-1,3,4-trione Derivatives—Under aerobic conditions and in the presence of 2 mm DTT, isoquinoline-1,3,4-trione derivatives potently inhibit caspase-3 in a time- and dose-dependent manner as shown in Fig. 1A. The activity of caspase-3, monitored continuously by the production of pNA from Ac-DEVD-pNA, decreased in a pseudo-first order process. Volume dilution and dialysis methods indicated that the inhibition is irreversible (23). The observed rate of inactivation increased nearly linearly with increased concentration of compound III, yielding an apparent inactivation rate constant (kobs/I) of 20,098 ± 867 s-1 m-1 within the linear response zone (Fig. 1B). In the presence of 0.45 μm compound III, the half-life of the inactivation was 1.33 min. At higher concentrations of compound, the inactivation rate was too rapid to be quantified accurately. As shown in Fig. 2, the inactivation rate constant is consistent well with the IC50 value that was used a guide in the structural modification of the inhibitors. Interestingly, compound V, which is structurally related to compound IV but has a disrupted ketone (trione), shows no inhibition for caspase-3 even at 400 μm.
FIGURE 1.

Time-dependent inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives under air at room temperature. A, progress curves for caspase-3 reaction with Ac-DEVD-pNA in the presence of increasing concentrations of compound III. The assay system containing 2% DMSO was defined for control. The progress curves were fitted by use of a first-order equation to determine the observed inactivation rate (kobs). B, a replot of kobs for the progress curves in A as a function of inhibitor concentration. The slope (kobs/I) of the linear plot was used to quantify the effectiveness of inactivation.
FIGURE 2.

Chemical structures of isoquinoline-1,3,4-trione derivatives. IC50 and kobs/I (mean ± S.E., n > 3) were obtained in 50 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, and 2 mm DTT. IC50 values were calculated from the residual activity of caspase-3 after incubation with 3 times dilution compounds for 15 min. ND, not determined.
DTT-dependent Inactivation of Caspase-3 by Isoquinoline-1,3,4-trione Derivatives—To evaluate the thiol requirement, the DTT concentration was increased from 0 to 20 mm in the assay system (Table 1). Within 0 to 0.05 mm DTT, the spontaneous loss of the caspase-3 activity had a varied half-life from 60 to 100 min. The presence of 0.1 μm compound III only perturbed slightly the loss of the activity with an apparent half-life of about 60–50 min (n = 3). However, when the DTT concentration was higher than 0.1 mm, the apparent half-life of caspase-3 increased gradually to about 2000 min. At the same time, the presence of 0.1 μm compound III dramatically reduced the apparent half-life to about 4 min (n = 3). These results indicate that the increased DTT concentrations significantly accelerated the rapid inactivation of caspase-3 mediated by isoquinoline-1,3,4-trione derivatives under aerobic conditions.
TABLE 1.
DTT dose-dependent inactivation of caspase-3 by 0.1 μm compound III The half-life (t½) for the inactivation at given condition was defined as t½ = 0.693/kobs.
|
DTT
|
t½
|
||
|---|---|---|---|
| 2% DMSO | 0.1 μm Compound III | ||
| mm | min | ||
| 0.00 | 61.8 ± 1.0 | 62.3 ± 3.6 | |
| 0.02 | 67.1 ± 4.4 | 60.8 ± 0.2 | |
| 0.04 | 90.8 ± 0.8 | 49.7 ± 3.8 | |
| 0.08 | 546.9 ± 43.7 | 41.2 ± 4.6 | |
| 0.16 | 719.6 ± 14.1 | 26.6 ± 1.8 | |
| 0.31 | 825.7 ± 19.0 | 12.0 ± 2.5 | |
| 0.63 | 933.6 ± 83.9 | 8.9 ± 0.6 | |
| 1.25 | 1352.8 ± 54.4 | 4.7 ± 0.2 | |
| 2.50 | 1428.9 ± 122.1 | 4.5 ± 0.1 | |
| 5.00 | 1799.0 ± 118.3 | 4.3 ± 0.3 | |
| 10.00 | 1940.6 ± 46.3 | 3.3 ± 0.5 | |
| 20.00 | 2528.1 ± 18.9 | 2.4 ± 0.0 | |
Effects of a series of thiol-containing reducing agents on the potency of isoquinoline-1,3,4-trione derivatives were also studied. The presence of 2 mm reduced glutathione (GSH), l-cystine, or β-mercaptoethanol protected caspase-3 from inactivation with a different potency. However, these agents had only weak effects on the inactivation by compound III (even in 100 μm) (Table 2). Lipoic acid and some other dithiol-containing agents (such as 1,2-butanedithiol, 1,4-butanedithiol, 1,2-benzenedithiol, and 4,4′-thiobisbenzenthiol) have been reported to be able to facilitate 1,3-dioxo-2,3-dihydro-1H-pyrrolo[3,4-c]quinolines to produce the same potent inhibition of caspase-3 as DTT (32). However, our results show that dihydrolipoic acid (reduced lipoic acid) but not lipoic acid has a great effect on the sensitivity of caspase-3 to isoquinoline-1,3,4-trione derivatives (Table 2). Hence, the effects of isoquinoline-1,3,4-trione derivatives on the caspase-3 inactivation are specific to DTT and dihydrolipoic acid at least for the agents under investigation.
TABLE 2.
Effects of different redox compounds on the inactivation of caspase-3 by compound III
|
t½
|
|||||||
|---|---|---|---|---|---|---|---|
| 2% DMSO | 0.1 μm Compound III | 10 μm Compound III | 100 μm Compound III | ||||
| min | |||||||
| Redox free | 145 ± 2 | 137 ± 7 | 142 ± 2 | 122 ± 9 | |||
| GSH | 1525 ± 44 | 1715 ± 18 | 1214 ± 225 | 1343 ± 49 | |||
| β-ME | 1693 ± 53 | 1673 ± 12 | 1639 ± 51 | 1762 ± 130 | |||
| l-Cystine | 1697 ± 35 | 1663 ± 85 | 1623 ± 16 | 1557 ± 99 | |||
| Lipoic acid | 1193 ± 222 | 1489 ± 31 | 1168 ± 47 | 633 ± 101 | |||
| Dihydrolipoic acid | 167 ± 5 | 1.3 ± 0.1 | NDa | ND | |||
| DTT | 1191 ± 60 | 4.1 ± 0.2 | ND | ND | |||
ND, not determined
Oxygen-dependent Inactivation of Caspase-3 by Isoquinoline-1,3,4-trione Derivatives—To determine the oxygen dependence of the caspase-3 inactivation, caspase-3 was incubated with compound III in the presence and absence of air. Anaerobic conditions were created by conducting the experiment under nitrogen in an oxygen-free buffer (see “Experimental Procedures”). As shown in Fig. 3, compound III mediates a time-dependent and complete inactivation of caspase-3 under aerobic conditions. The inactivation followed a first-order process with a half-life of 4.25 min at 0.1 μm compound III. At 0.4 μm compound III, the inactivation was reduced proportionally to a half-life of 1.37 min. In contrast to the case in the oxygenic conditions, inhibition in the oxygen-free conditions is negligible. Thus, our results indicate that the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives depends on the presence of oxygen.
FIGURE 3.

Oxygen-dependent inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives. Caspase-3 was incubated with 0.1 (□) or 0.4 μm (○) compound III in air (solid) or under nitrogen (open) at room temperature. The residual caspase-3 activity was determined after different incubation times and was normalized to control (caspase-3 activity that no incubation).
ROS Mediate the Inactivation of Caspase-3 by Isoquinoline-1,3,4-trione Derivatives—To investigate if the redox cycle participates in the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives, a chemically reduced form of H2DCFDA was used as an indicator for reactive oxygen species. The detected free radical signal displayed compound III and DTT in dose-dependent manners (Fig. 4, A and B). In 2 mm DTT-containing buffer, a strong ROS signal could be detected in the presence of 2 μm compounds I-IV but not compound V. There was no ROS signal produced by compounds I-IV for those reducing agents that cannot accelerate isoquinoline-1,3,4-trione derivatives-mediated caspase-3 inactivation (Fig. 4C). However, in the presence of 2 mm dihydrolipoic acid, the ROS signal was 10 times higher than that produced with 2 mm DTT (Fig. 4D). Electron spin resonance spin trapping with 5,5-dimethyl-1-pyrolline-N-oxide also confirmed the generation of free radicals (supplemental Fig. S1). The nature of the ROS signal detected in the electron spin resonance experiments is currently under investigation. Taken together, these results demonstrate that isoquinoline-1,3,4-trione derivatives can react with DTT or dihydrolipoic acid to generate ROS.
FIGURE 4.

Isoquinoline-1,3,4-trione derivatives react with DTT or dihydrolipoic acid to produce ROS. A, compound III dose-dependent production of free radical signals detected by H2DCFA. The assay system contained 1 mm DTT and various concentrations of compound III. B, DTT dose-dependent production of free radicals signal. The assay system contained 0.2 μm compound III and various concentrations of DTT. C, effect of different reducing agents on production of free radicals signal by isoquinoline-1,3,4-trione derivatives and compound V. The isoquinoline-1,3,4-trione derivative concentration was 2 μm, the reducing agent concentration was 2 mm. Assay in reducer-free condition was defined as control. D, effect of dihydrolipoic acid on production of free radicals signal by isoquinoline-1,3,4-trione derivatives. β-ME, β-mercaptoethanol.
To further confirm the generation of ROS, we examined the effects of the oxygen-free radical scavenger, catalase and superoxide dismutase on the caspase-3 inactivation by isoquinoline-1,3,4-trione derivatives. As shown in Fig. 5A, catalase (150 units/ml) or superoxide dismutase (35 units/ml) could significantly protect caspase-3 from inactivation at 0.2 μm compound III. Furthermore, concurrent with the protection of the caspase-3 activity, catalase or superoxide dismutase could also reduce the ROS signal generated by compound III (Fig. 5B), whereas catalase and superoxide dismutase alone had a negligible effect on the ROS signal.
FIGURE 5.

ROS mediates the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives. A, catalase (150 units/ml) or superoxide dismutase (35 units/ml) protected caspase-3 over 60 min from inactivation caused by 0.2 μm compound III with 2 mm DTT in air. The assay system contained 0.2 μm compound III and 2 mm DTT but no catalase or superoxide dismutase was defined as control. B, catalase (150 units/ml) or superoxide dismutase (35 units/ml) reduced ROS signal generated by compound III. Control was defined as A. C, caspase-3 reduced the ROS signal produced by 2 μm compound III with 2 mm DTT. The assay system contained 2 μm compound III or 1 mm H2O2 but no DTT or caspase-3 was defined as control. D, correlation between inhibition of caspase-3 (solid line) and generation of ROS (dashed line) by different concentrations of isoquinoline-1,3,4-trione derivatives.
To explore the effect of caspase-3 on the ROS signal, 100 nm caspase-3 was supplied to the assay system. Similar to that H2O2 is able to be consumed by caspase-3, the ROS signal generated by isoquinoline-1,3,4-trione derivatives can also be reduced by caspase-3 (Fig. 5C). Interestingly, although the ROS signal produced by compound III only emerged in the presence of DTT, the presence of DTT would reduce the ROS signal produced by H2O2. Furthermore, the intensity of the ROS signal produced by different isoquinoline-1,3,4-trione derivatives is positively correlated with their inactivation potential on caspase-3 (Fig. 5D). These results confirmed that ROS is involved in the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives.
Overall Structure of the Caspase-3-Inhibitor Complexes—The crystal structures of caspase-3 in complexes with inhibitors I–IV were solved using molecular replacement and refined to 2.5-, 2.8-, 2.6-, and 2.4-Å resolution, respectively. There are four caspase-3 molecules and one inhibitor molecule in the asymmetric unit (Table 3 and Fig. 6A). Each caspase-3 molecule is composed of a large and a small subunit, cleaved at the intradomain site, IETD. The four caspase-3 molecules in each structure are structurally identical with an average root mean square deviation of about 0.3–0.6 Å based on superimposition of all Cα atoms. The root mean square deviation of the four complex structures is about 0.7–0.9 Å. In all four structures the four caspase-3 molecules form two dimers and have a similar overall structure as that reported previously (16).
FIGURE 6.

Crystal structures of caspase-3 in complexes with inhibitors. A, a ribbon diagram shows the overall structure of caspase-3. B, molecular surface maps represent the hydrophobic pocket with the bound inhibitors. C, a stereo view shows the composition of the hydrophobic pocket at the dimer interface and the oxidation of the catalytic cysteine (Cys163) to sulfonic acid. D, the four inhibitors are superimposed at the binding site within bound compounds I, II, III, and IV colored in magenta, green, cyan, and yellow, respectively. E, a stereo view shows the catalytic active site of molecule C. The 2Fo - Fc sa_omit_map (1σ contour level) for the oxidized catalytic cysteine and the residues nearby are shown with cyan meshes.
Specific Oxidation of the Catalytic Cysteine to Sulfonic Acid—Previous work has shown that the conserved cysteine residue (Cys163 in human caspase-3) at the active site functions as the nucleophile during the catalytic reaction and should be kept in the reduced form to perform the enzymatic activity. In our structures, the evident electron density indicates that the thiol group of the catalytic Cys163 of all four caspase-3 molecules had been oxidized to sulfonic acid (Fig. 6, C and E), whereas the other cysteines, such as Cys47, Cys116, Cys148, Cys220, and Cys264 were not oxidized. Moreover, Cys170, which is located on the surface near the active site also exists in the reduced form in all four structures.
Inhibitor Binding Site and Mutagenesis of Residues Nearby—In all four structures, there was good electron density for the bound inhibitor. The inhibitors are bound at a hydrophobic pocket at the interface of the two dimers. The hydrophobic pocket is composed of residues Thr166, Leu168, Tyr204, Trp206, Thr255, and Phe256 from molecules A and C (Fig. 6, B and C) and is located near the active site. The bound inhibitors have mainly hydrophobic contacts with the surrounding residues, and vary slightly in their positions, orientations, and interactions due to their different chemical structures (Fig. 6D).
We also performed the mutagenesis study of the amino acids (Thr166, Leu168, Tyr204, Trp206, Thr255, and Phe256) at the inhibitor-binding site. Consistent with the previous report that Leu168, Tyr204, Trp206, and Phe256 are involved in the catalytic reaction and the substrate-recognition of caspases (33, 34), mutation of these residues to alanine or arginine would affect the activities and the inhibition constants of Ac-DEVD-CHO for caspase-3 (Table 4). However, the potency of compound III against both the wild-type and mutant caspase-3 was not obviously affected, except the Y204A mutant. Because the potency of H2O2-mediated inactivation of the wild-type and Y204A mutant caspase-3 was affected in a similar manner, these results indicate that the difference between the Y204A mutant and the wild-type capase-3 could be attributed to their varied sensitivity to ROS rather than different binding potency with compound III (Table 4).
TABLE 4.
Inhibition constants determined for the wide type and mutant caspase-3 of Ac-DEVD-CHO compound III and H2O2 All IC50 values were obtained in 50 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, and 2 mm DTT and calculated from the residual activity of caspase-3 after incubation with 3 times dilution compounds for 15 min.
| Specific activity of enzyme | Ac-DEVD-CHO IC50 | Compound III IC50 | H2O2 IC50 | ||
|---|---|---|---|---|---|
| μm/min/μg | nm | μm | |||
| WT | 105.55 | 15.12 | 10.54 | 84.4 | |
| T166A | 82.08 | 11.18 | 8.74 | 96.8 | |
| T166K | 53.82 | 20.85 | 8.06 | 96.7 | |
| L168A | 5.39 | 37.73 | 9.55 | 56.2 | |
| L168K | 15.52 | 37.60 | 8.49 | 41.6 | |
| Y204A | 10.93 | 54.50 | 23.57 | 300.6 | |
| Y204K | 40.75 | 30.99 | 12.37 | 69.6 | |
| W206A | No activity | NDa | ND | ND | |
| W206K | No activity | ND | ND | ND | |
| T255A | 34.71 | 13.86 | 8.01 | 99.5 | |
| T255K | 69.88 | 22.52 | 7.49 | 63.6 | |
| F256A | 11.18 | 22.79 | 11.03 | 67.7 | |
| F256K | 15.18 | 39.47 | 9.48 | 74.4 | |
ND, not determined
DISCUSSION
The biochemical data presented here indicate that under oxygenic conditions isoquinoline-1,3,4-trione derivatives could react with dithiol reagents (such as DTT) to generate free radicals, which could inactivate caspase-3 through oxidation of the thiol group of the catalytic cysteine (Cys163) into sulfonic acid (-SO3H). Based on both biochemical and structural data, we propose the possible mechanism as follows (Fig. 7).
FIGURE 7.

Proposed scheme for the catalytic inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives through redox cycling. In the presence of DTT in vitro and possibly dihydrolipoic acid in vivo, isoquinoline-1,3,4-trione derivatives rapidly undergo reduction to the corresponding semiquinone anion radicals (RQ-). The reaction is reversible in the presence of atmospheric oxygen by reduction oxygen to ROS. The farther oxidation of DTT and dihydrolipoic acid intermediate also could generate ROS (38, 39). The produced ROS catalyzes the step by step oxidation of the active site cysteine of caspase-3 to the sulfonic acid. The semiquinone anion radicals may also contribute to the specific oxidation of the catalytic cysteine via a intermediate (Caspase-SH/RQ-). Caspase-SH, caspase-SOH, caspase-SO2H, and caspase-SO3H represent the thiol, sulfenic, sulfinic, and sulfonic acid states of the catalytic cysteine.
The irreversible inhibition of caspase-3 by isoquinoline-1,3,4-trione derivatives in an oxygen and DTT or dihydrolipoic acid-dependent manner was supported by our kinetic data. The oxidized DTT could also be observed in the purification process of the caspase-3-inhibitor complex (data not shown) because the oxidized DTT has strong absorbance at about 280 nm, whereas the reduced form does not (35, 36). On the other hand, the free radical signal was detected using H2DCFA as the detection regent (Fig. 4) and further confirmed by electron spin resonance measurement (supplemental Fig. S1). Furthermore, the sulfonic acid product of the specific oxidation of the catalytic cysteine was also identified in the caspase-3-inhibitor complex structures (Fig. 6, C and E). In addition, the catalytic cysteine of caspase-3 is oxidized step by step in the proposed mechanism. Although we have not tried to identify the intermediate forms, the formation of the sulfenic intermediate and the subsequent sulfinic intermediate has been identified using mass spectrometry in ROS-mediated inactivation of the catalytic cysteine of PTP1B (37).
ROS generation in the process of the oxidation of DTT and dihydrolipoic
acid has also been reported previously. For example, the
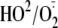 radicals could be produced within radical-induced oxidation of DTT but not its
monothiolic subunit 2-mercaptoethanol in the acidic aqueous solution
(38). Oxidation of
dihydrolipoic acid by the phenol radical induced via the phenol/peroxidase
system would form the thiol radical, and the primary thiol radical at pH 7.4
will be deprotonated to form the cyclic dithiol radical anion that can react
rapidly with oxygen to form the reactive radical superoxide
(
radicals could be produced within radical-induced oxidation of DTT but not its
monothiolic subunit 2-mercaptoethanol in the acidic aqueous solution
(38). Oxidation of
dihydrolipoic acid by the phenol radical induced via the phenol/peroxidase
system would form the thiol radical, and the primary thiol radical at pH 7.4
will be deprotonated to form the cyclic dithiol radical anion that can react
rapidly with oxygen to form the reactive radical superoxide
(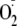 )
(39). Our data presented here
demonstrate that isoquinoline-1,3,4-trione derivatives also induce the
oxidation of DTT and dihydrolipoic acid to generate ROS
(Fig. 4).
)
(39). Our data presented here
demonstrate that isoquinoline-1,3,4-trione derivatives also induce the
oxidation of DTT and dihydrolipoic acid to generate ROS
(Fig. 4).
We supposed that the ROS including both O2-derived free
radicals, such as superoxide anion radical
(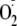 ),
hydroxyl radical (·OH), hydroperoxyl radical
(HO2), and hydrogen peroxide (H2O2), and
non-O2-derived RQ- radicals derived from
isoquinoline-1,3,4-trione derivatives can catalyze the redox cycling. The
sensitivities toward catalase and superoxide dismutase indicate that all these
reactive species could participate in redox cycle propagation and caspase-3
inactivation. Unfortunately, as for the RQ- radicals, although the
electron density for the bound inhibitor at the dimer interface of caspase-3
was evident, it was difficult to clarify whether it was the RQ-
radical or the compound itself because of the limited resolution of the
crystal structures.
),
hydroxyl radical (·OH), hydroperoxyl radical
(HO2), and hydrogen peroxide (H2O2), and
non-O2-derived RQ- radicals derived from
isoquinoline-1,3,4-trione derivatives can catalyze the redox cycling. The
sensitivities toward catalase and superoxide dismutase indicate that all these
reactive species could participate in redox cycle propagation and caspase-3
inactivation. Unfortunately, as for the RQ- radicals, although the
electron density for the bound inhibitor at the dimer interface of caspase-3
was evident, it was difficult to clarify whether it was the RQ-
radical or the compound itself because of the limited resolution of the
crystal structures.
We propose that it is the generated ROS rather than the direct interaction of the compounds with caspase-3 playing a key role in the process of the inactivation of caspase-3 by isoquinoline-1,3,4-trione derivatives. The reasons are as follows. First, in the crystal structures, there was only one compound molecule bound at the interface of two dimeric caspase-3 in the asymmetric unit and the inhibitor-binding site is located near two of the four active sites, but all four catalytic cysteines of the four caspase-3 molecules are in the oxidized form. Second, in the dithiol reagent (such as DTT)-free or oxygen-free conditions where no ROS would be generated, isoquinoline-1,3,4-trione derivatives could not inactivate caspase-3. In addition, compound V, which cannot react with the dithiol reagents to generate ROS also could not inactivate caspase-3. Thus, the key issue in the process of the caspase-3 inactivation by isoquinoline-1,3,4-trione derivatives is the ROS generation. Thus, the enhanced potency of compounds II, III, and IV over compound I could be explained by their stronger redox-cycling efficiency (Fig. 4C). This is supported by the obvious correlation between the intensity of the produced ROS signal and the inactivation potential on caspase-3 by different isoquinoline-1,3,4-trione derivatives (Fig. 5D).
The compounds exhibit no obvious difference in inactivation of the wild-type and mutant caspase-3 (Table 4), suggesting that interaction of the inhibitors with caspase-3 could be non-specific and does not play a critical role in caspse-3 inhibition. Kinetic analysis of compound III and H2O2 on caspase-3 inhibition further demonstrates that compound III itself does not directly inhibit caspase-3 nor facilitate the inhibition of caspase-3 by H2O2 (supplemental Fig. S2). However, as the inhibitors are indeed bound next to the active site, we cannot exclude the possibility that binding of inhibitor may facilitate the oxidation of caspase-3 by bringing the redox cycling process near the two active sites and transiently prompt the oxidation of the other two active sites.
The properties of the catalytic machinery of caspase-3 render the catalytic cysteine particularly sensitive to electrophiles (33). The crystal structures of caspases in complexes with substrates or inhibitors suggest that substrate hydrolysis of caspases involves the formation of a thiophosphate enzyme intermediate, which is further hydrolyzed in the second step (33). An invariant cysteine residue at the conserved active site of caspases functions as the catalytic nucleophile. A histidine and a glycine (His121 and Gly122 in human caspase-3) nearby surround the oxyanion to facilitate nucleophilic attack of the sulfur atom of cysteine on the electrophilic carbonyl carbon and stabilize the tetrahedral intermediate. These features of the catalytic machinery have led to the identification of more effective peptidyl N-nitrosoaniline inhibitors (13) and several peptidyl feature reduced caspase-3 inhibitors (40) also. In addition, the catalytic cysteine of caspase-3 is also remarkably sensitive to the oxidizing agents, for example, to form oxidized cysteine with hydrogen peroxide (41), superoxide anion radical, and nitric oxide (13). Consistently, structural results presented here indicate that only the catalytic cysteine but not the other cysteines in caspase-3 are oxidized to sulfonic acid in the crystal structures of caspase-3 treated with isoquinoline-1,3,4-trione derivatives and DTT. It is likely that the differences among the microenvironment of the active site of the caspases and other cysteine proteases surrounding the catalytic cysteine might render it with varied sensitivities to oxidation, which could explain the phenomena we found previously that these compounds have different selectivity against caspase-3, other caspases, and other proteases (22, 23).
Previously Okun et al. (32) found that in the presence of β-mercaptoethanol the inhibitory activity of 1,3-dioxo-2,3-dichloro-1H-pyrrolo[3,4-c]quinolines, which were identified as selective and reversible inhibitors of caspase-3 from screening in a DTT-containing buffer could not be measured. It was proposed that DTT and β-mercaptoethanol differentially affect the enzymatic parameters and the thermoinactivation of caspase-3 and thus define its sensitivity to different chemo types of small molecule inhibitors. However, our results indicate that the effects of most thiol-containing reducing agents on the enzymatic parameters of caspase-3 can be attributed to the slowdown of the inactivation rate of caspase-3 in air. Actually, it should be noted that the reducing agents (such as DTT, dihydrolipoic acid, 1,2-butanedithiol, and 1,4-butanedithiol), which were reported to be able to maintain the inhibition potency of 1,3-dioxo-2,3-dichloro-1H-pyrrolo[3,4-c]quinolines are all two thiol-containing redox reagents. Those are very similar to our results, in which both DTT and dihydrolipoic acid are dithiol reagents and can take part in the redox cycle catalyzed by isoquinoline-1,3,4-trione derivatives. It has also been shown that the DTT-participated redox cycle plays a central role in the inhibition of protein-tyrosine phosphatases, which also use a cysteine residue as the catalytic nuclephil (37, 42). Therefore, we conclude that the different potential of thiol-containing reducing agents in participating in the redox cycle to generate ROS results in the different effects on the inhibition potency of these chemo types.
ROS have long been implicated in regulating apoptosis. Although ROS, including H2O2, can induce cell death, there is evidence showing that it can also inhibit this process (43–45). Thus, ROS is potentially an important dual regulator of apoptosis. Furthermore, it has also been reported that the activity of caspases in cells can be directly inhibited by H2O2 or menadione-induced H2O2 (43, 44, 46). The potential participation of dihydrolipoic acid in isoquinoline-1,3,4-trione derivatives mediated inactivation of caspase-3 implies that the pro-oxidant actions of dihydrolipoic acid may play an important role in apoptosis, which raises the possibility of developing these chemo types for therapeutic purpose.
α-Lipoic acid, also known as thioctic acid, is an essential cofactor in the α-ketoacid dehydrogenase complex and the glycine cleavage system (47). It has been regarded as a powerful natural antioxidant used as a therapeutic agent in the treatment of diabetic neuropathy (48) and as a nutritional supplement in European countries and the United States. It has also been reported that depending on the concentration, α-lipoic acid could exert both antioxidant and pro-oxidant properties and protect human bone marrow stromal cells from tumor necrosis factor-α-induced apoptosis (49). On the other hand, similar to other vicinal thiols, dihydrolipoic acid (reduced lipoic acid) is more easily oxidized than monothiols, leading to high activity in -SH/-S-S-interchange reactions. Recently, dihydrolipoic acid thiyl radical, superoxide radical anion, and hydroxyl radical have been observed by aerobic electron spin resonance spin trapping with 5,5-dimethyl-1-pyrolline-N-oxide (39). The observation of thiyl radical and reactive oxygen species derived from dihydrolipoic acid suggests that this compound may act as a pro-oxidant under the right conditions. Our results demonstrate that in the presence of isoquinoline-1,3,4-trione derivatives, dihydrolipoic acid has a high tendency to react with oxygen to generate ROS, which could specifically oxidize the catalytic cysteine of caspase-3. This implies that DTT could be replaced with a high level of dihydrolipoic acid to take part in the redox cycle in vivo, which may explain partially the ability of these chemicals to attenuate cell apoptosis (19, 22), taking into account that dihydrolipoic acid exists in mitochondria of various cells. This suggestion does not exclude the possibility that other molecules in vivo could catalyze isoquinoline-1,3,4-trione derivatives to generate ROS, thereby changing the cellular redox state and the activity of thiol-enzymes besides caspases. Directly targeting other apoptosis related proteins in vivo may also contribute to their cellular effects.
In summary, through the specific oxidation of the catalytic cysteine, isoquinoline-1,3,4-trione derivatives are potential inactivators of caspase-3 under aerobic conditions. The features of this mechanism may provide useful information for designing caspase-3 inhibitors.
Supplementary Material
The atomic coordinates and structure factors (codes 3DEH, 3DEI, 3DEJ, and 3DEK) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The work was supported by Ministry of Science and Technology of China Grants 2006AA02Z112 and 2006AA02A313 and National Natural Science Foundation of China Grants 90713046 and 20730028. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: DTT, 1,4-dithiothreitol; H2DCFDA, 2′,7′-dichlorofluorescein diacetate; ROS, reactive oxygen species; SOD, superoxide dismutase; pNA, p-nitroanilide; DMSO, dimethyl sulfoxide; Ac, acetyl.
References
- 1.Vaux, D. L., Haecker, G., and Strasser, A. (1994) Cell 76 777-779 [DOI] [PubMed] [Google Scholar]
- 2.Steller, H. (1995) Science 267 1445-1449 [DOI] [PubMed] [Google Scholar]
- 3.Metzstein, M., Stanfiold, G., and Horvitz, H. (1998) Trends Genet. 14 410-416 [DOI] [PubMed] [Google Scholar]
- 4.Meier, P., Finch, A., and Evan, G. (2000) Nature 407 796-801 [DOI] [PubMed] [Google Scholar]
- 5.Thompson, C. B. (1995) Science 267 1456-1462 [DOI] [PubMed] [Google Scholar]
- 6.Nicholson, D. W. (2000) Nature 407 810-816 [DOI] [PubMed] [Google Scholar]
- 7.Reed, J. C. (2002) Nat. Rev. Drug Discov. 1 111-121 [DOI] [PubMed] [Google Scholar]
- 8.Salvesen, G. S., and Dixit, V. M. (1997) Cell 91 443-446 [DOI] [PubMed] [Google Scholar]
- 9.Cohen, G. M. (1997) Biochem. J. 326 1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornberry, N. A., and Lazebnik, Y. (1998) Science 281 1312-1316 [DOI] [PubMed] [Google Scholar]
- 11.Hengartner, M. O. (2000) Nature 407 770-776 [DOI] [PubMed] [Google Scholar]
- 12.Porter, A. G., and Jänicke, R. U. (1999) Cell Death Differ. 6 99-104 [DOI] [PubMed] [Google Scholar]
- 13.Guo, Z., Xian, M., Zhang, W., McGill, A., and Wang, P. G. (2001) Bioorg. Med. Chem. 9 99-106 [DOI] [PubMed] [Google Scholar]
- 14.Nobel, C. S., Kimland, M., Nicholson, D. W., Orrenius, S., and Slater, A. F. (1997) Chem. Res. Toxicol. 10 1319-1324 [DOI] [PubMed] [Google Scholar]
- 15.Scott, C. W., Sobotka-Briner, C., and Wilkins, D. E. (2003) J. Pharmacol. Exp. Ther. 304 433-440 [DOI] [PubMed] [Google Scholar]
- 16.Lee, D., Long, S. A., Adams, J. L., Chan, G., Vaidya, K. S., Francis, T. A., Kikly, K., Winkler, J. D., Sung, C. M., Debouck, C., Richardson, S., Levy, M. A., DeWolf, W. E., Jr., Keller, P. M., Tomaszek, T., Head, M. S., Ryan, M. D., Haltiwanger, R. C., Liang, P. H., Janson, C. A., McDevitt, P. J., Johanson, K., Concha, N. O., Chan, W., Abdel-Meguid, S. S., Badger, A. M., Lark, M. W., Nadeau, D. P., Suva, L. J., Gowen, M., and Nuttall, M. E. (2000) J. Biol. Chem. 275 16007-16014 [DOI] [PubMed] [Google Scholar]
- 17.Lee, D., Long, S. A., Murray, J. H., Adams, J. L., Nuttall, M. E., Nadeau, D. P., Kikly, K., Winkler, J. D., Sung, C. M., Ryan, M. D., Levy, M. A., Keller, P. M., and DeWolf, W. E., Jr. (2001) J. Med. Chem. 44 2015-2026 [DOI] [PubMed] [Google Scholar]
- 18.Chu, W., Zhang, J., Zeng, C., Rothfuss, J., Tu, Z., Chu, Y., Reichert, D. E., Welch, M. J., and Mach, R. H. (2005) J. Med. Chem. 48 7637-7647 [DOI] [PubMed] [Google Scholar]
- 19.Kravchenko, D. V., Kuzovkova, Y. A., Kysil, V. M., Tkachenko, S. E., Maliarchouk, S., Okun, I. M., Balakin, K. V., and Ivachtchenko, A. V. (2005) J. Med. Chem. 48 3680-3683 [DOI] [PubMed] [Google Scholar]
- 20.Hardy, J. A., Lam, J., Nguyen, J. T., O'Brien, T., and Wells, J. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 12461-12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nobel, C. S., Burgess, D. H., Zhivotovsky, B., Orrenius, S., and Slater, A. F. (1997) Chem. Res. Toxicol. 10 636-673 [DOI] [PubMed] [Google Scholar]
- 22.Chen, Y. H., Zhang, Y. H., Zhang, H. J., Liu, D. Z., Gu, M., Li, J. Y., Wu, F., Zhu, X. Z., Li, J., and Nan, F. J. (2006) J. Med. Chem. 49 1613-1623 [DOI] [PubMed] [Google Scholar]
- 23.Zhang, Y. H., Zhang, H. J., Wu, F., Chen, Y. H., Ma, X. Q., Du, J. Q., Zhou, Z. L., Li, J. Y., Nan, F. J., and Li, J. (2006) FEBS J. 273 4842-4852 [DOI] [PubMed] [Google Scholar]
- 24.Copeland, R. A. (2005) in Evaluation of Enzyme Inhibitors in Drug Discovery. A Guide for Medicinal Chemists and Pharmacologists, John Wiley & Sons, Hoboken, NJ [PubMed]
- 25.Afzal, M., Matsugo, S., Sasai, M., Xu, B., Aoyama, K., and Takeuchi, T. (2003) Biochem. Biophys. Res. Commun. 304 619-624 [DOI] [PubMed] [Google Scholar]
- 26.Trayner, I. D., Rayner, A. P., Freeman, G. E., and Farzaneh, F. (1995) J. Immunol. Methods 186 275-284 [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 207-326 [DOI] [PubMed] [Google Scholar]
- 28.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 29.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 30.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 31.Lovell, S. C., Davis, I. W., Arendall, W. B., 3rd, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 32.Okun, I., Malarchuk, S., Dubrovskaya, E., Khvat, A., Tkachenko, S., Kysil, V., Kravchenko, D., and Ivachtchenko, A. (2006) J. Biomol. Screen 11 694-703 [DOI] [PubMed] [Google Scholar]
- 33.Fuentes-prior, P., and Salvesen, S. G. (2004) Biochem. J. 384 201-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agniswamy, J., Fang, B., and Weber, I. T. (2007) FEBS J. 274 4752-4765 [DOI] [PubMed] [Google Scholar]
- 35.Henson, C. P., and Cleland, W. W. (1964) Biochemistry 3 338-345 [DOI] [PubMed] [Google Scholar]
- 36.Wunderlich, M., and Glockshuber, R. (1993) Protein Sci. 2 717-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, Q., Dubé, D., Friesen, R. W., LeRiche, T. G., Bateman, K. P., Trimble, L., Sanghara, J., Pollex, R., Ramachandran, C., Gresser, M. J., and Huang, Z. (2004) Biochemistry 43 4294-4303 [DOI] [PubMed] [Google Scholar]
- 38.Lal, M., Rao, R., Fang, X., Schchmann, H. P., and von Sonntag, C. (1997) J. Am. Chem. Soc. 119 5735-5739 [Google Scholar]
- 39.Mottley, C., and Mason, R. P. (2001) J. Biol. Chem. 276 42677-42683 [DOI] [PubMed] [Google Scholar]
- 40.Becker, J. W., Rotonda, J., Soisson, S. M., Aspiotis, R., Bayly, C., Francoeur, S., Gallant, M., Garcia-Calvo, M., Giroux, A., Grimm, E., Han, Y., McKay, D., Nicholson, D. W., Peterson, E., Renaud, J., Roy, S., Thornberry, N., and Zamboni, R. (2004) J. Med. Chem. 47 2466-2474 [DOI] [PubMed] [Google Scholar]
- 41.Smith, G. K., Barrett, D. G., Blackburn, K., Cory, M., Dallas, W. S., Davis, R., Hassler, D., McConnell, R., Moyer, M., and Weaver, K. (2002) Arch. Biochem. Biophys. 399 195-205 [DOI] [PubMed] [Google Scholar]
- 42.Guertin, K. R., Setti, L., Qi, L., Dunsdon, R. M., Dymock, B. W., Jones, P. S., Overton, H., Taylor, M., Williams, G., Sergi, J. A., Wang, K., Peng, Y., Renzetti, M., Boyce, R., Falcioni, F., Garippa, R., and Olivier, A. R. (2003) Bioorg. Med. Chem. Lett. 13 2895-2898 [DOI] [PubMed] [Google Scholar]
- 43.Hampton, M. B., and Orrenius, S. (1997) FEBS Lett. 414 552-556 [DOI] [PubMed] [Google Scholar]
- 44.Samali, A., Nordgren, H., Zhivotovsky, B., Peterson, E., and Orrenius, S. (1999) Biochem. Biophys. Res. Commun. 255 6-11 [DOI] [PubMed] [Google Scholar]
- 45.Lee, Y. J., and Shacter, E. (1999) J. Biol. Chem. 274 19792-19798 [DOI] [PubMed] [Google Scholar]
- 46.Borutaite, V., and Brown, G. C. (2001) FEBS Lett. 500 114-118 [DOI] [PubMed] [Google Scholar]
- 47.Reed, L. J. (1974) Acc. Chem. Res. 7 40-46 [Google Scholar]
- 48.Packer, L., Kraemer, K., and Rimbach, G. (2001) Nutrition 17 888-895 [DOI] [PubMed] [Google Scholar]
- 49.Byun, C. H., Koh, J. M., Kim, D. K., Park, S. I., Lee, K. U., and Kim, G. S. (2005) J. Bone. Miner. Res. 20 1125-1135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.