

Abstract
Mycobacterium tuberculosis (Mtb) cytochrome P450 gene CYP121 is shown to be essential for viability of the bacterium in vitro by gene knock-out with complementation. Production of CYP121 protein in Mtb cells is demonstrated. Minimum inhibitory concentration values for azole drugs against Mtb H37Rv were determined, the rank order of which correlated well with Kd values for their binding to CYP121. Solution-state spectroscopic, kinetic, and thermodynamic studies and crystal structure determination for a series of CYP121 active site mutants provide further insights into structure and biophysical features of the enzyme. Pro346 was shown to control heme cofactor conformation, whereas Arg386 is a critical determinant of heme potential, with an unprecedented 280-mV increase in heme iron redox potential in a R386L mutant. A homologous Mtb redox partner system was reconstituted and transported electrons faster to CYP121 R386L than to wild type CYP121. Heme potential was not perturbed in a F338H mutant, suggesting that a proposed P450 superfamily-wide role for the phylogenetically conserved phenylalanine in heme thermodynamic regulation is unlikely. Collectively, data point to an important cellular role for CYP121 and highlight its potential as a novel Mtb drug target.
The human pathogen Mycobacterium tuberculosis (Mtb)4 has made an alarming resurgence and again poses a global threat to human health (World Health Organization fact sheet on “Tuberculosis”; located on the World Wide Web). The worldwide spread of tuberculosis has been fuelled by the development and spread of drug- and multidrug-resistant Mtb strains (2). The growing numbers of Mtb strains resistant to front line antitubercular drugs (e.g. rifampicin and isoniazid) has revealed a dearth of effective second line agents and has highlighted a desperate need for the development of novel drugs (3).
Against this backdrop, the determination of the Mtb H37Rv genome sequence (and latterly the Mtb CDC1551 sequence) provided important new information toward a more detailed understanding of the biology of Mtb, its genome organization, and its protein repertoire (4, 5). Mtb H37Rv is a virulent strain that has been the most commonly used Mtb strain in laboratory and clinical studies for over 50 years. Its genome revealed an unexpectedly large number of genes encoding cytochrome P450 (CYP or P450) enzymes, 20 CYP genes in the 4.41-megabase Mtb H37Rv genome (4). The Escherichia coli genome (of similar size) is devoid of P450s, and the 57 P450s in the human genome are contained within a ∼3000-megabase genome. Thus, the CYP gene “density” in the Mtb genome is >200-fold that in the human genome, indicating important cellular roles for these Mtb oxygenases (6). In addition, the first prokaryotic example of a sterol demethylase P450 (CYP51B1) was identified in Mtb (4, 7). This raised the possibility that Mtb P450s could be novel drug targets, since fungal CYP51s are validated targets for azole drugs (e.g. fluconazole and clotrimazole) that coordinate the P450 heme iron and prevent oxidative transformation of lanosterol to ergosterol, with severe effects on fungal membrane integrity (8). It was shown subsequently that several imidazole and triazole-based antifungals were effective anti-mycobacterial agents, with MIC values for various azoles being on the order of 0.1–20 μg/ml for M. smegmatis (9–11).
However, CYP51B1 is not an essential gene for Mtb viability in culture and is unlikely to be a Mtb azole drug target (12). Instead, the Mtb CYP121 P450 was shown to bind very tightly to a range of azole antifungal drugs. Hitherto, there has been no evidence presented for the essentiality (or otherwise) of the CYP121 gene (Rv2276) in Mtb H37Rv (12). Although Mtb genome-wide transposon mutagenesis revealed that several P450 genes were not essential for viability or optimal growth in vitro, these studies did show that CYP128 was essential (12, 13). However, no data were presented regarding essentiality of CYP121 in these studies (12, 13) (see the Tuberculosis Animal Research and Gene Evaluation Taskforce (TARGET) site on the World Wide Web). Atomic structures of Mtb CYP51B1 and CYP121 were determined, both in complex with fluconazole and in ligand-free forms (15–18). The CYP51B1-fluconazole complex structure demonstrated direct coordination of P450 heme iron by a fluconazole triazole nitrogen (15). The ligand-free structure of CYP121 revealed a highly constrained active site, and major reorganization of CYP121 structure was predicted to be necessary to facilitate direct azole coordination of the P450 heme iron (18). However, the fluconazole-bound CYP121 structure showed a relative lack of structural perturbation and instead revealed an unusual mode of inhibitory coordination, with the azole nitrogen bridging to the heme iron via an interstitial water molecule (17). This type of binding was observed previously by Poulos and Howard (19) in studies of 2-phenylimidazole binding to P450cam. Recently, the structure of Mtb CYP130 (encoded by the Rv1256c gene absent from the genome of the Mycobacterium bovis BCG vaccine strain) was solved in complex with econazole, an effective antitubercular drug (20, 21). Econazole binds tightly to CYP130 (Kd = 1.93 μm) (22), although the Kd is higher than that for CYP121 (0.024 μm; this study). However, CYP130 is a nonessential gene in Mtb strain CDC1551 (23).
The ligand-free CYP121 structure was the highest resolution P450 atomic structure solved to date (1.06 Å) and also revealed novel aspects of P450 structure, including (i) a distorted heme macrocycle caused by the displacement of a pyrrole group by a proline side chain, (ii) heme bound in two distinct conformations related by a 180° flip, and (iii) a relatively rigid active site hydrogen-bonded network of residues above the heme plane that defined the active site geometry and might contribute to a proton relay pathway to the heme iron (18).
In this study, we validate the importance of CYP121 to Mtb H37Rv viability by establishing its essentiality through CYP121 gene knock-out. We investigate the active site structure and role of several residues in the vicinity of the heme through a systematic determination of CYP121 mutant atomic structures, allied to detailed spectroscopic, thermodynamic, and azole binding analysis of these active site variants. We also report the MIC values for inhibition of Mtb H37Rv growth by azole drugs and compare these with their affinity (Kd values) for CYP121 and active site mutants thereof. Our study defines a new gene (CYP121) essential for viability of Mtb H37Rv and details the structural and biophysical properties of key CYP121 active site mutants. These data reveal important determinants of heme iron coordination and heme conformational and thermodynamic properties of CYP121 and enable a more detailed understanding of roles of residues either unique to CYP121 or broadly conserved in the P450 superfamily.
EXPERIMENTAL PROCEDURES
Generation and Characterization of M. tuberculosis CYP121 Deletion Strain
A deletion delivery vector was generated by amplifying the upstream and downstream regions of CYP121 using primer pairs EUF5 (AAG CTT GAG ACG ACT CTG CTC CCA AC) and EUR5 (GGT ACC GCA CAG TGC ATA CGA GGA GA), and EUF6 (GGT ACC GCG CTG TTG AAA AAG ATG C) and EUR6 (GCG GCC GCA ACA CCG TTC TGG CGA TTA C) and cloning into p2NIL (24) to generate an unmarked in-frame deletion. Restriction sites used for cloning are underlined. The gene cassette from pGOAL19 (24) was cloned in as a PacI fragment to generate the final delivery vector pTACK121. A complementing vector (pCOLE121) was constructed by amplifying the CYP121 gene using primers CYP121D1 (CCT TAA TTA ATC GTT GAA TTG CTA CCA CCA) and CYP121D2 (CCT TAA TTA AGG TGC AAG GTC GAA ATT GTT) (PacI sites underlined) and subcloned into pAPA3 (25) under the control of the Ag85a promoter.
Attempts to construct the CYP121 deletion strain using a two-step homologous recombination method were made (24). A single crossover strain was generated by electroporating M. tuberculosis with 5 μg of UV-treated pTACK121, and recombinants were selected on 100 μg/ml hygromycin, 20 μg/ml kanamycin, and 50 μg/ml X-gal (24). A single strain was streaked out in the absence of any antibiotics to allow the second crossover to occur. Double crossovers were selected and screened for using 2% (w/v) sucrose and 50 μg/ml X-gal; white colonies were patch-tested for kanamycin and hygromycin sensitivity to ensure that they had lost the plasmid. PCR was used to screen for the presence of the wild type (WT) or deletion allele using primers CYP121D1 and CYP121D2.
To generate a merodiploid strain, pCOLE121 was electroporated into the single crossover strain, and recombinants were isolated on 10 μg/ml gentamicin, 100 μg/ml hygromycin, 20 μg/ml kanamycin, and 50 μg/ml X-gal. A single recombinant was streaked out without antibiotics to allow a second crossover to occur, and double crossovers were isolated as before, except that gentamicin was included at all stages. PCR was used to screen for the presence of the WT or deletion allele using primers CYP121D1 and CYP121D2. Southern blot analysis was used to confirm the genotype of strains.
Determination of Mtb H37Rv MIC Values with Selected Azoles
Azole susceptibility testing on M. tuberculosis H37Rv was done by radiometric measurements using the BACTEC 460 system (BD Biosciences). A standard protocol was followed, using a biosafety level 3 biocontainment facility (25). The BACTEC vials contained Middlebrook 7H12B medium (BD Biosciences) with 14C-labeled palmitic acid as a carbon source. Four different azole drugs were tested for bacterial growth inhibition (econazole, miconazole, ketoconazole, and clotrimazole). Concentrated stocks of the azoles were made in DMSO and stored at -70 °C until use. A range of azole drug concentrations were tested from 2 to 64 μg/ml. The principle of the BACTEC system is described in more detail in the supplemental material.
Immunodetection of Native CYP121 in Mtb
Short term culture filtrate and total cell lysate of M. tuberculosis H37Rv proteins were prepared as previously described (26). Antiserum 1426D used for identification of CYP121 was raised by immunizing rats with purified, recombinant CYP121 delivered in Montanide ISA720 adjuvant (Seppic, Paris, France). SDS-PAGE and Western blots were performed as described under reducing conditions. To reduce responses to potential E. coli contaminants 1426D serum was absorbed with an E. coli extract for Western blot experiments (26).
Generation of CYP121 Mutants
Mutagenesis of CYP121 was done using the Stratagene QuikChange® site-directed mutagenesis kit for mutants A233G, F338H, S237A, S279A, R386L, and P346L. Mutant clones were confirmed by DNA sequencing (MWG Biotech) using generic T7 and T7 terminator oligonucleotide primers, allowing verification of entire gene sequences. Full methods for mutant production are given in the supplemental material. Primers used for CYP121 mutant gene generation are detailed in Table S1.
Expression and Purification of WT and Mutant CYP121 Proteins
The Mtb Rv2276 gene encoding WT CYP121 protein was expressed from E. coli HMS174 (DE3)/pKM2b transformants (22). CYP121 protein was prepared from 10-liter cultures, as described previously (22). Mutant CYP121 proteins were prepared similarly. The purity of CYP121 proteins was assessed by spectral properties (ratio of Soret absorption, at 416.5 nm in most cases, to protein-specific absorption at 280 nm, with an A416.5/A280 ratio of >1.8 indicating pure protein), and by SDS-PAGE analysis of protein samples (on 12% denaturing gels). CYP121 concentration was determined from the Soret absorption of the ferric enzyme in its ligand-free low spin state using ε416.5 = 110 mm-1 cm-1, as described previously (27). For CYP121 mutants that displayed some high spin heme iron, a NO complex was generated by brief bubbling of ferric enzyme with NO gas. This gave rise to a single Fe(III)NO species (rather than a mixture of high spin and low spin Fe(III) species), and the absorption at the new Soret peak (437 nm; ε437 = 102 mm-1 cm-1) was then used to determine mutant enzyme concentration with reference to WT and as detailed in our previous work (27).
Crystallization and Structural Elucidation of CYP121 Mutant Enzymes
All mutant CYP121 enzymes were crystallized under the same conditions as reported previously for WT CYP121 (18). Complete data sets were obtained on single flash-cooled crystals at 100 K at the European Synchrotron Radiation Facility ID14 stations. Data were analyzed and merged using MOSFLM and SCALA (28, 29). Structures were refined using REFMAC5 (30), using the WT structure as the starting model. The solvent model was built automatically using ARP/wARP (31). The oxidation state of the CYP121 heme may be ferrous, by analogy with the observed effects of synchrotron radiation on other heme proteins (32). For final data and refinement statistics, see Table S2.
Ligand Binding Studies on CYP121 and Active Site Mutants
Optical titrations for determination of azole binding constants (Kd values) were done as previously described (22). Pure WT CYP121 and mutants (typically 1–3 μm) were suspended in Buffer A in a 1-cm path length quartz cuvette, and a spectrum for the ligand-free form was recorded (250–800 nm) at 25 °C on a Cary UV-50 Bio scanning spectrophotometer (Varian, UK). Azole ligands (clotrimazole, econazole, fluconazole, miconazole, ketoconazole, voriconazole, 2-phenylimidazole (2-PIM), and 4-phenylimidazole (4-PIM)) were titrated from concentrated stocks in DMSO solvent (apart from the phenylimidazoles, which were prepared in 60% ethanol) until no further optical perturbation was observed. Full information on titration methods is provided in the supplemental material. Induced optical change versus ligand concentration data were fitted using either a standard hyperbolic function (for data from 2-PIM and 4-PIM titrations) or to Equation 1, which provides a more accurate description of the binding of the antifungal azoles to WT and mutant forms of CYP121 (22, 27). Data were fitted using Origin software (OriginLab, Northampton, MA).
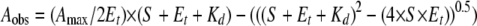 |
(Eq. 1) |
In Equation 1, Aobs is the observed absorbance change at ligand concentration S, Amax is the absorbance change at ligand saturation, Et is the P450 concentration, and Kd is the dissociation constant for the P450-ligand complex.
All other spectral measurements for enzyme quantification and for establishing features of CYP121 in various redox states and in complex with other ligands (i.e. CO and NO) were also performed using a Cary 50 UV-visible spectrophotometer, either aerobically or under anaerobic conditions in a glove box (Belle Technology, Portesham, UK) for ferrous enzymes.
Analysis of Redox Potentials of CYP121 Mutants
Redox potentials for WT and mutant CYP121 enzymes were determined by anaerobic spectroelectrochemical titration according to established methods and as detailed in our previous studies of CYP121 and other P450s (22, 27, 33–35). Full details are given in the supplemental material.
Spectroscopic Characterization of CYP121 Mutants
UV-visible Spectroscopy—All UV-visible absorption spectra for oxidized WT and mutant CYP121 enzymes were recorded on a Cary UV-50 spectrophotometer in Buffer A at 25 °C, as were spectra for azole ligand complexes and for Fe(III)NO and Fe(II)CO complexes.
Resonance Raman—Resonance Raman (RR) spectra were obtained using 15-milliwatt, 406.7-nm radiation at the sample, from a Coherent Innova 300 krypton ion laser, and acquired using a Renishaw micro-Raman system 1000 spectrophotometer. The sample in Buffer A was held in a capillary under the microscope at a concentration of ∼50 μm, and an extended scan was obtained from 200 to 1700 cm-1 (total exposure time of 30 s). Ligand-free and fluconazole (100 μm)-bound CYP121 samples were analyzed. Data processing, curve fitting, and band assignment was done using GRAMS/32 software (Thermo Scientific).
EPR Spectroscopy—EPR spectra for the ferric CYP121 WT and mutant enzymes were recorded on a Bruker ER-300D series electromagnet and microwave source interfaced with a Bruker EMX control unit and fitted with an ESR-9 liquid helium flow cryostat (Oxford Instruments) and a dual mode microwave cavity from Bruker (ER-4116DM). Spectra were recorded at 10 K with a microwave power of 2.08 milliwatts and a modulation amplitude of 10 gauss for the CYP121 enzymes. Oxidized CYP121 and mutant samples were prepared in Buffer A.
Reconstitution of a Mtb Class I Electron Transport Chain to CYP121 and Its Active Site Mutants
In order to establish if active site mutations disrupted electron transfer from NADPH to the P450 heme iron, kinetics of electron transport were analyzed using a Mtb H37Rv class I P450 redox system comprising the NAD(P)H-dependent flavoprotein reductase (FprA, encoded by Rv3106), the 3Fe-4S ferredoxin Fdx (encoded by Rv0763c adjacent to the CYP51B1 gene on the genome), and the relevant WT or mutant CYP121 protein. Conditions were 4 μm CYP121 protein, 18 μm Fdx, and 4 μm FprA in 1 ml of Buffer A at 25 °C. The buffer was deaerated and saturated with CO by extensive bubbling with the gas in a sealed quartz cuvette. Thereafter, the protein components were added (less than 20 μl of total additions), and the reaction was initiated by injection of 300 μm NADPH. Spectra (250–800 nm) were recorded regularly until no further change was detected and the P450 Fe(II)CO complexes were fully formed.
The kinetics of complex formation were determined by plotting extent of Fe(II)CO complex formation against reaction time and fitting the resultant data using an exponential function and Origin software.
Materials
Bacterial growth media (Tryptone, yeast extract) were from Melford Laboratories (Ipswich, Suffolk, UK). The 1-kb DNA ladder was from Promega. Azole drugs were from MP Biomedicals Inc. All other reagents were from Sigma and were of the highest grade available.
RESULTS AND DISCUSSION
Essentiality of the CYP121 Gene in M. tuberculosis H37Rv and Validation of CYP121 Expression in Mtb
We used a two-step homologous recombination strategy to demonstrate that CYP121 (Rv2276) is essential. A nonreplicating delivery vector carrying an unmarked in-frame deletion was constructed and used in a two-step homologous recombination process to isolate deletion mutants in either the WT or merodiploid. In the WT background, 40 double crossovers were screened by PCR, and all had the WT genotype. In contrast, we were able to isolate double crossovers with the deletion in the merodiploid background. Of eight strains screened, all had the deletion allele, and none had the WT allele. The genotype of the strains was confirmed by Southern analysis using two different restriction enzymes (Fig. 1).
FIGURE 1.

Southern hybridization of CYP121 delinquents. Genomic DNA was digested with BamHI and hybridized with the CYP121 probe. The WT (2.5 kb), integrated (4.2 kb), and deletion (1.3 kb) bands are indicated. Lane 1, 1-kb ladder; lane 2, WT; lanes 3–7, CYP121 delinquents; lane 8, 1-kb ladder. In lanes 3–7, the upper band reports the presence of the newly integrated CYP121 gene, whereas the lower band reports on the probe hybridization to the remnant of the original chromosomal copy of CYP121 following generation of the in-frame deletion.
In parallel studies, we established that the CYP121 protein was produced by Mtb using immunological methods. Anti-CYP121 serum was produced, and it was demonstrated that this serum recognized a band of the correct molecular weight from Mtb cell lysate (Fig. S1). Thus, expression and production of this essential P450 was also validated in Mtb.
Deletions encompassing CYP121 were reported in some clinical isolates (36). However, it was postulated that such gene deletions could offer short term advantages to Mtb in, for example, evading immune response or in curtailing latency (36). Thus, genetic background is important when considering gene essentiality. For example, CYP121 may play a role in a pathway that is wholly deleted in the clinical isolates. Recent studies by Gao et al. (37) demonstrated that CYP121 was among the 16% of Mtb genes consistently expressed across a selection of clinical isolates and was the only P450 (of 17 Mtb CYP genes in their data set) in this category. Our results show that CYP121 protein is produced in Mtb and are thus consistent with these data. Similarly, our demonstration of CYP121 gene essentiality is consistent with previous genome-wide transposition studies, which indicated that several Mtb CYP genes were not essential for growth in vitro but which did not provide any data relevant to CYP121 essentiality (12, 13).
Determination of MIC Values for Azole Drugs against M. tuberculosis H37Rv
Our previous studies showed that selected azole drugs were potent inhibitors of growth of M. smegmatis and various Streptomyces strains (actinobacteria that also have a large complement of CYP genes) (11). M. smegmatis MIC values for econazole (<0.1 μg/ml), clotrimazole (0.1 μg/ml), and miconazole (1.25 μg/ml) were comparable with (or superior to) that for the leading antitubercular drugs rifampicin (1.25 μg/ml) and isoniazid (5 μg/ml), with ketoconazole being less effective (20 μg/ml) and the more polar fluconazole substantially less effective (>100 μg/ml). Negligible effects on growth of E. coli were observed for these azoles.
To determine the effects of azole antifungal drugs against Mtb H37Rv, we determined MIC values using the BACTEC system (38). The more water-soluble azoles were not effective against M. smegmatis, and previous work has also indicated that fluconazole was ineffective against Mtb (39). For this reason, we determined Mtb H37Rv MIC values for the more hydrophobic azole compounds clotrimazole, econazole, ketoconazole, and miconazole (Azole structures are shown in Fig. S2). The data indicated that econazole (MIC = 8 μg/ml) and miconazole (8 μg/ml) were most effective, followed by clotrimazole (11 μg/ml) and ketoconazole (16 μg/ml). Although these MIC values are higher than for M. smegmatis, they indicate that the azoles retain activity against Mtb. Although this is the first report of MIC values against Mtb for most of these azoles, our MIC value for ketoconazole (16 μg/ml) is also consistent with MIC values reported recently by Byrne et al. for Mtb H37Rv (8–16 μg/ml) and for the avirulent H37Ra strain (8 μg/ml) in liquid culture (39). The rank order of potency of MIC values for the drugs characterized to have anti-Mtb effects is the same as that determined for their Kd values for CYP121, suggesting again that this enzyme could be an important target for these drugs in vivo. Econazole and miconazole had the lowest MIC values for Mtb H37Rv, and econazole was also the drug with lowest Kd value for CYP121.
In M. smegmatis, econazole and clotrimazole inhibited synthesis of glycopeptidolipids, a major component of the bacterial outer layer (40). Although CYP121 is refractory to optical change upon interaction with a variety of lipids and steroids, we have been able to demonstrate binding interactions between CYP121 and crudely fractionated lipids from Mtb (ongoing studies).5 These preliminary data thus also point to recognition of Mtb lipid substrate(s) by CYP121, and it is notable that the relevant fractions contain the complex long chain (60–90-carbon) mycolic acids as potential substrates.
It is also important to note that recent studies showed that econazole was effective in clearing Mtb infection in a mouse model, further validating the antitubercular therapeutic potential of this drug (41). In studies by Khuller and co-workers (21, 42), the MIC90 value for econazole (i.e. the lowest drug concentration at which 90% of bacterial growth was inhibited) was evaluated as 0.12–0.125 μg/ml for both Mtb H37Rv and for various multidrug-resistant Mtb strains. The reasons for the discrepancy between these values and those of ourselves (and others) are uncertain but are probably due to the different methods of analysis (colony enumeration versus growth rate analysis by metabolic measurements in our case).
Data presented here are thus consistent with an essential role for CYP121 in Mtb H37Rv viability and show that azole drugs, for which CYP121 has high affinity, are also highly effective in inhibiting Mtb growth.
Crystal Structures of CYP121 Mutants
Analysis of the high resolution (1.06 Å) structure of WT CYP121 revealed an unusual active site architecture and highlighted several residues that were postulated to be important for, for example, (i) regulation of active site structure (Ala233, Ser237, and Arg386), (ii) hydrogen bonding and proton relay networks (Ser237, Arg386, and Ser279), (iii) heme conformation and water ligand retention (Pro346, Ser237, and Arg386), and (iv) thermodynamic regulation of heme iron (Phe338). To address roles of these residues in P450 structure and azole binding, we determined crystal structures of all mutants.
All CYP121 mutants were crystallized under identical conditions to WT CYP121. Crystallographic data are presented in Table S2. The corresponding crystal structures of all CYP121 mutants revealed that all mutants retained a WT-like conformation and that differences were localized to the immediate environments of mutated residues. Recent studies by Schlichting's group (32) showed further x-ray photo-reduction of heme iron in oxyferrous P450cam at 140 K. However, the redox potential of the CYP121 heme iron is substantially more negative than that for the CYP121 oxycomplex (22). Given the highly similar protocol used for data collection, we assume that all CYP121 structures reported here (and for the previously reported WT structure) are in the same oxidation state following data collection (18). Hence, structural changes observed between WT CYP121 and CYP121 mutants should reflect effects of mutations rather than any oxidation state changes in the heme iron. In combination with biophysical analysis, important conclusions were drawn relating to structural and thermodynamic properties of CYP121, which have wider implications within the P450 superfamily.
For the F338H and R386L mutant structures, no significant changes, with the exception of the amino acid change itself, could be identified (Fig. 2, A and B; a representative electron density map illustrating quality of the data is also presented as Fig. S3). However, the P346L mutation has considerable effects on the heme macrocycle conformation. In the WT structure, the Pro346 side chain is in close contact with the heme group and appears to be responsible for the extreme distortion from planarity for pyrrole ring D. Removal of the Pro side chain in the P346L mutant leads to a more planar heme conformation. In this case, the multiple conformations observed for the Leu346 side chain appear linked to the multiple conformations of the heme D pyrrole, confirming that the nature and conformation of the residue at position 346 largely govern the position of heme pyrrole D (Fig. 2C and Fig. S4).
FIGURE 2.

Active site structures of CYP121 mutant enzymes. Atomic structures were determined for each of six CYP121 point mutants in the vicinity of the heme cofactor. The figure shows an overlay of the CYP121 mutant structures with the WT structure (Protein Data Bank code 1N40). Mutant structures are atom-colored with green carbons, whereas the WT structure is in grayscale. With the exception of C, the heme is only shown for the WT structure for clarity. In C (as for other panels) the WT heme is in gray, whereas the heme group of the P346L mutant is shown with green carbons. The P346L heme and Leu346 side chain are observed in two distinct conformations. A more detailed view is presented in Fig. S4 in the supplemental material. Selected side chains are displayed in sticks and associated water molecules in spheres for each overlaid structure. A–F show the overlays of F338H, R386L, P346L, A223G, S237A, and S279A mutants, respectively, with WT CYP121.
In contrast, the A233G and S237A mutants have little effect on the heme conformation but significantly alter the environment of the heme sixth ligand (Fig. 2, D and E). Ala233 is conserved as Ala or Gly in virtually all P450s, and A233G CYP121 was generated in view of steric constraints to azole drug binding noted in previous CYP121 structural studies (17, 18). However, affinity for azoles was not substantially altered from WT CYP121 (see below and Table S3). For the A233G mutant structure, a new water molecule is present in the space formerly occupied by the Ala233 side chain. The pitch of the I helix segment of the protein (in which Ala233 is located) is not significantly altered in the A233G mutant. The S237A mutant structure reveals an altered position of the sixth ligand itself, which is now in close contact with the Ala233 carboxyl backbone. This increases the apparent heme iron-to-distal water distance considerably from 2.15 to ∼3.1 Å.
Finally, the S279A mutation leads to a series of modest changes that occur along residues previously postulated to be part of a proton transfer pathway. The conformations of Asn277, Ser240, and Thr244 are changed, in addition to an altered water molecule network. This confirms that Ser279 plays a pivotal role in the local conformation of these hydrogen bonding networks and that it could serve to relay protons from the solvent to the P450 active site (Fig. 2F).
The atomic structure of WT CYP121 revealed that heme was bound in two orientations related by a 180° rotation about an axis of symmetry across the CHα-Fe-CHδ atoms of the molecule (18). It is thus rather difficult to define heme substituent group changes in the CYP121 mutant crystal structures due to this heme heterogeneity and in view of the facts that mutant structures are at different levels of resolution (1.08–1.90 Å) and that photoreduction of heme iron may occur during data collection. However, ligand/substrate effects on heme substituent conformations have been reported previously for certain P450 enzymes (e.g. see Ref. 43). At least in the case of the P346L CYP121 mutant, substantial perturbations of the propionate group attached to the distorted pyrrole ring are observed by comparison with WT CYP121 and are consistent with RR data (Fig. 2C).
UV-visible Spectroscopic Analysis of CYP121 Variants and Their Interactions with Azole Drugs
UV-visible absorption spectra were collected for WT and all CYP121 variants. In their oxidized forms, the S279A, F338H, and P346L mutants had spectra almost identical to WT CYP121, with the heme Soret maximum at 416.5 nm. However, differences were noted for the R386L variant (Soret at 418 nm) and for the A233G and S237A mutants. As shown in Fig. 3, the optical spectrum for the R386L mutant has a less prominent shoulder (at ∼393 nm) on the Soret peak. Previous studies of WT CYP121 indicated that there was a minor proportion of high spin heme iron present at ambient temperature (22). The data for the R386L mutant are consistent with its near complete conversion to the low spin form. The latter two mutants both bound heme iron in a mixture of low spin and high spin forms (Fig. 3). The A233G mutant has a Soret maximum at 413 nm and a large high spin feature at ∼396 nm, and the S237A mutant has comparable features at 414 and 396 nm. The A233G mutant has slightly greater high spin content compared with the S237A mutant. Evidently, there are perturbations in heme environment in the A233G and S237A mutants that disrupt the axial coordination by water. These findings are consistent with structural data for these mutants and suggest that the additional water molecule in the vicinity of the heme iron observed in the A233G mutant is actually an alternative position for the sixth aqua ligand and that the A233G distal water is in an equilibrium between these positions, explaining the shift toward high spin. A similar aqua ligand switch was suggested for WT P450 BM3 in complex with its substrate N-palmitoylglycine (44).
FIGURE 3.

UV-visible spectroscopic properties of selected CYP121 mutants. The electronic absorption spectra for the purified, ferric forms of CYP121 mutants R386L (solid line), S279A (dashed line), and A233G (dotted line) are shown. The R386L mutant is almost completely low spin with a Soret maximum at 418 nm. The S279A mutant has WT-like spectral properties with a small proportion of high spin heme iron and a Soret maximum at 416.5 nm. The A233G mutant has a large component of high spin heme iron, and the Soret band is split between low spin and high spin components, with Soret absorption maximal at 413 nm. CYP121 mutant enzyme concentration was 9 μm in each case.
In light of (i) our previous work showing very tight binding of azole drugs to WT CYP121 and the effectiveness of these agents against M. smegmatis (11, 22) and (ii) work presented here that highlights the potency of these compounds against Mtb H37Rv itself (i.e. MIC values), we evaluated the binding of a series of azole and phenylimidazole drugs to CYP121 and its mutants by optical titration. The Kd values for binding of clotrimazole, econazole, fluconazole, miconazole, voriconazole, ketoconazole, 2-PIM, and 4-PIM to WT and CYP121 mutants are detailed in Table S3. In each case, the Soret maximum for the azole complex is at ∼423 nm. Imidazole binding was very weak (Kd >50 mm) and more than 6 orders of magnitude greater than the econazole Kd for WT and most mutants.
The Kd values determined reveal that mutations do not substantially affect binding of the azoles in almost all cases. This is consistent with the lack of any gross disruption of the active site structure in the mutants revealed from crystallographic studies (Fig. 2). The data show that econazole/clotrimazole bind extremely tightly (Kd values of <0.2 μm for WT CYP121), followed in rank order by miconazole (<0.5 μm) and then ketoconazole (3.44 μm). Thus, the order of affinity of these azoles for CYP121 follows the rank order of MIC values against Mtb H37Rv. Fluconazole and voriconazole bind more weakly (Kd values of 8.61 and 16.3 μm for WT CYP121), and the phenylimidazoles bind more weakly still (Kd values of 101.8 and 32.3 μm for WT CYP121 with 2-PIM and 4-PIM, respectively). Exemplary spectral data for the binding of econazole to the S237A CYP121 mutant are shown in Fig. 4A, along with the relevant fit for these data (Kd <0.2 μm) and for the binding of ketoconazole to the same mutant (7.66 μm) (Fig. 4B).
FIGURE 4.

Analysis of azole antifungal binding to the CYP121 S237A mutant. A, absorption spectra are shown for CYP121 S237A (3.6 μm) in the absence of ligand (thin solid line) and following the addition of 0.21, 0.41, 1.02, 1.22, 1.84, and 2.45 μm econazole (dotted lines). The spectrum shown as a thick solid line is at near saturation with econazole (3.68 μm) and has its Soret maximum at 423 nm. Inset, difference spectra generated by subtraction of the spectrum for the ligand-free S237A mutant from those spectra collected at the various stages of the econazole titration. Maximum and minimum absorption values are at 426 and 393 nm, respectively. B, overlaid data fits for the binding of econazole (open circles) and ketoconazole (open triangles) to CYP121 S237A. Data fitting was done using Equation 1 as described under “Experimental Procedures.” The Kd values for econazole and ketoconazole were 0.043 and 7.66 μm, respectively.
Mutant CYP121 proteins displayed UV-visible spectra with Soret maxima similar to WT for ferric adducts with cyanide (438 nm) and nitric oxide (437 nm). Data for ferrous-CO adducts are described under “Reconstruction of a Mtb CYP121 Redox System.”
Determination of Heme Iron Reduction Potentials in CYP121 Active Site Mutants
Previous studies of WT CYP121 indicated a rather negative reduction potential for the heme iron (more negative than -400 mV versus standard hydrogen electrode). To establish influence on heme potential of (i) disruption of the aforementioned hydrogen-bonding network linking the distal water molecule to active site residues and (ii) active site structure and heme distortion, we determined heme iron potential by spectroelectrochemical titration for mutants on both the distal face (R386L, S237A, S279A, and A233G) and the proximal side (P346L and F338H) of the heme. For P346L, we sought to probe influence of heme pyrrole distortion on heme iron potential. For F338H, previous studies on P450 BM3 established that the corresponding, phylogenetically conserved, phenylalanine residue (Phe393) is located in a similar position with respect to the iron-Cys linkage and that mutations at this position have a profound influence on BM3 heme iron reduction potential and reactivity of the reduced enzyme with dioxygen (33).
Redox potentials of CYP121 mutants were not substantially altered in most cases. Under anaerobic conditions and with sodium dithionite as reductant, WT and most mutants were reduced to an extent of ∼70–80% without requirement for the additions of substantial excesses of reductant that otherwise affected absorption spectra and promoted enzyme aggregation. The extent of reduction achieved was sufficient to enable accurate fitting of data and to obtain good estimates of heme iron redox potential. Values determined for WT and variant CYP121 enzymes were as follows: WT (-467 ± 5 mV versus standard hydrogen electrode), A233G (-420 ± 6 mV), F338H (-469 ± 6 mV), P346L (-458 ± 7 mV), S237A (-430 ± 8 mV), and S279A (-479 ± 6 mV). These data indicate that there are small increases in heme iron reduction potential in the A233G and S237A mutants, consistent with the partial conversions of these enzymes toward the high spin form, with the increased distance of the distal water ligand from the heme iron in S237A, and with previous data on, for example, the P450 BM3 and P450cam enzymes (33, 45, 46).
There is no major change in heme potential of the P346L mutant by comparison with WT CYP121, indicating that distortion of the heme pyrrole D ring does not have a major effect in regulating CYP121 heme iron potential (Fig. 2C). The P346L heme iron is reduced by dithionite to a slightly greater extent (∼5%) than is the WT under similar conditions, confirming that the small positive shift in heme iron potential determined for P346L is valid. In addition, the P346L redox spectra are noticeably different from those for WT or other CYP121 mutants, in that the predominantly reduced form has a more asymmetric Soret feature with maximal absorption at ∼420 nm (compared with ∼407 nm for the other proteins) and a visible region peak at 559 nm (Fig. S5). This probably indicates that reduction of the P346L heme iron to the ferrous state results in protonation of the heme thiolate to thiol in a proportion of the molecules, consistent both with the position of the mutation (directly adjacent to the fifth heme ligand) and with previous conclusions from our study of Mtb CYP51B1 (27, 47).
Similarly, the F338H mutation has a negligible effect on heme potential, in
contrast to the positive shift in heme potential (∼95 mV) previously
observed in a F393H mutant of P450 BM3
(46,
48). Also, our studies of the
reduced CYP121 F338H mutant indicated that it did not stabilize a
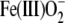 species,
whereas stabilization of this species was seen for the BM3 F393H mutant
(47). In the CYP121 F338H
structure, the side chain of His338 was in almost exactly the same
position as the WT Phe338 side chain, and no interaction between
His338 and the proximal cysteinate was evident. In previous work,
Schelvis and co-workers (43)
reported that conformational orientations of heme vinyl groups (which became
more coplanar with the P450 BM3 heme on iron reduction) and the dependence of
these alterations on the nature of the side chain at position 393 could be
major determinants regulating heme iron reduction potential in this system.
However, RR spectra for the CYP121 F338H enzyme (see supplemental material)
suggest that heme vinyls may already be more coplanar with the heme than in
WT. Thus, these data indicate that there may not be an implicit role for the
conserved phenylalanine in controlling heme thermodynamics and oxygen
reactivity across the P450 enzyme superfamily.
species,
whereas stabilization of this species was seen for the BM3 F393H mutant
(47). In the CYP121 F338H
structure, the side chain of His338 was in almost exactly the same
position as the WT Phe338 side chain, and no interaction between
His338 and the proximal cysteinate was evident. In previous work,
Schelvis and co-workers (43)
reported that conformational orientations of heme vinyl groups (which became
more coplanar with the P450 BM3 heme on iron reduction) and the dependence of
these alterations on the nature of the side chain at position 393 could be
major determinants regulating heme iron reduction potential in this system.
However, RR spectra for the CYP121 F338H enzyme (see supplemental material)
suggest that heme vinyls may already be more coplanar with the heme than in
WT. Thus, these data indicate that there may not be an implicit role for the
conserved phenylalanine in controlling heme thermodynamics and oxygen
reactivity across the P450 enzyme superfamily.
However, a major effect was observed for the R386L mutant, in which heme iron potential (-189 ± 5 mV) was substantially more positive than for WT and other mutants. Despite the absence of major structural change, the R386L redox potential was increased by ∼280 mV. As far as we are aware, this is an unprecedented increase in heme potential effected by mutation in any P450, and the magnitude is also much greater than that induced by conversion of P450s to the high spin state on type I substrate binding (e.g. see Refs. 33, 35, and 45).
Spectral data accompanying the R386L redox titration and the relevant data fit to the Nernst function are shown in Fig. 5, A and B, respectively. The absorption versus potential data fit for P346L is compared with that for R386L CYP121 to emphasize the substantial change in heme iron redox potential induced in the latter (Fig. 5B). The UV-visible spectrum for ferrous R386L CYP121 shows a broad absorption band with Soret maximum close to 407 nm (Fig. 5A). A small feature at ∼559 nm also suggests a small proportion of thiol-coordinated ferrous iron in R386L (but much less than in P346L; see Fig. S5). The R386L structure indicated only marginal conformational perturbations, mainly limited to small changes in positions of active site waters and with no significant alteration of the heme aqua ligand detectable (Fig. 2B). The ferric R386L mutant was almost completely low spin from optical spectra. The large R386L redox potential change indicates the critical nature of Arg386 in CYP121 thermodynamic regulation and suggests its displacement (or shielding of its charge) on substrate binding is a major determinant in triggering CYP121 activity by thermodynamically favoring electron transport from Mtb redox partners (likely to be a class I, ferredoxin reductase/ferredoxin system) (6).
FIGURE 5.

Redox potential analysis of CYP121 mutants R386L and P346L. A, selected spectra from the potentiometric titration of CYP121 R386L mutant (9.3 μm) are shown. The most intense spectrum (solid line; Soret at 418 nm) is that for the oxidized (ferric) enzyme. Successive spectra shown as dotted lines were taken at regular intervals during the redox titration. The spectrum for the reduced (ferrous) enzyme is also shown as a solid line, exhibiting a broad Soret absorption band centered at ∼407 nm. The arrows indicate the direction of absorption change occurring during the reductive phase of the titration in regions of the spectrum at which major absorption changes occur. B, overlaid fits of absorption versus applied potential for the R386L mutant (open triangles) and for the P346L mutant (closed circles). Data were fitted using the Nernst function, as described under “Experimental Procedures” and in the supplemental material. Midpoint reduction potential values were -189 ± 5 mV and -458 ± 7 mV(versus standard hydrogen electrode), respectively.
Spectroscopic Studies of CYP121 Mutants
Resonance Raman—RR spectroscopy is a sensitive method for detecting heme iron spin, oxidation, and coordination state and also for analyzing heme geometry and substituent group conformations (49, 50). RR was used to examine each CYP121 mutant (in ligand-free and fluconazole-bound forms) to probe for conformational and/or electronic variations from the WT enzyme. The ν4 band is the major feature in all of the spectra and is an important heme iron oxidation state marker. In both ligand-free and fluconazole-bound CYP121 proteins, this feature is consistently at 1372 ± 1 cm-1, consistent with a ferric oxidation state and with previous data on ferric P450s (51–53). In most cases, RR shows that the ligand-free CYP121 mutants are predominantly low spin in their resting state. The ν3 marker band, which is highly diagnostic for heme iron spin state, has bands at ∼1488 cm-1 for high spin and at ∼1501 cm-1 for low spin ferric heme iron for CYP121. For the majority of the CYP121 mutants, the dominant ν3 species is located at 1501 ± 1 cm-1, with a minor species at 1488 ± 2 cm-1. Exceptions are A233G and S237A, where the intensity of the high spin ν3 feature is greater (40–45% of overall ν3 peak intensity), and R386L, where there is negligible spectral contribution from the high spin ν3 species (Fig. S6). These data are thus consistent with predictions based on optical spectroscopy, and confirm heme iron spin state modulation in these three mutants.
In the fluconazole complexes of WT and mutant CYP121 proteins, the major difference compared with ligand-free forms is the shift of the ν3 bands to a single (low spin) species at 1500 ± 1 cm-1. Positions of the ν2 and ν4 bands in the fluconazole complexes are also consistent with all CYP121 proteins being in a 6-coordinate, mainly low spin ferric state (49).
RR spectral assignments and comparisons for ligand-free and fluconazole-bound forms of WT/mutant CYP121 P450s are presented in Tables S4 and S5. Perturbations to heme substituent group conformations in ligand-free and fluconazole-bound forms of CYP121 mutants were also detected by RR and are discussed in more detail in the supplemental material.
EPR—EPR spectra were recorded for oxidized WT and mutant CYP121 P450s, as described under “Experimental Procedures.” EPR g-values in all cases were similar to those for WT CYP121 (gx = 2.47, gy = 2.25, and gz = 1.90) and indicative of a predominantly low spin, ferric, cysteinate-coordinated heme iron (see Table S6). Thus, the proportion of high spin heme iron observed for the A233G and S237A mutants by both electronic absorption and RR spectroscopy was not detected at 10 K. This is consistent with the high spin heme iron populations in these mutants being frozen down to the low spin state at 10 K.
Some minor variations in the line width of the outer gz and gx g-tensor elements of the rhombic trio are noted between WT and mutant CYP121 P450s (see Fig. S7). The g-values are very sensitive to the nature of the heme axial ligands and to perturbations such as ligand orientations. These line width variations may indicate slight changes in the flexibility of the axial linkages to the heme iron (as might be expected for mutations in the direct environment of the heme ligands, such as P346L and F338H). Otherwise, the EPR data report that WT and all CYP121 mutants are isolated in their native (thiolate-coordinated) forms.
Reconstitution of a Mtb CYP121 Redox System
To examine whether mutations gave rise to structural or other effects that perturb interactions with a Mtb redox partner, we examined Fe(II)CO complex formation in WT/mutant CYP121 enzymes, with electron transfer to the P450s driven by NADPH via a Mtb class I redox system (the NAD(P)H-dependent reductase FprA and the 3Fe-4S ferredoxin Fdx) (27, 54, 14), as described under “Experimental Procedures.” The rate of Fe(II)CO complex formation was determined by fitting absorption change data at 450 nm (i.e. the peak for the Fe(II)CO complex) using an exponential function. Fig. 6 shows spectral changes accompanying the development of the Fe(II)CO complex for the CYP121 F338H mutant. Fig. 6 (inset) shows a plot of absorption change (450 nm) versus time overlaid for the R386L and F338H mutants.
FIGURE 6.

Reconstitution of a Mtb redox system and electron transfer to CYP121. A Mtb redox system of FprA (4 μm), Fdx (18 μm), and WT/mutant CYP121 (4 μm) was set up in CO-saturated buffer (see “Experimental Procedures”). CYP121 complex formation was initiated by NADPH addition (300 μm). Shown is spectral accumulation of the Fe(II)CO form of the F338H CYP121 mutant over time. The initial spectrum (thick line) is prior to NADPH addition and has contributions from oxidized FprA/Fdx proteins. The dashed line spectrum is after NADPH addition and shows bleaching of reductase proteins. Later spectra (dotted lines) were collected at 2, 5, 10, 15, 25, and 35 min. The final spectrum (thin solid line) at 50 min shows a predominantly thiolate-coordinated Fe(II)CO enzyme with Soret at 448 nm. The inset shows a plot of A448 (percentage of P450 formed) versus time, with data fitted using an exponential function for the F338H (open triangles) and R386L (open circles) CYP121 mutants. Rate constants determined for Fe(II)CO complex formation were 0.060 ± 0.001 and 0.089 ± 0.002 min-1, respectively.
The data demonstrate successful reconstitution of a class I P450 redox chain composed entirely of Mtb H37Rv proteins. The Fdx ferredoxin gene (Rv0763c) is adjacent to that for the CYP51B1 P450 (Rv0764c) on the Mtb H37Rv genome, and Fdx was also shown to be a viable redox partner for CYP51B1 (27). Electron transfer to CYP121 is relatively slow (in the absence of an oxidizable substrate), with a rate of Fe(II)CO complex formation of 0.066 ± 0.002 min-1 for WT CYP121. The most notable variations were observed for R386L (0.089 ± 0.002 min-1) and for S237A (0.076 ± 0.003 min-1). Other mutants exhibited rates that were similar to WT CYP121: A233G (0.069 ± 0.003 min-1), F338H (0.060 ± 0.001 min-1), P346L (0.057 ± 0.001 min-1), and S279A (0.064 ± 0.003 min-1). Thus, the more positive redox potential for the R386L mutant appears to be a dominant factor in enhancing electron transfer rate in this system, whereas the P346L mutation in the vicinity of the proximal cysteinate ligand has retarded the rate somewhat, possibly as a consequence of disruption to the Fdx binding site and/or the electron transfer pathway to the heme iron. Although there is a modest increase in the rate for the S237A mutant (which has an increased proportion of high spin heme iron in the ferric form), the A233G mutant (with a similar proportion of high spin heme iron) has a rate identical within error to WT CYP121.
In previous studies, we showed that an exquisite pH dependence of the P450/P420 equilibrium exists in CYP121 and demonstrated that thiolate protonation occurs readily in the Fe(II)CO complex for this enzyme (1). However, thiolate protonation occurs instead in the Fe(II) ligand-free form for Mtb CYP51B1, such that CO binding merely provides a convenient spectral signature for an event that occurs at the preceding reduction step (27). In addition, we have previously demonstrated that CO binding to ferrous CYP121 occurs at ∼103 s-1 under our conditions and clearly does not influence interprotein electron transfer rates observed here (1). As is evident from the spectral data shown in Fig. 6, the P450 (thiolate-coordinated) form of the Fe(II)CO complex dominates over the P420 form for the F338H mutant and also for WT CYP121 and all other mutants. Once the Fe(II)CO complex is formed, the P450/P420 ratio remains constant. The relative quantity of the CYP121 P450 species formed using the homologous redox partner system is consistently greater than that achieved using the reductant dithionite as a surrogate for the class I system (and despite ensuring that pH conditions were identical), indicating that the P450/P420 equilibrium can be stabilized in favor of the thiolate-coordinated form through the use of native-like redox partners. This fact that the homologous redox system is superior in maintaining the thiolate-coordinated state of the ferrous CYP121 heme iron also highlights potential problems with using artificial reductants for generation of P450 Fe(II)CO complexes.
Conclusions—Key conclusions from this study are the demonstration that CYP121 is an essential gene for viability of Mtb H37Rv, the validation of production of CYP121 protein by Mtb, and the determination of MIC values for a variety of azole-based P450 inhibitors, whose values correlate well with the respective Kd values for their binding to CYP121 protein. Structural studies of CYP121 highlighted several residues predicted to be important determinants of heme geometry, thermodynamic properties, active site structure, and hydrogen bonding networks possibly relevant to proton relay pathways to heme iron. Six different mutants at key positions around the heme site were generated, and the proteins were isolated and structurally and biophysically characterized. Structural data revealed a robust active site architecture, and mutations on both proximal and distal faces of the heme have only small effects on overall structure, confined to the immediate vicinity of the mutations. A role was revealed for Arg386 in thermodynamic regulation, with an unprecedented ∼280-mV increase in P450 heme potential in the R386L mutant. A class I P450 electron transfer system comprising FprA, Fdx, and CYP121 P450s was reconstituted. An enhanced electron transfer rate was observed to the CYP121 R386L mutant, consistent with its more positive heme potential. The residue at position Pro346 was shown to control conformation of heme pyrrole D. However, changes to heme distortion had little effect on P346L heme iron potential, although EPR indicated a “looser” proximal ligand coordination environment, consistent with perturbing the residue next to the iron proximal ligand. EPR studies also showed retention of cysteinate coordination in all mutants. An alternative position for the heme distal water was seen for the A233G mutant, consistent with changes in heme iron spin-state equilibrium established spectroscopically.
In summary, our studies provide detailed analysis of active site structure in CYP121, a P450 that we demonstrate to be essential for Mtb viability and to bind potent anti-Mtb azole drugs with high affinity. Ongoing studies are directed toward establishing CYP121 substrate selectivity and biological function, with preliminary data pointing to interactions with complex mycobacterial lipids.
Supplementary Material
This work was supported by United Kingdom Biotechnology and Biological Sciences Research Council Grants BBS/B/06288/2 and C19757/2 and by European Union FP6 Project NM4TB. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S6 and Figs. S1–S7.
Footnotes
The abbreviations used are: Mtb, M. tuberculosis; X-gal, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; WT, wild type; PIM, phenylimidazole; RR, resonance Raman.
K. J. McLean, P. Carroll, D. G. Lewis, A. J. Dunford, H. E. Seward, R. Neeli, M. R. Cheesman, L. Marsollier, P. Douglas, W. E. Smith, I. Rosenkrands, S. T. Cole, D. Leys, T. Parish, M. Jackson, and A. W. Munro, unpublished results.
References
- 1.Dunford, A. J., McLean, K. J., Sabri, M., Seward, H. E., Heyes, D. J., Scrutton, N. S., and Munro, A. W. (2007) J. Biol. Chem. 282 24816-24824 [DOI] [PubMed] [Google Scholar]
- 2.Matsumoto, M., Hashizume, H., Tsubouchi, H., Sasaki, H., Itotani, M., Kuroda, H., Tomishige, T., Kawasaki, M., and Komatsu M. (2007) Curr. Top. Med. Chem. 7 499-507 [DOI] [PubMed] [Google Scholar]
- 3.Spigelman, M. K. (2007) J. Infect. Dis. 196 S28-S34 [DOI] [PubMed] [Google Scholar]
- 4.Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry, C. E., III, Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M. A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S., and Barrell, B. G. (1998) Nature 393 537-544 [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann, R. D., Alland, D., Eisen, J. A., Carpenter, L., White, O., Peterson, J., DeBoy, R., Dodson, R., Gwinn, M., Haft, D., Hickey, E., Kolonay, J. F., Nelson, W. C., Umayam, L. A., Ermolaeva, M., Salzberg, S. L., Delcher, A., Utterback, T., Weidman, J., Khouri, H., Gill, J., Mikula, A., Bishai, W., Jacobs, Jr., W. R., Venter, J. C., and Fraser, C. M. (2002) J. Bacteriol. 184 5479-5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLean, K. J., Clift, D., Lewis, D. G., Sabri, M., Balding, P. R., Sutcliffe, M. J., Leys, D., and Munro, A. W. (2006) Trends Microbiol. 14 220-228 [DOI] [PubMed] [Google Scholar]
- 7.Aoyama, Y., Horiuchi, T., Gotoh, O., Noshiro, M., and Yoshida, Y. (1998) J. Biochem. (Tokyo) 124 694-696 [DOI] [PubMed] [Google Scholar]
- 8.Odds, F. C., Brown, A. J., and Brown, A. J. (2003) Trends Microbiol. 11 272-279 [DOI] [PubMed] [Google Scholar]
- 9.Guardiola-Diaz, H. M., Foster, L. A., Mushrush, D., and Vaz, A. D. (2001) Biochem. Pharmacol. 61 1463-1470 [DOI] [PubMed] [Google Scholar]
- 10.Jackson, C. J., Lamb, D. C., Kelly, D. E., and Kelly, S. L. (2000) FEMS Microbiol. Lett. 192 159-162 [DOI] [PubMed] [Google Scholar]
- 11.McLean, K. J., Marshall, K. R., Richmond, A., Hunter, I. S., Fowler, K., Kieser, T., Gurcha, S. S., Besra, G. S., and Munro, A. W. (2002) Microbiology 148 2937-2949 [DOI] [PubMed] [Google Scholar]
- 12.Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003) Mol. Microbiol. 48 77-84 [DOI] [PubMed] [Google Scholar]
- 13.Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 12712-12717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer, F., Raimondi, D., Aliverti, A., and Zanetti, G. (2002) Eur. J. Biochem. 269 3005-3013 [DOI] [PubMed] [Google Scholar]
- 15.Podust, L. M., Poulos, T. L., and Waterman, M. R. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 3068-3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Podust, L. M., Yermalitskaya, L. V., Lepesheva, G. I., Podust, V. N., Dalmasso, E. A., and Waterman, M. R. (2004) Structure 12 1937-1945 [DOI] [PubMed] [Google Scholar]
- 17.Seward, H. E., Roujeinikova, A., McLean, K. J., Munro, A. W., and Leys, D. (2006) J. Biol. Chem. 281 39437-39443 [DOI] [PubMed] [Google Scholar]
- 18.Leys, D., Mowat, C. G., McLean, K. J., Richmond, A., Chapman, S. K., Walkinshaw, M. D., and Munro, A. W. (2003) J. Biol. Chem. 278 5141-5147 [DOI] [PubMed] [Google Scholar]
- 19.Poulos, T. L., and Howard, A. J. (1987) Biochemistry 26 8165-8174 [DOI] [PubMed] [Google Scholar]
- 20.Ouellet, H., Podust, L. M., and Ortiz de Montellano, P. R. (2008) J. Biol. Chem. 283 5069-5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad, Z., Sharma, S., Khuller, G. K., Singh, P., Faujdar, J., and Katoch, V. M. (2006) Int. J. Antimicrob. Agents 28 543-544 [DOI] [PubMed] [Google Scholar]
- 22.McLean, K. J., Cheesman, M. R., Rivers, S. L., Richmond, A., Leys, D., Chapman, S. K., Reid, G. A., Price, N. C., Kelly, S. M., Clarkson, J., Smith, W. E., and Munro, A. W. (2002) J. Inorg. Biochem. 91 527-541 [DOI] [PubMed] [Google Scholar]
- 23.Lamichhane, G., Zignol, M., Blades, N. J., Geiman, D. E., Dougherty, A., Grosset, J., Broman, K. W., and Bishai, W. R. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 7213-7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parish, T., and Stoker, N. (2000) Microbiology 146 1969-1975 [DOI] [PubMed] [Google Scholar]
- 25.Parish, T., and Stoker, N. (2000) J. Bacteriol. 182 5715-5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenkrands, I., Aagaard, C., Weldingh, K., Brock, I., Dziegiel, M. H., Singh, M., Hoff, S., Ravn, P., and Andersen, P. (2008) Tuberculosis 88 335-343 [DOI] [PubMed] [Google Scholar]
- 27.Mclean, K. J., Warman, A. J., Seward, H. E., Marshall, K. R., Girvan, H. M., Cheesman, M. R., Waterman, M. R., and Munro, A. W. (2006) Biochemistry 45 8427-8443 [DOI] [PubMed] [Google Scholar]
- 28.Leslie, A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, No. 26
- 29.Evans, P. R. (2005) Acta Crystallogr. Sect. D 62 72-82 [DOI] [PubMed] [Google Scholar]
- 30.Dodson, E. J. (1997) Acta Crystallogr. Sect. D 53 240-255 [DOI] [PubMed] [Google Scholar]
- 31.Morris, R. J., Perrakis, A., and Lamzin, V. S. (2003) Methods Enzymol. 374 229-244 [DOI] [PubMed] [Google Scholar]
- 32.Beitlich, T., Kühnel, K., Schulze-Briese, C., Shoeman, R. L., and Schlichting, I. (2007) J. Synchrotron Radiat. 14 11-23 [DOI] [PubMed] [Google Scholar]
- 33.Daff, S. N., Chapman, S. K., Turner, K. L., Holt, R. A., Govindaraj, S., Poulos, T. L., and Munro, A. W. (1997) Biochemistry 36 13816-13821 [DOI] [PubMed] [Google Scholar]
- 34.Dutton, P. L. (1978) Methods Enzymol. 54 411-435 [DOI] [PubMed] [Google Scholar]
- 35.Lawson, R. J., Leys, D., Sutcliffe, M. J., Kemp, C. A., Cheesman, M. R., Smith, S. J., Clarkson, J., Smith, W. E., Haq, I., Perkins, J. B., and Munro, A. W. (2004) Biochemistry 43 12410-12426 [DOI] [PubMed] [Google Scholar]
- 36.Tsolaki, A. G., Hirsh, A. E., DeRiemer, K., Enciso, J. A., Wong, M. Z., Hannan, M., Goguet de la Salmoniere, O. L., Aman, K., Kato-Maeda, M., and Small, P. M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4865-4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao, Q., Kripke, K. E., Saldanha, A. J., Yan, W., Holmes, S., and Small, P. M. (2005) Microbiology 151 5-14 [DOI] [PubMed] [Google Scholar]
- 38.Saint-Joanis, B., Demangel, C., Jackson, M., Brodin, P., Marsollier, L., Boshoff, H., and Cole, S. T. (2006) J. Bacteriol. 188 6669-6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byrne, S. T., Denkin, S. M., Gu, P., Nuermberger, E., and Zhang, Y. (2007) J. Med. Microbiol. 56 1047-1051 [DOI] [PubMed] [Google Scholar]
- 40.Burguière, A., Hitchen, P. G., Dover, L. G., Dell, A., and Besra, G. S. (2005) Microbiology 151 2087-2095 [DOI] [PubMed] [Google Scholar]
- 41.Ahmad, Z., Sharma, S., and Khuller, G. K. (2006) FEMS Microbiol. Lett. 261 181-186 [DOI] [PubMed] [Google Scholar]
- 42.Ahmad, Z., Sharma, S., and Khuller, G. K. (2005) FEMS Microbiol. Lett. 251 19-22 [DOI] [PubMed] [Google Scholar]
- 43.Chen, Z., Ost, T. W., and Schelvis, J. P. (2004) Biochemistry 43 1798-1808 [DOI] [PubMed] [Google Scholar]
- 44.Haines, D. C., Tomchick, D. R., Machius, M., and Peterson, J. A. (2001) Biochemistry 40 13456-13465 [DOI] [PubMed] [Google Scholar]
- 45.Sligar, S. G., and Gunsalus, I. C. (1976) Proc. Natl. Acad. Sci. U. S. A. 73 1078-1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munro, A. W., Leys, D. G., McLean, K. J., Marshall, K. R., Ost, T. W., Daff, S., Miles, C. S., Chapman, S. K., Lysek, D. A., Moser, C. C., Page, C. C., and Dutton, P. L. (2002) Trends Biochem. Sci. 27 250-257 [DOI] [PubMed] [Google Scholar]
- 47.Perera, R., Sono, M., Sigman, J. A., Pfister, T. D., Lu, Y., and Dawson, J. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 3641-3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ost, T. W., Miles, C. S., Munro, A. W., Murdoch, J., Reid, G. A., and Chapman, S. K. (2001) Biochemistry 40 13421-13429 [DOI] [PubMed] [Google Scholar]
- 49.Hildebrandt, P., Greinert, R., Stier, A., and Taniguchi, H. (1989) Eur. J. Biochem. 186 291-302 [DOI] [PubMed] [Google Scholar]
- 50.Hu, S., Smith, K. M., and Spiro, T. G. (1996) J. Am. Chem. Soc. 118 12638-12646 [Google Scholar]
- 51.Miles. J. S., Munro, A. W., Rospendowski, B. N., Smith, W. E., McKnight, J. E., and Thomson, A. J. (1992) Biochem. J. 288 503-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith S. J., Munro, A. W., and Smith, W. E. (2003) Biopolymers 70 620-627 [DOI] [PubMed] [Google Scholar]
- 53.Matsuura, K., Yoshioka, S., Tosha, T., Hori, H., Ishimori, K., Kitagawa, T., Morishima, I., Kagawa, N., and Waterman, M. R. (2005) J. Biol. Chem. 280 9088-9096 [DOI] [PubMed] [Google Scholar]
- 54.McLean, K. J., Scrutton, N. S., and Munro, A. W. (2003) Biochem. J. 372 317-327 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.