

Abstract
Calcium oscillations exert physiological control on mitochondrial energy metabolism and can also lead to mitochondrial membrane permeabilization and cell death. The outcome of the mitochondrial calcium signaling is altered by stress factors such as ceramide or staurosporine. However, the mechanism of this pro-apoptotic switch remains unclear. Using genetic, biochemical, pharmacological and functional approaches we here show that ceramide and staurosporine target PP2A and protein kinases A and C, respectively in a mitochondria-associated signaling complex to induce dephosphorylation of the BH3-only protein Bad. Dephosphorylated Bad sensitizes the mitochondrial permeability transition pore (PTP) to Ca2+ through a Bcl-xL sensitive and VDAC-mediated process. Furthermore, the Bad-induced sensitization of the PTP to Ca2+ does not require Bax or Bak. Thus, phospho-regulatory mechanisms converge on Bad to switch between the survival and apoptotic functions of mitochondrial calcium signaling by activating a previously unforeseen mechanism, whereby a BH3-only protein bypasses Bax/Bak and engages the PTP.
Introduction
Propagation of cytoplasmic calcium oscillations to the mitochondria provides a mechanism to stimulate energy metabolism, coordinating the production of ATP with the cellular demand (Hajnoczky et al., 1995; Jouaville et al., 1999; Robb-Gaspers et al., 1998). However, under a variety of stress conditions calcium oscillations also serve as the trigger for mitochondrial membrane permeabilization, and cell death through apoptosis or necrosis (Demaurex and Distelhorst, 2003; Pinton et al., 2001; Szalai et al., 1999). The stress agents include lipid metabolites (ceramide, gangliosides, arachidonic acid (Pinton et al., 2001; Scorrano et al., 2001; Szalai et al., 1999)), viral proteins (Chami et al., 2003), reactive oxygen species (ROS) (Gerasimenko et al., 2002; Madesh and Hajnoczky, 2001; Scorrano et al., 2003), and various drugs (Orrenius et al., 2003; Zamzami et al., 2005). All these factors initiate mechanisms that converge on the mitochondrial PTP to set the threshold for its activation by mitochondrial matrix [Ca2+] ([Ca2+]m) (Bernardi and Forte, 2007; Green and Kroemer, 2004; Hajnoczky et al., 2003; Kroemer et al., 2007). However, it remains unclear what mechanisms link the stress factor effect to the sensitization of the PTP and whether these mechanisms utilize the pro-apoptotic Bax and Bak known as “master regulators” of mitochondrial membrane permeabilization during apoptosis.
C2 ceramide (Cer) can alter ER Ca2+ mobilization (Pinton et al., 2001), the activity of the mitochondrial respiratory complexes I and III (France-Lanord et al., 1997; Gudz et al., 1997) and can also form a channel in the outer mitochondrial membrane (OMM) (Siskind et al., 2002). However, in several cell types Cer sensitizes the PTP in the absence of increased Ca2+ transfer to the mitochondria or any mitochondrial depolarization (Pacher and Hajnoczky, 2001; Szalai et al., 1999). Evidence has also accumulated that the pro-apoptotic effect of Cer can be suppressed by protein kinase C (PKC) activation (Obeid et al., 1993) and that Cer activates both protein phosphatase I (PP1) (Kishikawa et al., 1999) and protein phosphatase 2A (PP2A) (Kowluru and Metz, 1997; Ruvolo et al., 1999). Like Cer, the pan-protein kinase inhibitor staurosporine (ST) was found to sensitize the PTP (Szalai et al., 1999), implicating protein phosphor-regulation as an important factor in determining the specificity of mitochondrial calcium signaling. Following this lead, we have now discovered a signaling pathway whereby PP2A, protein kinase A and C control the phosphorylation of the BH3-only Bcl-2 family protein Bad, and that dephosphorylated Bad influences the functional interaction of the PTP with anti-apoptotic Bcl-xL and VDAC. This is the first example for a Bax/Bak independent role of a BH3-only protein in sensitizing the PTP and inducing mitochondrial membrane permeabilization.
Results
Cer-induced sensitization of the PTP to Ca2+ requires PP2A
We and others have previously described that Cer and other stress factors sensitize mitochondria to Ca2+-induced PTP opening that manifests as Cer-induced enhancement of the ΔΨm loss and [Ca2+]c rise also referred to as delayed mitochondrial dysregulation of ΔΨm and [Ca2+]c during Ca2+ pulsing or IP3-induced Ca2+ release (Chami et al., 2003; Demaurex and Distelhorst, 2003; Gerasimenko et al., 2002; Pacher and Hajnoczky, 2001; Szalai et al., 1999). As Cer has been shown to induce activation of both purified protein phosphatases (PP) and protein kinases (PK) (PP1: (Kishikawa et al., 1999), PP2A: (Dobrowsky et al., 1993), PKC α and δ: (Huwiler et al., 1998); PKC ξ: (Bourbon et al., 2000); PKA: (Cabral et al., 2007)), we undertook a pharmacological approach to characterize the PP and PK in the Cer-dependent delayed mitochondrial impairment defined by Cer-induced enhancement of the ΔΨm loss and [Ca2+]c rise. We performed simultaneous fluorometric measurements of ΔΨm and [Ca2+]c in suspensions of permeabilized HepG2 cells. In the absence of Cer, addition of the catalytic subunit of protein kinase A (PKAc) or the PP inhibitor okadaic acid (OA) did not alter the Ca2+ pulsing-induced transient depolarization and [Ca2+]c rise (Fig1AB). However, the Cer-induced enhancement and prolongation of the depolarization and the [Ca2+]c rise was greatly suppressed by both PKAc and OA (Fig1AB). On average ∼70% protection was obtained either by PKAc (ΔΨm at 900s, 71±2% protection and [Ca2+]c half time, 73±4% protection, n=6) or by OA (ΔΨm at 900s, 63±4% protection and [Ca2+]c half time, 71±4% protection, n=7) (Fig1C). Similar to OA, calyculin A (Caly A), another PP inhibitor also selectively inhibited the Cer-dependent ΔΨm and [Ca2+]c responses (56±10% and 67±8%, respectively, n=4, Fig1C). Thus, the mitochondrial handling of Ca2+ was not affected by the PP inhibitors and by PKAc in control cells, but the Cer-dependent delayed mitochondrial dysregulation was suppressed by these agents indicating a role for potential PP and PKA substrates in the Cer-induced sensitization of the PTP to Ca2+.
Fig1. Cer+Ca2+-induced ΔΨm loss and delayed Ca2+ dysregulation is suppressed by PP inhibitors and PK activators.

Simultaneous measurements of ΔΨm and [Ca2+]c were carried out in suspensions of permeabilized HepG2 cells using JC1 and fura2FF, respectively. Permeabilized cells were treated with Cer (40 uM, purple) or DMSO (Cont, black) before addition of Ca2+ pulses (30 μM each). (A) PKAc (50units/ml, Sigma) and (B) OA (50nM) were added before the addition of Cer. Representative analog traces showing the Cer-induced enhancement of the ΔΨm loss and [Ca2+]c rise and their modulation by PKA and OA (PKAc alone, dark green; PKAc+Cer, light green; OA alone, dark cyan; OA+Cer, light cyan). (C,D) Summary of the effect of various PP inhibitors and kinase activators (cptcAMP (5μM), 8BrcAMP (5μM) and 8BrcGMP (5μM) and TPA (50nM)) on delayed Ca2+ dysregulation.
OA exerted protection at a very low concentration (5nM, not shown) that selectively inhibits PP2A (Hardie, 1990), suggesting that the effect of Cer on the ΔΨm and [Ca2+]c dysregulation was mediated by PP2A. Specific pharmacological activators of PKs were next employed to identify endogenous PKs that may counteract PP2A-mediated sensitization of PTP to Ca2+ in the presence of Cer. The PKA activators, cptcAMP and 8BrcAMP selectively inhibited the Cer effect similar to PKAc (SFig1, Fig1D, cptcAMP: ΔΨm, 63±4% and [Ca2+]c half time, 70±5% protection, n=6; 8BrcAMP: ΔΨm, 62±4% and [Ca2+]c half time, 73±4% protection, n=3)). Furthermore, the pan-PKC activator, tetradecanoyl-12-phorbol-13-acetate (TPA), also exerted protection (Fig1D, ΔΨm, 60±8% and [Ca2+]c half time, 66±4% protection, n=5), while cGMP, a PKG activator failed to alter the Cer effect (Fig1D). In addition, a specific PKA inhibitor, PKI (6-22) abolished the protective effect of the cptcAMP and a specific PKC inhibitor, Ro 31 inhibited the protective effect of TPA (SFig2). In the presence of ST, none of the kinase activators could establish any protection (Fig1D). Thus, protein phosphorylation mediated by both PKA and PKC isoforms can suppress the mechanism of the Cer-induced sensitization of the PTP to Ca2+. It is less likely that Ca2+ sensitization of PTP is due to inhibition of PKA or PKC by Cer in light of published work demonstrating that Cer activates rather than inhibits recombinant PKA and PKC isoforms [PKC α and δ: (Huwiler et al., 1998); PKC ξ: (Bourbon et al., 2000); PKA: (Cabral et al., 2007)]. Consistent with this argument, addition of PK inhibitors in place of Cer (180s before Ca2+) does not recapitulate the effect of Cer PTP sensitization (SFig2). It is, however, possible that inhibition of PKA and PKC enzymes may account for PTP sensitization during prolonged incubation with ST (1h) (Szalai et al., 1999).
Dependence of the Cer+Ca2+-induced cytochrome c (cyto c) release on PP2A
Although the amount of Ca2+ taken up by the mitochondria in intact cells is much smaller than in permeabilized cells exposed to Ca2+ pulsing, intracellular Ca2+ oscillations have also been found to induce PTP in Cer-treated intact cells (Pacher and Hajnoczky, 2001; Szalai et al., 1999). PTP activation by [Ca2+]c oscillations in the presence of Cer is followed by cyto c release and caspase-3 activation in intact cells (Pacher and Hajnoczky, 2001; Szalai et al., 1999). To investigate whether PP inhibitors can also suppress the Ca2+-induced cyto c release we used cyto c-GFP expressing HepG2 cells (Fig2). To induce [Ca2+]c oscillations, cells were stimulated with ATP that evokes IP3 formation through P2y receptor activation (Szalai et al., 1999). Cyto c-GFP was confined to the mitochondria in cells that were untreated or were exposed to ATP alone (Fig2). In cells treated with Cer alone, quantitative analysis revealed a moderate decrease in the mitochondrial cyto c-GFP and a small increase in the cytoplasmic cyto c-GFP fluorescence (Fig2B). However, Cer+ATP-treatment resulted in significant redistribution of cyto c-GFP from mitochondria to the cytoplasm (Fig2AB) that was inhibited by Bcl-xL overexpression (SFig3). Strikingly, addition of either Caly A or OA resulted in strong inhibition of the Cer+ATP-induced appearance of cyto c-GFP in the cytoplasm (Fig2AB). Importantly, cyclosporin A (CsA) an inhibitor of the PTP abolished the Cer+ATP-induced cyto c-GFP redistribution. Collectively, these results show that PTP-dependent cyto c release in response to Cer+ATP was effectively prevented by PP2A inhibitors, suggesting that breakdown of the OMM barrier was initiated by Cer at least in part through PP2A.
Fig2. Cer+Ca2+-induced cyto c-GFP release is attenuated by PP inhibitors and CsA.
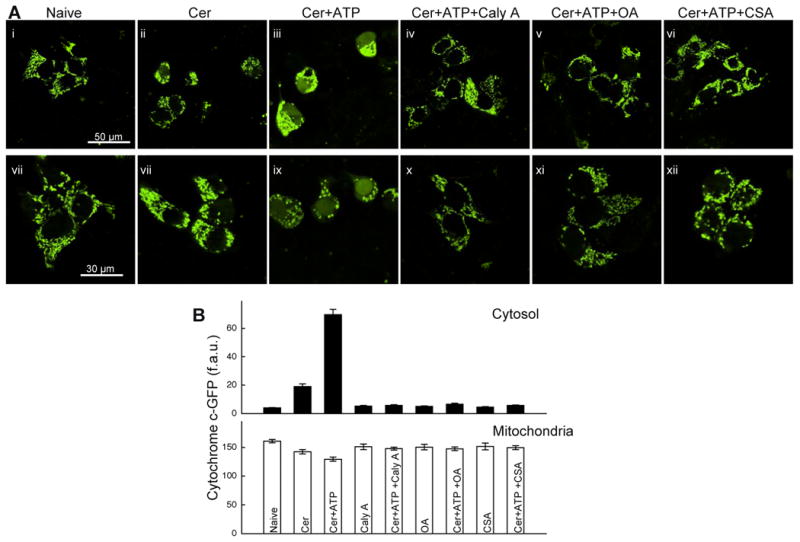
(A) Confocal images of cyto c-GFP–transfected HepG2 cells treated with Cer, Cer +ATP (100μM) and Cer and ATP in the absence or presence of Caly A (50nM), OA (50nM) or CsA (2μM). (B) Quantification of cyto c–GFP. Since the nucleus is devoid of mitochondria and released cyto c–GFP entered the nuclear matrix, the amount of cyto c–GFP released into the cytosol was assessed by quantitation of fluorescence over the nucleus.
Cer induces PP2A-mediated dephosphorylation of Bad
The integrity of the OMM barrier and the sensitivity of the PTP to various inducers are regulated by Bcl-2 family proteins (Green and Kroemer, 2004; Kroemer et al., 2007). Previous studies have shown that phosphorylation neutralizes the pro-apoptotic activity of Bad, a BH3-domain only Bcl-2 family protein (Chiang et al., 2003; Datta et al., 1997; Zha et al., 1996). Depending on the cellular context, multiple PKs and PPs have been implicated in dynamic regulation of Bad phosphorylation. For example, calcineurin, PP1 and PP2A can dephosphorylate Bad (Ayllon et al., 2001; Chiang et al., 2003; Danial et al., 2003; Wang et al., 1999). Western blot analysis for phospho-Bad revealed that Cer treatment decreased the phosphorylation of endogenous Bad in both permeabilized and intact HepG2 cells (Fig3AB) and in ST-treated intact cells as well as in mitochondria isolated from Bad transfected 293T cells (Fig3E). Dephosphorylation occurred at Ser112, Ser136, and Ser155 (Fig6AB). Furthermore, recombinant PP2A also caused dephosphorylation of Bad in permeabilized HepG2 cells (Fig3A). Both the Cer-and the PP2A-induced Bad dephosphorylation were prevented by PP inhibitors, PKAc, and pharmacologic activators of PKA or PKC (Fig3CDE). However, the Cer-induced Bad dephosphorylation was not affected by either depletion of Ca2+ (Fig3D) or by the addition of CsA that inhibits both calcineurin and PTP opening (Fig3CD), suggesting that Bad dephosphorylation in this setting does not depend on calcineurin, mitochondrial Ca2+ uptake or PTP opening. Moreover, the Cer-induced increase in the total mitochondria-associated Bad and its prevention by PP2A inhibitor OA, (Fig3E) further support that the phosphorylation status of Bad was shifted towards dephosphorylation in the mitochondria.
Fig3. Phosphoregulation of Bad in intact and permeabilized HepG2 cells.

(A) Immunoblot showing Cer-induced dephosphorylation of Bad in permeabilized cells. Exogenous PP2A also evoked dephosphorylation of Bad, which was prevented by PP2A inhibitor OA. Pretreatment of the permeabilized cells with Cer and OA was performed as described in the Legend to Fig1. (B) Cer-induced dephosphorylation of Bad in intact cells treated with Cer, Cer+Caly A and ST for 1hr shows. (C&D) Cumulative quantitative analysis of immunoblot signals obtained in experiments similar to A and B. (E) Immunoblot of mitochondria prepared from Bad-overexpressing 293T cells treated with Cer (40μM) in the absence or presence of OA (50nM) for 1.5 hrs before isolation of mitochondria. The mitochondrial protein prohibitin serves as a loading control.
Fig6. Bad is part of a signaling complex that also contains PKA, PKC, PP2A and Bcl-xl.

(AB) Regulation of Bad phosphorylation by endogenous PKA and PKC and by ST treatment. Whole cell lysates (A) or isolated mitochondria (B) were prepared from 293T cells transiently transfected with GST-Bad or control GST-Vector that were treated for 1hr with TPA (200nM), ST (1μM) and cptcAMP(5μM) (C) GST pull-down assay performed in overexpressing 293T cells shows that the Bad immunoprecipitate also contains PKA, PKC, PP2A and Bcl-xL and VDAC. The pull-down was done in whole cell lysates for PKA, PKC and PP2A, and in lysates of permeabilized cells for Bcl-xL and VDAC. (D) FLAG-Bcl-xl and FLAG-Vector overexpressing 293T cells were treated for 1.5 hrs with Cer(40uM). FLAG pull-down assay was done in permeabilized cell lysates.
Cer-induced sensitization of PTP opening requires Bad
To directly address the possible role of Bad in PTP opening, recombinant Bad protein was added to permeabilized HepG2 cells. In the absence of Ca2+ pulses, recombinant Bad caused only a small and reversible depolarization without affecting [Ca2+]c, while massive and irreversible depolarization and [Ca2+]c rise was observed when a series of Ca2+ pulses were applied (Fig4A). Immunoblotting of the cytoplasmic fractions prepared from the same samples revealed marked cyto c increase in samples treated with Bad+Ca2+ (Fig4B). These results endorse a role for Bad in sensitization of the PTP to Ca2+ and subsequent release of cyto c. As an independent approach, HEK293 cells overexpressing Bad were similarly evaluated for Ca2+-induced depolarization and [Ca2+]c rise (Fig4CD). Immunoblotting of cell lysates confirmed the overexpression and the PKA-and PKC-mediated phosphorylation of Bad (Fig6A). Ca2+-induced depolarization and cytoplasmic Ca2+ retention was increased in Bad-overexpressing cells and, in particular, when ST was added to inhibit Bad phosphorylation (Fig4CD). Thus, both exogenously added Bad and endogenously expressed protein sensitize cells to Ca2+-induced PTP opening.
Fig4. Effect of Bad on Ca2+-induced mitochondrial membrane permeabilization.

(AB) Ca2+ pulsing (3×30uM)-induced ΔΨm loss and [Ca2+]c retention (A) and cyto c release (B) in the presence or absence of recombinant Bad (300nM) in suspensions of permeabilized HepG2 cells. (CD) Simultaneous recordings of [Ca2+]c (C) and ΔΨm (D) in 293T cells transiently transfected with control or Bad constructs that were untreated or pretreated with ST. Half recovery time from last Ca2+ pulse is shown in C. (EF) Delayed Ca2+ dysregulation (E) and mitochondrial swelling (F) in suspensions of permeabilized WT and bad-/- MEFs treated with Cer or solvent before addition of Ca2+ pulses. (G) Cell death in WT and bad-/- MEFs treated with Cer (40μM) and Tg (2uM) for 12hrs. Symbols (square and circle) represent the result of 2 separate experiments each was carried out in duplicates.
To test whether Bad was required for the Cer-induced sensitization, we used primary mouse embryonic fibroblasts (MEFs) isolated from WT and bad-/- mice (Ranger et al., 2003). We performed simultaneous fluorometric measurements of [Ca2+]c and light scattering measurements to examine mitochondrial swelling induced by PTP opening. In the absence of Cer, application of Ca2+ pulses was associated with comparable [Ca2+]c rise and light scattering in both WT and Bad-deficient cells (Fig4EF). Strikingly, upon addition of Cer, bad-/- cells showed diminished [Ca2+]c dysregulation and smaller decrease in light scattering (Fig4EF). The protection afforded by Bad ablation was on average 50% in the Ca2+ dysregulation and 70% in the mitochondrial swelling assay. These values are similar to the protection attained by PP2A inhibition and by PKA/PKC activation. Our findings provide genetic evidence that Bad is responsible for a major component of the Cer-induced sensitization of the PTP.
As Cer-induced sensitization of PTP can influence the apoptotic response to Ca2+-mobilizing agonists, we further tested the genetic requirement of Bad in cell death evoked by the combination of Cer and thapsigargin (Tg), a Ca2+ mobilizing agent. The Cer+Tg-induced cell death was practically abolished in the bad-/- MEFs (Fig4G), indicating the genetic requirement of BAD in this death paradigm. Notably, suppression of the Cer-dependent Ca2+ dysregulation and apoptosis in bad-/- cells cannot be attributed to a change in the amplitude of [Ca2+]c signal as WT and bad-/- cells treated with Cer and stimulated with Tg or ATP, an IP3-linked agonist, showed similar [Ca2+]c dynamics (SFig4).
Pro-survival effect of PP2A inhibitors and PKA and PKC activators is Bad dependent
Cer has been proposed to engage multiple ER and mitochondrial pro-apoptotic mechanisms that may also be relevant for the control of PTP opening, including Ca2+ release from the ER (Pinton et al., 2001), direct inhibition of respiratory complex III (Gudz et al., 1997), stimulation of complex I-mediated ROS formation (France-Lanord et al., 1997) and formation of a channel in the OMM (Siskind et al., 2002), which can be disassembled by Bcl-xL (Siskind et al., 2008). Here, we have described the Cer-PP2A-Bad pathway as a new mechanism for the regulation of the PTP and cyto c release and provided evidence for an important role of this pathway in HepG2, HEK and MEF cells. To determine whether the regulation of phosphorylation events by PP2A, PKA and PKC is confined to the Bad-dependent mechanisms, we tested the effect of PP and PK inhibitors both in WT and bad-/- MEFs. In WT MEFs, OA, caly A, cptcAMP and TPA exerted strong protection against the Cer-dependent [Ca2+]c dysregulation and mitochondrial swelling in manner similar to HepG2 cells (Fig5AB). However, in the absence of Bad the Cer effect was small and OA, Caly A, cptcAMP and TPA did not afford protection against Cer-dependent [Ca2+]c dysregulation and mitochondrial swelling, rather they enhanced the effect of Cer (Fig5CD). It is possible that Bad is the preferred substrate of PP2A and PKA and PKC, however, in its absence other substrates are targeted that may have reverse effects. To further validate the role of Bad phosphorylation in Cer-dependent [Ca2+]c dysregulation, fluorometric measurements of [Ca2+]c were performed in Cer-pretreated suspensions of permeabilized MEFs expressing WT Bad or a phosphomimetic Bad mutant, BadS155D (Danial et al., 2008) (Fig5EF). Cer-induced sensitization to Ca2+ dysregulation was greatly inhibited in WT MEFs expressing BadS155D. Furthermore, the remaining effect of Cer was insensitive to the PP2A inhibitor (Fig5E). Genetic reconstitution of bad-/- MEFs with BadS155D did not significantly improve the Ca2+ retention, but showed a tendency for attenuation of the effect of OA (Fig5F). In comparison, re-expression of WT Bad allowed a large Cer+Ca2+ effect and restored a robust protective effect mediated by OA (Fig5F). Thus, the phosphorylation-mediated protection is restricted to the Bad-dependent sensitization of the PTP in Cer-treated cells.
Fig5. Bad-dependent phospho-regulation of Ca2+-induced mitochondrial membrane permeabilization.
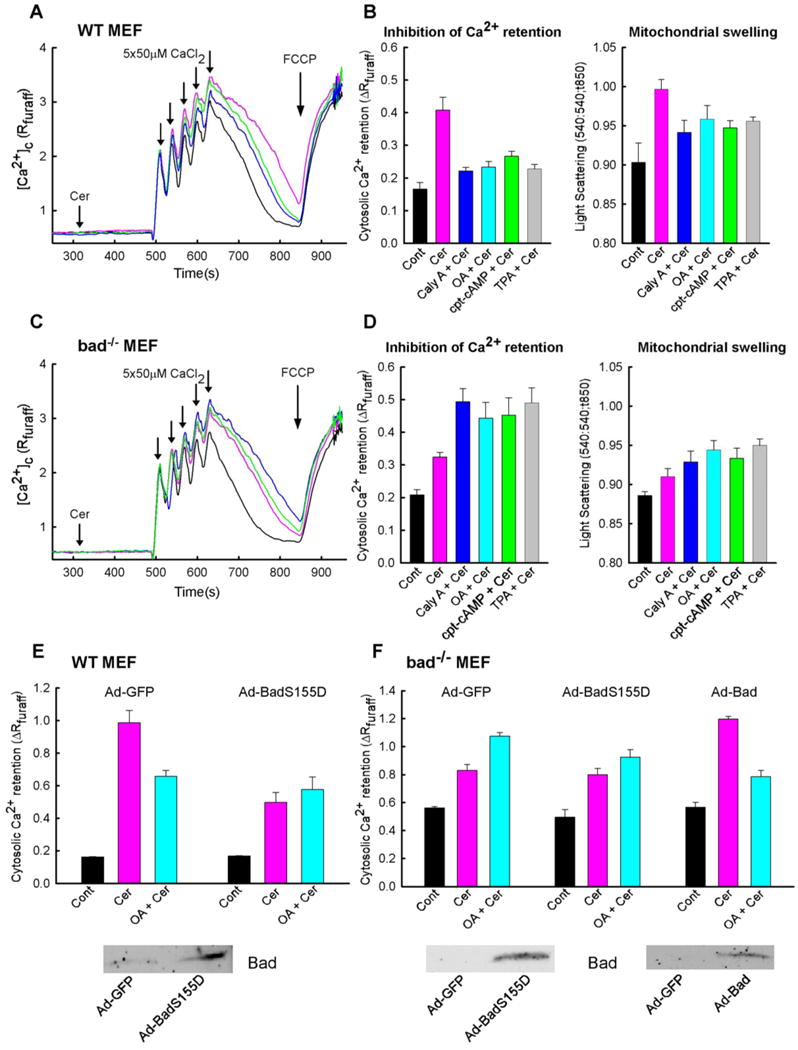
(A-D) Simultaneous measurements of [Ca2+]c and mitochondrial swelling in suspensions of permeabilized WT and bad-/- MEFs pretreated with Caly A (50nM), OA (50nM), TPA (200nM) and cptcAMP (5 uM) followed by addition of Cer (40 μM) or solvent. (EF) Analysis of delayed Ca2+ dysregulation in suspensions of permeabilized WT and bad-/- MEFs. WT MEFs were infected with either control Ad-GFP or Bad-expressing adenovirueses (Ad-Bad or Ad-BadS155D), permeabilized and pretreated with OA (50nM) and/or Cer (40 uM). Immunoblots in E and F show BAD expression in WT and bad-/- MEFs infected by Bad adenoviruses, respectively.
Cer and ST target a signaling complex that allows the interaction of Bad with PP2A, PKA, PKC, Bcl-xL and VDAC
Recent studies have reported the presence of Bad in distinct signaling complexes containing select PKs and PP in charge of its modification (Danial et al., 2003; Djouder et al., 2007). In the mitochondrial signaling complexes, interaction of Bad with glucokinase (Danial et al., 2003) and with Bcl-2 and Bcl-xL (Yang et al., 1995) has also been documented. In HepG2 cells and MEFs, stable interactions between Bad and the endogenous PP2A, PKA and PKC were predicted as the enzymes were able to regulate Bad phosphorylation in permeabilized cells after more than thousand fold dilution of the cytoplasm (Figs1,3,5 and SFig1). Indeed, it was surprising that PKAc-dependent phosphorylation was maintained in permeabilized cells since PKA activation is generally believed to cause liberation of PKAc from the regulatory subunit that anchors the holoenzyme. Notably, recent structural studies indicate that PKAc also has a scaffold function (Taylor et al., 2008). Immunocytochemistry performed in permeabilized HepG2 cells indicated that at least a fraction of the catalytic subunit of PKAc, PKC and PP2A is associated with the mitochondria (SFig5). Side-by-side comparison of intact and permeabilized cells showed that ∼50% of PKAc was retained after permeabilization (SFig6). To test the possibility that dynamic regulation of Bad phosphorylation in response to Cer occurred through a signaling complex containing PP2A, PKA and PKC pull down assays were performed (Fig6). In HEK293 cells transiently transfected with GST-Bad, treatment with cptcAMP or TPA induced Bad phosphorylation in both whole cell lysates and isolated mitochondria, whereas ST induced dephosporylation, indicating phospho-regulation of Bad by the endogenous enzymes (Fig6AB). Pull down assays using lysates prepared from cells transiently expressing Bad indicated co-presence of PKA, PKC, PP2A and Bcl-xL together with Bad (Fig6C). Interestingly, VDAC was also present in GST-Bad pull downs (Fig6C). This is especially intriguing in light of recent NMR structural studies indicating a direct interaction between Bcl-xL and VDAC (Malia and Wagner, 2007). To further examine if the interaction between Bad, Bcl-xL and VDAC depends on Bad phosphorylation, Bcl-xL immune complexes were interrogated in the presence or absence of Cer for the presence of Bad and VDAC. In the Cer-treated cells, more Bad and less VDAC was associated with Bcl-xL than in the control (Fig6D). These findings provide evidence that Bcl-xL is displaced from VDAC upon binding to dephosphorylated Bad. Taken together, formation of the Bad-, PP2A-, PKA-and PKC-containing signaling complex, which could be potentially coupled to Bcl-xL and VDAC, may offer a mechanism for integrating phospho-regulatory mechanisms to sensitization of the PTP.
Bax and Bak are dispensible for the Cer+Ca2+-mediated PTP opening and cell death
In many paradigms, BH3 only pro-apoptotic proteins induce cell death by direct or indirect activation of Bax and Bak (Chipuk and Green, 2008; Oh et al., 2006; Willis et al., 2007; Youle, 2007). Bad may interact with Bcl-xL and release Bax from Bcl-xL/Bax heterodimers, revealing its apoptotic activity (Willis et al., 2005; Yang et al., 1995). Binding of Bad to Bcl-xL has also been reported to free activator BH3-only molecules such as Bim, Bid and Puma to directly activate Bax (Kim et al., 2006). We next utilized bax-/-/bak-/- MEFs to test whether Bax and Bak are components of the Cer-PP2A-Bad pathway of PTP sensitization (Fig7A). Unlike bad-/- MEFs (Fig4E), bax/bak-/- MEFs showed substantial Cer-dependent [Ca2+]c dysregulation that was effectively suppressed by OA, Caly A and cptcAMP (Fig7A). Furthermore, in striking contrast to bad-/- MEFs (Fig4G), the combination of Cer and Tg induced substantial cell death in bax-/-/bak-/- MEFs, which was indistinguishable from WT controls (Fig7B), suggesting that Bad promotes the PTP activation and cell death independent of Bax and Bak. To interrogate the requirement of Bax and Bak in Cer-induced delayed mitochondrial dysregulation during physiological calcium mobilization, the synergism between Cer and agonist-induced [Ca2+]c oscillations were evaluated in bax/bak-/- MEFs using a similar approach we have previously reported for HepG2 and H9c2 cells (Pacher and Hajnoczky, 2001; Szalai et al., 1999). Quantification of the mitochondrial Ca2+ storage during prolonged stimulation by ATP (5h), an IP3 linked agonist, in bax/bak-/- MEFs showed reduction by 35±12% in the presence of Cer similar to WT MEFs. This result indicates that the Cer-dependent delayed mitochondrial dysregulation may occur during physiological Ca2+ mobilization independent of Bax and Bak.
Fig7. Cer+Ca2+-induced PTP opening and cell death independent of Bax and Bak.

(A) Delayed Ca2+ dysregulation in permeabilized bax-/-/bak-/- MEFs pretreated with Cer (40 uM) or solvent. Caly A(50nM), OA(50nM) and cptcAMP(5 uM) were added respectively before the addition of Cer. (B) Cell death in intact WT and bax-/-/bak-/- MEFs treated with Cer (40uM) and Tg (2uM) (CD) tBid sensitivity of the ΔΨm, cyto c release and [Ca2+]c retention in permeabilized bax-/-/bak-/- and WT MEFs. (E) Scheme depicting the distinct Bad-PTP and Bid-Bax/Bak mitochondrial membrane permeabilization pathways. The drawing emphasizes the phospho-regulatory elements of the control of mitochondrial membrane permeabilization without including details on the mechanism of the Bax/Bak-and PTP-dependent release of intermembrane space proteins. Small light blue dots illustrate the mitochondrial intermembrane space proteins (including cyto c, Smac/DIABLO, AIF, OMI/HtrA2, endonuclease G, pro-caspases) that are released to the cytoplasm to promote the execution of apoptosis. CypD, cyclophilin D; UP, Ca2+ uniporter.
To exclude the possibility that the lack of protection against Cer in bax-/-/bak-/- MEFs may have resulted from any phenotypic reversion or secondary changes in mitochondria, we examined the mitochondrial effects of tBid and found that bax-/-/bak-/- MEFs were insensitive to cyto c release and loss of ΔΨm (Fig7C) as previously reported (Wei et al., 2001). When Ca2+ pulsing followed the tBid-pretreatment, the added Ca2+ was poorly taken up in WT MEFs (Fig7D). Notably, tBid-dependent [Ca2+]c dysregulation was absent in bax-/-/bak-/- MEFs, indicating that the tBid effect in WT MEFs was due to the release of cyto c and the ensuing loss of the driving force of the mitochondrial Ca2+ uptake and was not dependent on activation of the PTP. tBid, in contrast to Bad (Fig4A), failed to exert any potentiation of the Ca2+-induced PTP opening in bax-/-/bak-/- MEFs (Fig7D). Our results point to previously unappreciated differences between the mechanisms and effects of select BH3-only molecules on mitochondria.
To determine if the Bad BH3 domain alone is competent to evoke sensitization of the PTP, we first applied a recombinant 15-mer minimal BH3 peptide (Holinger et al., 1999; Petros et al., 2004). This peptide (3-16μM) did not augment the Ca2+ pulsing induced depolarization or [Ca2+]c rise and attenuated the effect of Cer (SFig7AB). Subsequently, a 25-mer human Bad BH3 peptide with a greater affinity to Bcl-xL/Bcl-2 (Petros et al., 2004; Walensky et al., 2006) was used (0.25-5μM) that caused cyto c release and mitochondrial Ca2+ dysregulation similar to tBid in HepG2 cells (SFig7CE). The Bad 25-mer peptide did not cause cyto c release in mitochondria isolated from FL5.12 cells (Letai et al., 2002). This difference can be explained by our use of permeabilized HepG2 cells that are significantly more sensitive to tBid-induced cyto c release [<1nM tBid caused complete cyto c release in 5min (Madesh et al., 2002)]. By contrast, in bax-/-/bak-/- MEFs, the 25-mer peptide failed to cause cyto c release (SFig6E) and Ca2+ dysregulation and exerted protection against the Cer effect similar to the 15-mer peptide (SFig7D). Thus, the Bad BH3 domain is necessary for the interaction with Bcl-xL but is not sufficient to cause sensitization of PTP. These results further support a model whereby tBid and Bad engage two distinct pathways of mitochondrial membrane permeabilization and cell death, through Bax/Bak and PTP opening, respectively (Fig7E). Combination of the BH3 domain and an additional domain of the full length Bad is required for enabling signaling towards the PTP.
Discussion
Our findings delineate a molecular pathway that enables an apoptotic switch in mitochondrial calcium signaling. The cascade is mediated through Bad and leads to sensitization of PTP opening, providing the first evidence that a BH3 only protein can bypass Bax/Bak to engage the PTP. We propose a model in which stress agents such as ceramide and staurosporine lead to Bad dephosphorylation, possibly by activating PP2A or inhibiting PKA/PKC in a signaling complex. Dephosphorylation and firm association of Bad with the mitochondrial membrane enable an interaction with Bcl-xL and displacement of VDAC that leads to the sensitization of the PTP to Ca2+.
Cer is produced in the ER, including the mitochondria associated ER membranes (Morales et al., 2007) and can activate multiple effector mechanisms to alter the mitochondrial function (France-Lanord et al., 1997; Gudz et al., 1997; Pinton et al., 2001; Siskind et al., 2002). Using a variety of pharmacological and biochemical tools we have demonstrated that the coupling of Ca2+-dependent PTP activation and cyto c release to the Cer effect is largely dependent on PP2A activity in several cell types (Fig7E). This result emphasizes the pro-apoptotic effect of Cer-induced PP activation (Kishikawa et al., 1999; Kowluru and Metz, 1997; Ruvolo et al., 1999). Our data have also shown that the Cer-PTP coupling is suppressed by the activation of PKA and PKC but not PKG. Furthermore, the inhibition of PKA and PKC seems to underlie the ST-PTP coupling (Fig7E). In ST-induced apoptosis, the PTP activation may complement the predominant Bax pathway (Scorrano et al., 2003; Wei et al., 2001).
Dephosphorylation of Bad has been identified as a pro-apoptotic signal attributed to the activation of calcineurin (Wang et al., 1999), PP1 (Ayllon et al., 2001; Danial et al., 2003) and PP2A (Chiang et al., 2003). Here we show dephosphorylation of Bad in Cer and ST-treated cells, which is due to PP2A activation and PKA/PKC inhibition, respectively. We have directly visualized the phosphorylation state of Ser112, Ser136 and Ser155. Phosphorylation of Ser136 may be a prerequisite for the phosphorylation of Ser155 uncovering another important regulatory site of Bad (Danial et al., 2008; Datta et al., 2000; Zhou et al., 2000). Since massive dilution of the cytoplasm did not prevent the Cer-induced dephosphorylation of Bad, we suspected that the enzymes were anchored in the vicinity of Bad. Notably, a mitochondrial multimolecular complex has been described in liver and beta cells, which contains Bad, PP1, PKA, glucokinase and the WAVE1 PKA anchoring scaffold protein (Danial et al., 2003; Danial et al., 2008). Along this line, the present studies showed pull-down of PP2A, PKC and PKA with Bad, indicating that the functional interaction among these factors is supported by the formation of a signaling complex (Fig7E).
Our studies with recombinant Bad, overexpressed Bad, bad-/- MEFs and genetically reconstituted bad-/- cells provide unequivocal evidence that Bad is required for Cer-PTP coupling, cyto c release and cell death in several cell lines (Fig7E). This result was unexpected since, Bad, like many other BH3-only Bcl-2 family pro-apoptotic proteins is thought to bind and neutralize Bcl-xL (Yang et al., 1995). However, our data showed that both the Cer-PTP coupling and the Cer+Ca2+-induced cell death were preserved in bax-/-/bak-/- MEFs. Notably, suppression of the Cer-induced cell death has been shown in bax-/-/bak-/- MEFs but SERCA overexpression enhanced the Ca2+ available for mobilization and restored the Cer-induced cell killing (Scorrano et al., 2003). The effect of Cer alone required Bax/Bak in the ER to enhance the Ca2+ load however, consistent with our results, the combination of Cer+Ca2+ was able to activate a Bax/Bak independent mitochondrial death pathway. We have shown that the mitochondrial membrane permeabilization by tBid, another BH3 only protein, is insensitive to PTP inhibition (Madesh et al., 2002) and is absent in bax-/-/bak-/- MEFs. Thus, there is a clear separation between Bad-and tBid-dependent mitochondrial membrane permeabilization pathways (Fig7E).
Previous studies have shown that the ceramide-PTP coupling was suppressed by overexpression of Bcl-xL (Pacher and Hajnoczky, 2001). In the present paradigm, the Bad pull-down assay confirmed the interaction of Bad with Bcl-xL. Recent studies also described a direct interaction between Bcl-xL with VDAC (Malia and Wagner, 2007), which integrates a range of signaling inputs for the control of the PTP (Galluzzi and Kroemer, 2007; Pastorino and Hoek, 2003). Until recently, VDAC was also considered as a core component of the pore but permeability transition has been documented in VDAC1 and 3-lacking and VDAC2-depleted cells, suggesting that VDAC is dispensable for the PTP formation (Baines et al., 2007). Here, the pull-down assay revealed an interaction of Bad with VDAC. Furthermore, we found that in vdac1-/- MEFs (Wu et al., 1999), the Cer-dependent sensitization of the PTP towards Ca2+ was greatly suppressed and the protective effect of OA and PKA activators was abrogated similar to bad-/- MEFs (S Sinha Roy, WJ Craigen and G Hajnoczky, unpublished data). Collectively, these results are consistent with the idea that Bad sensitizes the PTP, at least in part, by displacing Bcl-xL from VDAC1. Since the common consequence of the BH3-only protein-Bcl-xL interaction is direct or indirect activation of Bax/Bak (Leber et al., 2007), the manner in which Bad binding to Bcl-xL may initiate a distinct mechanism to PTP activation is intriguing. Previous studies have shown that the BH3 domain of human Bad was sufficient to either activate (Li et al., 2007; Shangary and Johnson, 2002) or sensitize (Letai et al., 2002; Walensky et al., 2006) cyto c release. Here, we showed that the 25-mer Bad BH3 peptide evoked Bax/Bak-dependent cyto c release and the ensuing mitochondrial depolarization, further supporting that the BH3 domain alone is sufficient to activate the Bax/Bak membrane permeabilization pathway in certain experimental settings. However, the BH3 peptide was not competent to cause sensitization of the PTP to Ca2+, indicating that engagement of the Bcl-xL-VDAC pathway is not confined to the BH3 domain of the full length Bad. An intriguing possibility is that dephosphorylation of Bad in the mitochondria associated signaling complex is sufficient to disengage the Bad-dependent stimulation of metabolism (Danial et al., 2008) and to induce sensitization of the PTP, whereas recruitment of more dephosphorylated Bad from the bulk cytoplasm to the mitochondria might be needed to trigger the Bax/Bak-mediated permeabilization. Sensitization of the PTP would trigger cell death only if it is paired with enhanced calcium oscillations, and Ca2+ transfer to the mitochondria, whereas Bax/Bak activation alone ensures the execution of apoptosis. Collectively, our findings provide evidence for phospho-regulatory mechanisms that converge on Bad to regulate PTP sensitization and uncover a Bax/Bak independent mechanism for mitochondrial regulation by a BH3-only protein.
Methods
Recombinant proteins
Bad with a 6 histidine tag at the N-terminus was expressed in the pET23d vector in E.coli. The recombinant protein was recovered in the soluble bacteria fraction and was purified through chromatography on Ni-NTA-Agarose followed by Q-Sepharose and gel filtration on Superdex 75. The purified protein was stored in 25 mM Tris-HCl, 0.2 mM DTT, 30 % glycerol, pH 7.5 at −80 C°. Recombinant caspase-8-cleaved Bid (tBid) was produced as described earlier (Desagher et al., 1999).
Cell culture
HepG2, HEK293T cells (ATCC, Rockville, MD, USA), WT, bax-/-/bak-/- MEFs (kindly provided by Dr. SJ Korsmeyer, Harvard Univ.) and bad-/- MEFs were cultured in DMEM or EMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 mM pyruvate and 100 U/ml penicillin and 100 μg/ml streptomycin in humidified air (5% CO2) at 37°C. For imaging experiments cells were plated onto poly-D-lysine-treated glass coverslips at a density of 20,000–25,000/cm2 and were grown for 3–4 days. For cell suspension studies, cells were cultured for 4–6 days in flasks (75 cm2). For immunoprecipitation (IP) studies cells were grown on cell culture dish (10cm).
Cell transfection and viral infection
Cells were transfected with plasmid DNA (cyto c-GFP (kindly provided by Dr. AL Niemien (Heiskanen et al., 1999)), pEBG-vector and pEBG-Bad, Bcl-xL, FLAG-vector and FLAG-Bcl-xL, using Lipofecta-AMINE 2000 (Invitrogen) with 2 μg of plasmid DNA. Cells were infected with Adenoviral vectors containing GFP (Ad-GFP), a Bad phosphomimetic mutant BadS155D (Ad-BadS155D) and wild type Bad (Ad-Bad) (Danial et al., 2008). Infection was performed in 2% FBS containing medium for 7-8 hrs using 2000 viral particles per cell. Cells were further grown for 48 hrs post infection in normal 10% FBS containing medium before the experiments.
Evaluation of ΔΨm, [Ca2+]c and swelling in permeabilized cell suspension
Cells harvested using 0.25% trypsin–EDTA were washed in an ice cold Ca2+-free extracellular buffer, containing 120 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 0.2 mM MgCl2, 0.1 mM EGTA, 20 mM HEPES-NaOH pH 7.4. Equal aliquots of cells (2.4mg/ml) were resuspended and permeabilized with 40 μg/ml digitonin in 1.5 ml of an intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris pH 7.2 supplemented with 1 μg/ml of each of antipain, leupeptin and pepstatin. All the measurements were carried out in the presence of 2 mM MgATP and ATP regenerating system composed of 5 mM phosphocreatine, 5 U/ml creatine kinase. Mitochondria were energized with 2 mM succinate. Cer (Alexis) and ST (Alexis) were dissolved in DMSO added to the incubations in small volumes of DMSO (<0.2%) and the same amount of DMSO was added to the control samples. For simultaneous measurements of [Ca2+]c and ΔΨm, the permeabilized cells were supplemented with 0.5 μM fura2FF/FA (Kd∼4μM, Teflabs) and 800 nM JC1 (Molecular Probes). Fluorescence was monitored in a fluorimeter (Delta RAM, PTI) using 340 and 380 nm excitation and 535 nm emission for fura2FF whereas 490 nm excitation/535 nm emission and 570 nm excitation/595 nm emission were used for JC1. [Ca2+]c is shown as the excitation ratio (340 nm/380 nm) of fura2FF/FA fluorescence whereas ΔΨm is shown as the ratio of the fluorescence of J-aggregate (ex: 570 nm, em: 595 nm) and monomer (ex: 490 nm, em: 535 nm) forms of JC1. Delayed Ca2+ dysregulation was either calculated as the half recovery time from the last Ca2+ pulse (Figs1CD, 4C) or as the elevation of [Ca2+]c 5 min after the last pulse (Figs 4E, 5BDEF, 7A). For evaluation of PTP opening-induced mitochondrial swelling light scattering was measured using wavelengths ex: 540 nm, em: 540 nm. At the end of the fluorimetric measurements of ΔΨm, cytosol was separated from the membranes by centrifugation at 14,000 × g. Supernatants and membranes were rapidly frozen in liquid nitrogen and were kept at -80°C until use.
Subcellular fractionation
Cells were resuspended in a hyposmotic buffer (water (80%), ICM (20%), 5mM MgCl2, 200μM EGTA and 1X Complete protease inhibitors (Roche)) on ice for 15-20 min. Cells were disrupted by passing through 26-gauge needles 20 times. Cell lysates were centrifuged at 750 g for 10 min at 4°C to eliminate unlysed cells and nuclei. The supernatant was centrifuged at 10,000 g for 20 min at 4°C. The pellet served as the mitochondrial fraction. Pellets were lysed in RIPA buffer and used for immunoblotting.
Confocal imaging analysis of cyto c-GFP release
For imaging of cyto c distribution, cells transfected with cyto c–GFP construct were treated with Cer (40μM), Cer + ATP(100μM) and Cer and ATP in presence of Caly A (50nM, Alexis) or OA (50nM, Biomol) or CsA (2μM). After 3 h, cells were washed twice with 2.0% BSA/ECM consisting of 121 mM NaCl, 5 mM NaHCO3, 10 mM NaHepes, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, and 2% BSA, pH 7.4. The coverslips were transferred to the microscope stage and cyto c–GFP distribution was monitored with confocal imaging (488 nm excitation). Images were analyzed with customized software.
Intact cell imaging of [Ca2+]c
The cells were first loaded with fura2/AM (5 μM) for 30 min at 37 °C in 2.0% BSA/ECM, in the presence of 0.003% pluronic acid and 100 μM sulfinpyrazone. After dye loading, the cells were rinsed with ECM, and the coverslip was mounted to a heated (35 °C) incubator chamber in ECM containing 0.25% BSA and sulfinpyrazone. Fluorescence imaging was carried out using a Leica DMIRE2 inverted microscope fitted with a 40X (UApo, NA 1.35) objective and a cooled CCD camera (PXL, Photometrics). Excitation at 340 and 380nm was used with a broad band emission filter passing 460-600nm.
When the interaction of IP3-linked Ca2+ mobilization and Cer on delayed mitochondrial dysregulation was studied the bax-/-/bak-/- MEFs the cells were pretreated with ATP in the presence and absence of Cer 40μM for 5h and subsequently, were loaded with fura2/AM. During the [Ca2+]c time course measurement, FCCP was added to completely discharge the Ca2+ that was accumulated by the mitochondria during the stimulation. The FCCP-induced [Ca2+]c elevation was quantitated in each cell and was used as a measure of the mitochondrial Ca2+ retention. FCCP addition to non-stimulated cells caused no significant [Ca2+]c rise (Hajnoczky et al., 1995).
Flow cytometry
Cell death was evaluated by forward laser light scatter and side laser light scatter, which parameters are correlated to cell size and granularity, respectively.
GST pull-down assay
Cell extracts were prepared from 293T cells transfected with GST-Bad or GST-vector by scraping and lysing 1×107 cells in 200μL GST binding buffer (20mM Hepes pH7.4, 1.5mM MgCl2, 100mM NaCl, 1% Triton X-100, freshly added DTT1mM, 1mM PMSF, and complete protease inhibitors). Cell extracts were incubated overnight at 4°C with Glutathione Sepharose 4B beads (GE Healthcare, USA). Beads were washed thrice with GST-binding buffer and proteins were eluted with SDS sample buffer for western analysis.
Co-IP experiments
Cell extracts were prepared from the membrane fractions of FLAG-Bcl-xL or FLAG-transfected permeabilized 293T cells (1×107 cells) by lysing in 500μl RIPA buffer. The cleared lysates were then subjected to IP for overnight at 4°C with anti-FLAG M2 antibody (Sigma) and Agarose A/G beads (Santa Cruz) were added for an additional 4-5 hrs at 4°C. Beads were washed 4 times with RIPA buffer and proteins were eluted with SDS sample buffer for western analysis.
Immunoblot analysis
For western blotting, 20–30 μg proteins were loaded onto each lane of a 15% SDS-PAGE and electrophoretically transferred to nitrocellulose filters. Filters were blocked with blocking buffer (Pierce) overnight, followed by incubation with primary antibodies according to the dilutions recommended by the manufacturers. Primary antibodies were anti–cyto c (BD Pharmingen), anti-Bad, Bad Ser-112P and Bad Ser-136P and Bad Ser-155P (Cell Signaling), anti-PP2A and anti-GST (Santa Cruz), and anti-Bcl-xl, anti-PKAc, anti-PKC (BD Transduction). After incubation with the primary antibody, bound antibodies were visualized using horseradish peroxidase–coupled secondary antibody (GE Healtcare, UK) and Dura Signal chemiluminescence–developing kit (Pierce).
Statistics
The data are shown as mean±SE (n≥3). Significance of differences from the relevant controls was calculated by Student's t test.
Supplementary Material
Acknowledgments
The authors thank Eric Smith for creation of the scheme in Fig7E. This work was supported by an NIH grant (KO1CA10659) and a Burroughs Welcome Fund Career Award in Biomedical Sciences to N.D. and an NIH grant (GM59419) to G.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ayllon V, Cayla X, Garcia A, Roncal F, Fernandez R, Albar JP, Martinez C, Rebollo A. Bcl-2 targets protein phosphatase 1 alpha to Bad. J Immunol. 2001;166:7345–7352. doi: 10.4049/jimmunol.166.12.7345. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Forte M. The mitochondrial permeability transition pore. Novartis Found Symp. 2007;287:157–164. discussion 164-159. [PubMed] [Google Scholar]
- Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- Cabral LM, Wengert M, da Ressurreicao AA, Feres-Elias PH, Almeida FG, Vieyra A, Caruso-Neves C, Einicker-Lamas M. Ceramide is a potent activator of plasma membrane Ca2+-ATPase from kidney-promixal tubule cells with protein kinase A as an intermediate. J Biol Chem. 2007;282:24599–24606. doi: 10.1074/jbc.M701669200. [DOI] [PubMed] [Google Scholar]
- Chami M, Ferrari D, Nicotera P, Paterlini-Brechot P, Rizzuto R. Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J Biol Chem. 2003;278:31745–31755. doi: 10.1074/jbc.M304202200. [DOI] [PubMed] [Google Scholar]
- Chiang CW, Kanies C, Kim KW, Fang WB, Parkhurst C, Xie M, Henry T, Yang E. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol Cell Biol. 2003;23:6350–6362. doi: 10.1128/MCB.23.18.6350-6362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 2008 doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, Datta SR, Pitter KL, Bird GH, Wikstrom JD, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C. Cell biology. Apoptosis--the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouder N, Metzler SC, Schmidt A, Wirbelauer C, Gstaiger M, Aebersold R, Hess D, Krek W. S6K1-mediated disassembly of mitochondrial URI/PP1gamma complexes activates a negative feedback program that counters S6K1 survival signaling. Mol Cell. 2007;28:28–40. doi: 10.1016/j.molcel.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- France-Lanord V, Brugg B, Michel PP, Agid Y, Ruberg M. Mitochondrial free radical signal in ceramide-dependent apoptosis: a putative mechanism for neuronal death in Parkinson's disease. J Neurochem. 1997;69:1612–1621. doi: 10.1046/j.1471-4159.1997.69041612.x. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol. 2007;9:487–489. doi: 10.1038/ncb0507-487. [DOI] [PubMed] [Google Scholar]
- Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJ. Menadione-induced apoptosis: roles of cytosolic Ca(2+) elevations and the mitochondrial permeability transition pore. J Cell Sci. 2002;115:485–497. doi: 10.1242/jcs.115.3.485. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hardie DG. Roles of protein kinases and phosphatases in signal transduction. Symp Soc Exp Biol. 1990;44:241–255. [PubMed] [Google Scholar]
- Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Holinger EP, Chittenden T, Lutz RJ. Bak BH3 peptides antagonize Bcl-xL function and induce apoptosis through cytochrome c-independent activation of caspases. J Biol Chem. 1999;274:13298–13304. doi: 10.1074/jbc.274.19.13298. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Fabbro D, Pfeilschifter J. Selective ceramide binding to protein kinase C-alpha and -delta isoenzymes in renal mesangial cells. Biochemistry. 1998;37:14556–14562. doi: 10.1021/bi981401i. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kishikawa K, Chalfant CE, Perry DK, Bielawska A, Hannun YA. Phosphatidic acid is a potent and selective inhibitor of protein phosphatase 1 and an inhibitor of ceramide-mediated responses. J Biol Chem. 1999;274:21335–21341. doi: 10.1074/jbc.274.30.21335. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Metz SA. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 1997;418:179–182. doi: 10.1016/s0014-5793(97)01379-3. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Li R, Boehm AL, Miranda MB, Shangary S, Grandis JR, Johnson DE. Targeting antiapoptotic Bcl-2 family members with cell-permeable BH3 peptides induces apoptosis signaling and death in head and neck squamous cell carcinoma cells. Neoplasia. 2007;9:801–811. doi: 10.1593/neo.07394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madesh M, Antonsson B, Srinivasula SM, Alnemri ES, Hajnoczky G. Rapid kinetics of tBid-induced cytochrome c and Smac/DIABLO release and mitochondrial depolarization. J Biol Chem. 2002;277:5651–5659. doi: 10.1074/jbc.M108171200. [DOI] [PubMed] [Google Scholar]
- Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malia TJ, Wagner G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry. 2007;46:514–525. doi: 10.1021/bi061577h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales A, Lee H, Goni FM, Kolesnick R, Fernandez-Checa JC. Sphingolipids and cell death. Apoptosis. 2007;12:923–939. doi: 10.1007/s10495-007-0721-0. [DOI] [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- Oh KJ, Barbuto S, Pitter K, Morash J, Walensky LD, Korsmeyer SJ. A membrane-targeted BID BCL-2 homology 3 peptide is sufficient for high potency activation of BAX in vitro. J Biol Chem. 2006;281:36999–37008. doi: 10.1074/jbc.M602341200. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Pacher P, Hajnoczky G. Propagation of the apoptotic signal by mitochondrial waves. Embo J. 2001;20:4107–4121. doi: 10.1093/emboj/20.15.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10:1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. Embo J. 2001;20:2690–2701. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME, Korsmeyer SJ. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9324–9329. doi: 10.1073/pnas.1533446100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. Embo J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274:20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem. 2001;276:12035–12040. doi: 10.1074/jbc.M010603200. [DOI] [PubMed] [Google Scholar]
- Shangary S, Johnson DE. Peptides derived from BH3 domains of Bcl-2 family members: a comparative analysis of inhibition of Bcl-2, Bcl-x(L) and Bax oligomerization, induction of cytochrome c release, and activation of cell death. Biochemistry. 2002;41:9485–9495. doi: 10.1021/bi025605h. [DOI] [PubMed] [Google Scholar]
- Siskind LJ, Feinstein L, Yu T, Davis JS, Jones D, Choi J, Zuckerman JE, Tan W, Hill RB, Hardwick JM, Colombini M. Anti-apoptotic Bcl-2 Family Proteins Disassemble Ceramide Channels. J Biol Chem. 2008;283:6622–6630. doi: 10.1074/jbc.M706115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–26803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. Embo J. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Kim C, Cheng CY, Brown SH, Wu J, Kannan N. Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim Biophys Acta. 2008;1784:16–26. doi: 10.1016/j.bbapap.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Wu S, Sampson MJ, Decker WK, Craigen WJ. Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochim Biophys Acta. 1999;1452:68–78. doi: 10.1016/s0167-4889(99)00120-2. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Youle RJ. Cell biology. Cellular demolition and the rules of engagement. Science. 2007;315:776–777. doi: 10.1126/science.1138870. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Larochette N, Kroemer G. Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 2005;12 Suppl 2:1478–1480. doi: 10.1038/sj.cdd.4401682. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhou XM, Liu Y, Payne G, Lutz RJ, Chittenden T. Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155. J Biol Chem. 2000;275:25046–25051. doi: 10.1074/jbc.M002526200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.