

Abstract
The position of the a subunit of the membrane-integral F0 sector of Escherichia coli ATP synthase was investigated by single molecule fluorescence resonance energy transfer studies utilizing a fusion of enhanced green fluorescent protein to the C terminus of the a subunit and fluorescent labels attached to specific positions of the ε or γ subunits. Three fluorescence resonance energy transfer levels were observed during rotation driven by ATP hydrolysis corresponding to the three resting positions of the rotor subunits, γ or ε, relative to the a subunit of the stator. Comparison of these positions of the rotor sites with those previously determined relative to the b subunit dimer indicates the position of a as adjacent to the b dimer on its counterclockwise side when the enzyme is viewed from the cytoplasm. This relationship provides stability to the membrane interface between a and b2, allowing it to withstand the torque imparted by the rotor during ATP synthesis as well as ATP hydrolysis.
F0F1-ATP synthases are the membrane-embedded rotary enzymes in mitochondria, chloroplasts, and bacteria that provide ATP through oxidative and photophosphorylation (1). In these enzymes, ATP synthesis from ADP and phosphate is driven by the flow of ions, usually H+, down an electrochemical potential difference across the plasma membrane (2). Ion flow through the membrane-integral F0 sector drives the rotation of the turbine-like ring of c subunits. Extensive analysis of the accessibility of sites within the adjacent a subunit supports the model that this subunit provides two half-channels allowing the proton to access the H+ binding site on the c subunit from either side of the membrane (3). Rotation of the c ring by one subunit relative to a is required for net ion translocation by the system. The γ and ε subunits of the membrane-peripheral F1 sector interact with the c ring and turn with it. Rotation of these subunits relative to the catalytic sites housed in the three αβ pairs drive conformational changes that are linked to the binding of substrates as well as the synthesis and release of ATP.
The overall structure of the Escherichia coli F0F1-ATP synthase has been visualized by electron microscopy (4). Single particle analysis and three-dimensional image reconstruction reveal that in addition to the central γε stalk connecting F1 and F0 a second peripheral stalk links the two sectors. A number of lines of evidence show this peripheral stalk to be composed to two copies of the highly extended b subunit that interact with the single δ subunit near the top of the α3β3 hexamer. Because the function of the peripheral stalk is to hold the a subunit and α3β3 stationary to one another while the γεc10 rotor turns, it is sometimes called the stator stalk. Although the details of interaction of b2δ with α3β3 and with the a subunit are unknown, their arrangement and strength must be adequate to withstand the torque imparted by the turning rotor.
Limited high resolution structural information is available for the F0 part of the enzyme. The crystal structure of the c ring from a Na+-driven enzyme of Ilyobacter tartaricus has been solved at 2.4-Å resolution (5). Additional information for the N-terminal parts of the b subunits connecting the membrane portion with the F1 headpiece was obtained by NMR (6), and the dimerization domain of the b2 subunits has been crystallized (7). A crystal structure of much of the stator stalk of bovine mitochondrial ATPase has been reported (8), but this assembly is of a much different design, containing single copies of four different subunits. Structural details of subunit a are unavailable for any system, and the relative position of the a subunit in the membrane with respect to the b subunit remains unclear (see Fig. 1). In electron microscopic images, the hydrophobic membrane-embedded a subunit is covered by detergent molecules. Cross-linking experiments show the a subunit to be in contact with at least one of the b subunits, but the arrangement is uncertain. Mutational analyses imply that b2 makes contact with both the a and the c subunits (9, 10).
FIGURE 1.

Model of F0F1-ATP synthase. Rotating subunits γ, ε, and c10 are colored in black. The static subunits of F1, α3β3δ, are shown in gray. The b subunit dimer is shown as semitransparent gray double tubes, and the possible positions for the membrane-embedded subunit a are shown as a light gray boxes positioned according to cross-linking and stable subcomplex data, that is attached either to the left (gray box labeled A) of the b subunits, between the two b subunits (position B) and the ring of c subunits, or to the right (position C) of b2.
Here to determine the relative positions of the a and b2 subunits and to discriminate between a symmetric arrangement between the two b subunits (indicated as position B in Fig. 1) or an asymmetric position, we used a triangulation approach using a set of intersubunit distance measurements derived from single molecule analyses of ATP synthase. Previously we established the relationship of the b subunit dimer to the positions that can be occupied by ε using a similar method (11). We use Förster-type fluorescence resonance energy transfer (FRET)3 as an appropriate method for distance measurements in the range of 2-8 nm (12, 13). It is based on the distance-dependent, non-radiative energy transfer between two fluorophores. Bulk FRET analysis has been used successfully to predict the subunit arrangement in a number of proteins, but this approach is limited by problems of ensemble averaging. For example, achieving specific and complete labeling of a single site is often challenging. Multiple conformations of a protein or conformational fluctuations during the measurement time require resolution of more than one distance simultaneously. These limitations may be overcome by observing only a single molecule in a given time interval (14). Single molecule FRET measurements were introduced about a decade ago, and the method has now matured as a standard fluorescence technique (15, 16). Using several pairs of amino acid positions, single molecule triangulation methods result in an accuracy of about 0.5 nm for the three-dimensional localization of the fluorophores (17, 18).
F0F1-ATP synthase is a non-synchronizable enzyme with an intrinsic 3-fold pseudosymmetry arising from the number and arrangement of the three αβ pairs and the three associated positions of the central γ and ε subunits during catalysis. In the current work, we used the three-stepped rotation of γ and ε to obtain a set of three distance measurements to a-EGFP within a single enzyme. To assign the three-dimensional position of the FRET donor EGFP fused to subunit a, we made use of the distance constraints between the EGFP and the N terminus of the c subunits and of the previously demonstrated distinct dwell times of the three positions of the rotating ε subunit (11).
EXPERIMENTAL PROCEDURES
Construction of a Plasmid Carrying the a-EGFP Fusion—Plasmid pSD166 is a derivative of pACWU1.2 (19) with EGFP fused to the C terminus of the a subunit. To construct this plasmid, a sequence encoding EGFP was amplified using the following oligonucleotide primers: GCTCTGATCATGGATCCATGGTGAGC and GCTCAGATCTTTACTTGTACAGCTCGTCCATGCC (restriction sites for BclI and BglII are indicated in bold). The product was cut with BclI and BglII and cloned into pVF172, which carries part of the unc operon with a BglII site introduced at the 3′-end of uncB, which encodes the a subunit (20). A transformant bearing the insert in the proper direction for expression of an a-EGFP fusion protein was called pSD165. The 1573-bp PflMI/AvaI fragment of pSD165 was ligated with the 366-bp AvaI/PpuMI fragment of pSC5 (19, 21) and the 8.0-kbp PflMI/PpuMI fragment of pACWU1.2 in a three-part ligation to obtain plasmid pSD166. This plasmid encodes a cysteineless ATP synthase with the C terminus of the a subunit fused to EGFP through a GSMV linker. The sequence of the fusion region, MASEDHGSMVSKGEEL, can be related to the C-terminal sequence of a, MASEEH, and the N-terminal sequence of EGFP, MSKGEEL.
Expression, Purification and Spectral Characterization of F0F1—F1 carrying either the εH56C or γT106C mutation was prepared as described previously (11, 22) after expressing plasmid pRAP100 or pRA114 (23), respectively, in strain RA1 (24). Specific labeling of ε56 or γ106 with Alexa568-maleimide (Molecular Probes) was carried out to a labeling efficiency of about 30% (25). The specificity of the labeling was checked by the fluorogram of a SDS-PAGE gel.
Plasmid pSD166 carrying the EGFP fusion protein was expressed in strain RA1, and cell growth kinetics were similar to those of strain RA1 expressing pRA114. ATP synthase was purified as described previously (26). The isolated F1F0-EGFP was reconstituted into preformed liposomes (diameter, ∼120 nm), and F1 was exchanged with Alexa568-labeled F1 carrying the εH56C or γT106C mutations as described in Reference 11, resulting in membrane-integrated F0F1 labeled with both the FRET donor (EGFP) and the acceptor (Alexa568). Spectral analysis of the original preparation of ATP synthase carrying the a-EGFP fusion revealed impurities with a pH-dependent absorption band around 425 nm that could correspond to porphyrins or cytochromes. These impurities were non-fluorescent at the emission wavelengths used for the subsequent FRET measurements and were not anticipated to disturb the single molecule measurements for two reasons. (i) In the subsequent step of the preparation, the F1 parts were removed and replaced by Alexa568-labeled F1 to yield the intramolecular FRET system in the F0F1-ATP synthase. (ii) Remaining impurities in the lipid membrane that, in principle, could act as FRET acceptors will not show a stepping FRET level sequence during ATP hydrolysis and therefore would be discarded automatically as non-rotating FRET sources in the single molecule data analysis. Rates of ATP synthesis and hydrolysis were measured at 23 and 37 °C, respectively, as described previously (11). In the presence of 60 μm 1,3-dicyclohexylcarbodiimide, ATP synthesis activity of reconstituted F0F1-ATP synthase was completely abolished.
Confocal Single Molecule FRET Measurements—The single molecule measurements were carried out on a home-built confocal microscope (27-29). Proteoliposomes were excited continuously at 488 nm (argon ion laser, model 2020, Spectra Physics) or with picosecond pulses at 80 MHz (PicoTA490, Picoquant, Berlin, Germany), respectively (30). The laser beam was attenuated to 150 microwatts on the back aperture of the microscope objective and focused by a water immersion objective (UPlanSApo 60×, numerical aperture 1.2, Olympus) into a droplet of buffer solution placed on a microscope coverslide. The fluorescence emission was separated from the excitation light by a dichroic beam splitter (c488RDC, AHF analysentechnik AG, Tübingen, Germany). Emission of the two fluorophores was split by a second dichroic beamsplitter (HQ 575, AHF analysentechnik AG). Single photons were detected separately by two avalanche photodiodes (SPCM-AQR-14, EG&G, Quebec, Canada) after passing an interference filter (HQ 532/70, AHF analysentechnik AG) for EGFP or a long pass filter (LP595, AHF analysentechnik AG) for Alexa568. Detection efficiencies were ηD = 0.364 for EGFP and ηA = 0.404 for Alexa568. A cross-talk of 6.3% from donor fluorescence in the acceptor channel was corrected. Photons were counted by a TCSPC card (SPC630, Becker & Hickl, Berlin, Germany) in first-in-first-out (FIFO) mode with 50-ns time resolution using the router electronics (HRT-82, Becker & Hickl) to assign the FRET channel information for each photon. The TCSPC data also contained the arrival time information for the laser pulse for FRET donor fluorescence lifetime measurements with picosecond resolution. Fluorescence anisotropies of single EGFP-a-F0F1 (linear polarized excitation with 488 nm) or F0-Alexa568-ε-F1 (linear polarized excitation with 561 nm; continuous wave laser Jive, Cobolt, Stockholm, Sweden) in liposomes were measured separately in the confocal microscope using a polarizing beamsplitter in the fluorescence pathway. Signals of the two avalanche photodiodes were recorded by two synchronized, fast TCSPC cards (SPC152, Becker & Hickl). Intensity thresholds were applied to identify individual photon bursts, and fluorescence anisotropy values were calculated for each burst after correction for the avalanche photodiode detection efficiencies. Solutions of rhodamine 110, erythrosine, rhodamine 101, and a new perylene dye (31) in water were used as anisotropy references.
Data Analysis—Fluorescence intensity time trajectories were binned to 1 ms using the custom-made software “Burst_ Analyzer” (32). Photon bursts of single F0F1-ATP synthases were identified by intensity thresholds for the FRET donor and acceptor channels after substraction of a background signal of about 2-5 counts/ms. FRET levels and changes of FRET levels were assigned manually within each photon burst as described previously (33). Only FRET levels from photon bursts showing two or more FRET transitions were included in the FRET efficiency histograms. Fitting the histogram by Gaussian distributions yielded the mean values for the distinct distances assuming the previously determined fluorescence quantum yields of EGFP and Alexa568. The regular sequence of FRET transitions upon ATP hydrolysis was revealed from the maxima in the two-dimensional FRET transition density plot according to Ref. 34. Lower and upper FRET efficiency limits for the three mean FRET levels were assigned from the FRET transition density plot. Subsequently each FRET level within a photon burst was attributed to one of the three main FRET states. The subset of photon bursts with three or more FRET levels in the regular FRET transition sequence was selected to calculate the dwell times (from the intermediate FRET levels only) and to triangulate one single position of the EGFP chromophore by each set of three consecutive FRET levels.
RESULTS
The single molecule FRET triangulation approach to localize subunit a of F0 relies initially on determination of distances to the three stopping positions of the rotating ε or γ subunits of F1 during ATP hydrolysis. In a subsequent step, the dwell times of previous single molecule FRET measurements between ε and the b subunit dimer are compared with the dwell times obtained for the new FRET experiments to allow correlation of the three stopping positions so that the relationship of the positions of a and b2 may be established. This approach requires that four specific goals be achieved: (a) specific labeling of subunit a,(b) functional reassembling of the labeled F0 with a specifically labeled ε or γ subunit of F1,(c) discrimination of the three FRET efficiencies depending on the relative orientations of γ or ε, and (d) correlation of the different dwell times obtained in the FRET experiments of ε rotation versus subunit b to the dwell times obtained in the FRET experiments of the rotation of ε or γ versus subunit a.
The Fusion of EGFP to the C terminus of Subunit a—To specifically label the subunit a of F0F1 with a fluorophore for intramolecular FRET distance measurements we fused the autofluorescent protein EGFP to the C terminus of subunit a, which is located on the cytosolic side of the membrane. In fluorescence microscopic images, the EGFP fusion to F0F1 is located at the cell membrane (see supplemental Fig. S1). Few cells showed fluorescent inclusion bodies. The ATP synthase plasmid carrying the a-EGFP fusion supported growth of cells on non-fermentable carbon sources (acetate and succinate) at rates comparable to those of the wild type. We conclude that the fusion of EGFP did not impair the functionality of the F0F1-ATP synthase in E. coli.
The enzyme was isolated from the plasma membranes and reconstituted into lipid vesicles to yield a ratio of not more than one ATP synthase per liposome. ATP synthesis (vS = 32 s-1 at 23 °C) and hydrolysis (vH = 330 s-1 at 37 °C) activities were measured.
The fluorescence spectra of EGFP fused to subunit a of F0F1 in the presence of 0.1% dodecylmaltoside showed a small (2-nm) red shift of the fluorescence excitation maximum at pH 8.8 compared with free EGFP, and the emission maximum at 510 nm was nearly identical to published data (35). The fluorescence quantum yield for the EGFP fusion, ΦFl = 0.4, was measured previously (36). The single molecule brightness of EGFP on subunit a was found to be lower than that of rhodamine 110 in solution using fluorescence correlation spectroscopy. Nevertheless a mean number of detected photons n = 55 kHz per EGFP was achievable by excitation at 488 nm with a laser power of 150 microwatts at the back focal plane of the microscope objective. The fluorescence lifetime of EGFP fused to F0F1 was found to consist of two components in the absence of a FRET acceptor. The major fraction had a lifetime τ ∼2.7 ns, whereas a minor fraction showed a lifetime τ ∼2.2 ns in accordance with the literature (37). To summarize, the fluorescence properties of the EGFP fused to the a subunit were as expected, and the fluorophore seemed to be suitable for the single molecule FRET distance measurements.
Single Molecule FRET Levels from a-EGFP to Positions ε56 or γ106 during ATP Hydrolysis—The quantitative removal of the F1 part from F0F1-ATP synthase in lipid membranes was accomplished in the absence of Mg2+. Afterwards the enzyme was reassembled in the presence of Mg2+ with F1 labeled with Alexa568 at position ε56 or γ106. The mean ATP synthesis activity was measured as vS = 40 ATP s-1 at 23 °C; this is comparable to previously measured synthesis rates for labeled enzymes, which show modest reductions compared with unlabeled forms (33, 38).
For the single molecule measurements, proteoliposomes carrying single FRET-labeled ATP synthase molecules were diluted to ∼100 pm. Confocal excitation in a femtoliter-sized volume was achieved by focusing the 488 nm laser line of an argon ion laser (or 476 nm of a krypton ion laser for F0F1-ATP synthase labeled with the FRET acceptor at the γ subunit) into a microscope objective. As a single ATP synthase diffused freely into the confocal volume, a burst of photons was generated that was registered in the two spectral ranges corresponding to the FRET donor and FRET acceptor. Photon bursts of single liposome-embedded F0F1, containing a-EGFP and ε56-Alexa568, are shown in Fig. 2. In the presence of 1 mm ATP, the rotation of subunit ε with the attached FRET acceptor relative to EGFP on the non-rotating a subunit caused a stepwise change of the relative fluorescence intensities (Fig. 2, lower panels) with the rotor stopping at the catalytic dwell (39). We calculated the proximity factor P for each time bin as follows.
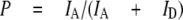 |
(Eq.1) |
where IA and ID are the background-corrected fluorescence intensities in the FRET acceptor or donor channel, respectively. We manually assigned the switching points of the FRET levels (Fig. 2, upper panels). These changes are related to the distance changes between the two fluorophores and were analyzed as FRET efficiency changes. Three main FRET levels were identified in photon bursts showing fluctuations: the mean low FRET efficiency (L) level was found at P = 0.2 ± 0.03, the mean medium FRET efficiency (M) was at P = 0.5 ± 0.08, and the mean high FRET efficiency (H) was at P = 0.86 ± 0.03 (Fig. 3A). For more than 79% of the FRET-labeled ATP synthases showing subunit rotation during ATP hydrolysis, the sequence of the FRET level transitions was → L → M → H → L →.
FIGURE 2.

Photon bursts of single FRET-labeled F0F1-ATP synthases upon ATP hydrolysis. Lower panels show fluorescence intensities of FRET donor EGFP fused to subunit a (green trace) and FRET acceptor Alexa568 (red trace) bound to ε56. Upper panels show the calculated proximity factor P (blue trace) with 1-ms time resolution and the mean P value for each assigned FRET level (black line). A, single photon burst recorded with continuous wave excitation at 488 nm. The FRET level transition sequence is L → M → H. B, single photon burst recorded with pulsed excitation at 488 nm. The FRET donor (EGFP) fluorescence lifetimes for the three FRET levels are τ = 2.22 ns (L), τ = 1.87 ns (M), and τ = 0.56 ns (H).
FIGURE 3.

Proximity factor distribution (A) and FRET transition density plot (B) of F0F1-ATP synthases during ATP hydrolysis. At least two distinct FRET levels had to be detected within a photon burst to be added to the histograms. A, proximity factors for FRET level L as white bars, for Mas light gray bars, and forHas dark gray bars (825 FRET level in total). B, FRET transition density plot with chromophore distances between EGFP and Alexa568 bound to ε56.
In addition to measuring FRET between a-GFP and position ε56, we measured single molecule FRET to Alexa568 at residue 106 of the γ subunit of F0F1 during ATP hydrolysis, yielding an independent set of distances (see supplemental Figs. S2 and S3). As with the samples containing labeled ε, three distinct FRET efficiency levels were found in fluctuating photon bursts according to the stepwise rotation of the γ subunit. The sequence of FRET level transitions was → L → M → H for about 80% of the photon bursts in accordance with the sequence observed above. In contrast, during proton-driven ATP synthesis in the presence of Mg2+, ADP, and phosphate the direction of rotation was reversed (see supplemental material) as deduced from the opposite order of FRET level transitions.
The sequence of FRET levels observed here during ATP hydrolysis are apparently opposite to the sequence observed previously when the enzymes were labeled at γ and the b subunit dimer (38) and also for the single molecule FRET measurements between ε and b (11). This difference must reflect the different positions of a-EGFP and b2 in the enzyme. A position of the FRET donor EGFP on one side of the peripheral b subunit dimer but not a position symmetrically in between the two b subunits, position “B” in Fig. 1, results in the observed change in sequence.
FRET distances for a-EGFP versus ε56—Assuming a fluorescence quantum yield of 0.66 (40) for Alexa568 at ε56, the proximity factor values can be transformed to FRET efficiencies, EFRET.
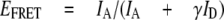 |
(Eq.2) |
where γ is a correction factor for spectral detection efficiencies and fluorescence quantum yields of the FRET donor and acceptor. Alternatively the FRET efficiency is calculated from the FRET donor lifetime in the absence of an acceptor, τD, and in the presence of an acceptor, τDA.
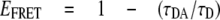 |
(Eq.3) |
The distances rDA between the fluorophores are related to EFRET by
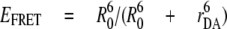 |
(Eq.4) |
according to the Förster theory with R0 being the Förster radius, that is the distance for 50% EFRET. We calculated R0 = 4.9 nm for EGFP and Alexa568 using the fluorescence quantum yields given above. For each FRET level, the distance values can be plotted in pairs to obtain the FRET transition density plot shown in Fig. 3B. The maximum of each FRET level was obtained by fitting the histograms shown in Fig. 3A with three Gaussians distributions, and the corresponding mean distances between the labels were calculated to be 6.8 nm for the low FRET orientation, 5.4 nm for the medium FRET orientation, and 4.0 nm for the high FRET orientation. As a control we applied pulsed excitation at 488 nm to analyze the FRET donor lifetimes in the presence of a FRET acceptor for each FRET level in a single ATP synthase. Three distinct FRET efficiencies were found. The mean L-level with an EGFP lifetime τL ∼2.45 ns corresponded to a 6.8-nm distance, the mean M-level with τM ∼1.76 ns corresponded to a 5.3-nm distance, and the mean H-level with τH ∼0.89 ns corresponded to a 4.3-nm distance (36). Thus, intensity-based and lifetime-based FRET efficiency measurements were in good agreement.
At this point, the EGFP position at the C terminus of a was defined by the three distances with respect to ε56. For the triangulation we assume that the three stopping positions of the rotary ε (or γ) subunit can be placed on a circle. According to the E. coli γε structure (41), the amino acid position of ε56 is probably 2.5 nm distant from the expected axis of rotation, and the γ106 position is about 3 nm off-axis. Given the stopping positions of ε56 (or γ106) separated by 120°, triangulation of EGFP from the mean FRET distances yields six possible positions. Three positions are above the plane of stopping positions of ε56 and γ106, and three are below the plane toward the F0 part. For all positions, the height from the plane ranged from 2.5 to 4.0 nm, depending on the chosen radius of the circle, with smaller radii corresponding to larger heights.
Dwell Time Analysis of FRET Distances—For an unequivocal three-dimensional positioning we now have to discriminate the six possible positions of the EGFP. We start with identifying a specific orientation of ε and γ with respect to the b subunit dimer as the external reference in the following analysis.
Cross-linking experiments (21, 42) and stable ab2 subcomplex formation (43, 44) between the transmembrane helices of subunit a and the N-terminal membrane portion of subunit b have established the nearby arrangement between these two subunits. Therefore, a theoretical position of subunit a on the opposite side of the ring of c subunits without direct contacts to the b subunit can be excluded, and only four possible positions for EGFP fused to subunit a remain, that is to the left or to the right of the b subunits and above or below the plane of γ or ε rotation.
Previously we have shown that the dwell times of the three stopping positions of the ε subunit in F0F1-ATP synthase are not identical, providing a basis for distinguishing between them (11, 32). In Fig. 4 the distributions of dwell times of the three stopping positions observed in 381 rotating single F0F1-ATP synthases with three and more FRET levels in the FRET level sequence → L → M → H → L → (between a-EGFP and ε) are shown. Dwell times were binned to 5-ms intervals and fitted by a monoexponential decay function. The low FRET efficiency orientation exhibited the shortest dwell time, 9 ± 1 (dwell time ± S.D. σ) ms; the medium FRET efficiency orientation had the longest dwell time, 11 ± 1 ms; and the high FRET position had an intermediate dwell time of 10 ± 1 ms (Table 1). Prebinning the dwell times to 3-ms intervals resulted in apparently higher time resolution (see supplemental material) but confirmed the shortest dwell time of the low FRET orientation. Dwell times for the holoenzyme F0F1-ATP synthase reconstituted in lipid vesicles were much longer compared with the high rotational speed reported for single F1 fragments (39, 45) that was interpreted as an indication of a low proton leakage of the liposomes (11). For the case of FRET between a-EGFP and γ, the shortest dwell time of 16 ms was associated with a high FRET orientation (see supplemental Fig. S4). To associate these FRET orientations with the different dwell times found previously for the ε versus b FRET measurements, we refer to the γ/ε crystal structure of the E. coli ATP synthase (41). Based on this structure, during counterclockwise stepped rotation of γ and ε, the γ106 position will be 120° behind position ε56. The bottom view of the F0F1-ATP synthase in Fig. 5 shows the three stopping positions of ε labeled I-III.
FIGURE 4.

Dwell time distributions of FRET levels L (blue bars, 63 level) (A), M (yellow bars, 247 level) (B), and H (red bars, 71 level) (C) during ATP hydrolysis in the presence of 1 mm ATP are shown. F0F1-ATP synthases were labeled with EGFP at the C terminus of a and with Alexa568 at ε56. Dwell times were binned in 5-ms intervals and fitted by monoexponential decays (black curves).
TABLE 1.
FRET distances and corresponding dwell times for the FRET pairs a-EGFP versus ε56 and a-EGFP versus γ106 on F0F1-ATP synthase during ATP hydrolysis
The shortest dwell times are highlighted (bold); dwell times are given with errors (± σ S.D.) for the fitting. Labeling γ106 slightly reduced the catalytic activities as reported previously (11, 33). Distances between fluorophore positions I, II, and III (or I′, II′, and III′ for γ106, respectively) and C refer to positions denoted in Fig. 5.
| Distance | Dwell time | |
|---|---|---|
| nm | ms | |
| Alexa568 at ε56 | ||
| Low FRET (I ↔ C) | 6.8 | 9 ± 1 |
| Medium FRET (II ↔ C) | 5.4 | 11 ± 1 |
| High FRET (III ↔ C) | 4.0 | 10 ± 1 |
| Alexa568 at γ106 | ||
| Low FRET (I′ ↔ C) | 7.6 | 20.3 ± 3 |
| Medium FRET (II′ ↔ C) | 5.2 | 20.2 ± 1 |
| High FRET (III′ ↔ C) | 3.3 | 16.0 ± 1 |
FIGURE 5.

Scheme for allocating the FRET efficiencies during ATP hydrolysis in the model for F1 when viewed from the membrane. Black arrows pointing to positions I, II, and III indicate the stopping positions of Alexa568 on ε56 (or on γ106) following 120° rotation on a circle with radius ∼2.5 nm (semitransparent gray circular area). The position of the b dimer according to previous FRET measurements (11) is shown as a gray ellipse labeled “b64.” Boxes A and C depict the possible positions of the chromophore in EGFP fused to the C terminus of subunit a.
Position I for ε56 was the medium FRET position in the previous FRET experiments of ε56 with respect to position b64, which exhibited the shortest dwell. Position II for ε56 was the low FRET orientation for ε56 versus b64 with the longest dwell. Now we evaluate which position for the EGFP matches the dwell time behavior. If we assume that the likely position of the EGFP at the a subunit is the “C” position for EGFP in Figs. 1 and 5, then both findings of the shortest dwell time for the low FRET orientation of ε56 (position I) as well as the longest dwell time for the medium FRET orientation of ε56 (position II) would fit. In addition, the high FRET orientation of γ106 (i.e. position III) with the shortest dwell time corresponds to the low FRET position of ε56. In contrast, if we would assume that the “A” position of EGFP with respect to the b subunits is correct, than the dwell time behavior cannot be explained. We conclude that the “C” position is the most likely one for EGFP.
Three-dimensional Model with Allocated C Terminus of Subunit a—Given the three a-ε FRET distances of 6.8, 5.3, and 4.0 nm, the likely radius of rotation between 3.5 and 2.5 nm for the FRET acceptor Alexa568 at ε56 yielded an EGFP-chromophore position between 3 and 3.5 nm above or below this plane of rotation. A final decision has to be made of which of the two remaining EGFP positions is appropriate, that is the “up” position toward the F1 headpiece or the “down” position at the membrane level. The expectation that the EGFP has to be placed at the membrane level because of the short 4-amino acid linker to the C terminus of subunit a, is supported by preliminary single molecule FRET distance measurements between EGFP at the cytoplasmic side of the a subunit and the N terminus of the c subunit on the periplasmic side of the membrane with shortest distances of about 4 nm during proton-driven rotation of the c ring (46). Therefore, it appears that EGFP takes the down position, likely making contact with the membrane surface.
Following the localization of EGFP at subunit a from mean distance values obtained by single molecule FRET, we reselected those F0F1-ATP synthases that showed three or more FRET levels in a single photon burst. From each triple set of sequential FRET levels in single photon bursts (or distances, respectively) in ATP hydrolysis order, the individual position of EGFP was reconstructed. The three-dimensional distribution of the EGFP positions was found to be highly consistent with only minor deviations. Thereby the error of the FRET triangulation approach could be estimated. The largest uncertainty was related to the determination of the height of EGFP with respect to the membrane and the plane of ε56 rotation (Fig. 6). However, compared with the determination of the b64 positions with respect to ε56 using the same FRET triangulation approach of successive FRET levels in single bursts, the absolute error was smaller for the EGFP positioning. This is because of the differences in the Förster radii for the two FRET pairs. The smaller R0 for EGFP-Alexa568 compensated for the larger relative errors because of significantly lower brightness or quantum yields, respectively.
FIGURE 6.

Individual positions of EGFP (small green balls) according to single molecule FRET triangulation using the FRET pair a-EGFP-ε56-Alexa368 (left side) or a-EGFP-γ106-Alexa568 (right side). The small yellow balls are the individual positions for the FRET acceptor Cy5 at b64 with respect to TMR at ε56 (positions recalculated from previous FRET data of Zimmermann et al. (11)). The γ subunit is shown in red, the ε subunit is in blue. Large light blue balls represent the apparent ε56 stopping positions upon 120° rotation of γε during catalysis. Large orange balls represent the three γ106 positions. Arrows indicate the fluorophore positions at the ε or γ subunit, respectively, for the rotor orientation shown in the images. Upper images show F1 when viewed from the membrane. In the lower images F0F1 are oriented with b2 subunits to the left side.
DISCUSSION
We applied single molecule FRET measurements to triangulate the position of subunit a with respect to the b2 dimer in F0F1-ATP synthase. As a prerequisite, specific labeling of a was achieved by fusion of the autofluorescent protein EGFP to the C terminus of a. The size of EGFP is about 5 nm in height and 3 nm in diameter (47). However, the additional protein mass on the proton-translocating subunit did not derogate the catalytic rates for ATP hydrolysis or ATP synthesis according to the biochemical ensemble measurements. Therefore the large size of EGFP could also be used for an alternative triangulation approach as shown for the oligomycin sensitivity-conferring protein (OSCP) subunit of the mitochondrial ATP synthase. Comparison of electron microscopic images of F0F1 with and without an added protein yielded the three-dimensional position of the C terminus with a small error of about 1 nm (48).
As the FRET acceptor we used a small rhodamine dye, Alexa568, which was bound specifically to cysteines at the ε or the γ subunit in the F1 part. These cysteine mutations did not affect the enzyme activities (38). During ATP hydrolysis in the presence of 1 mm ATP, the 120° stepped rotation of ε or γ moved the FRET acceptor fluorophore on a circle with ∼2.5-3.5-nm radius as expected from the size of the c ring consisting of 10 subunits. Thereby the distances to the FRET donor placed slightly outside the circle will change between 3 (shortest distance) and 7 nm (longest distance). FRET distance measurements are most accurate around the Förster distance R0 for 50% energy transfer. For the FRET fluorophores EGFP and Alexa568 we calculated R0 = 4.9 nm assuming an orientation factor κ2 = 2/3 (justified by single fluorophore anisotropies r = 0.3 for EGFP and r < 0.1 for Alexa568 at F0F1). Accordingly we estimated a maximum distance error of about 1 nm for the short as well as the long distances. However, the narrow three-dimensional distribution for individual EGFP positions shown in Fig. 6B results from the single molecule FRET triangulations that clearly indicate that only a small deviation for the mean EGFP position is expected.
Because of the size of EGFP, the distance between the C terminus of subunit a and the internal position of the chromophore is about 2.5 nm. This raises the possibility that changes in FRET could reflect motions, on the millisecond time scale, of EGFP between two or three metastable positions relative to ATP synthase rather than rotation. In this case, however, a random order of changes in FRET efficiency would be expected (see supplemental material). Any signals with changes that did not match the standard pattern (→ L → M → H → L →) were therefore omitted from the triangulation calculation and dwell time analysis. Our results showing the clustered positions of the EGFP chromophore (Fig. 6B) calculated from single F0F1-ATP synthases rotating unidirectionally support a stable position of EGFP. As we do not know the orientation of EGFP with respect to the membrane plane, the apparent large distance between the b subunits (represented as yellow dots for residue b64 in Fig. 6) and the EGFP chromophore might be explained by an orientation of EGFP with the long axis parallel to the membrane. Thereby the proposed compact helix arrangement in a with the C-terminal transmembrane helix 5 adjacent to transmembrane helix 3 (49) is not contradictory to the FRET localization presented here. A mean distance between 3 and 3.5 nm from the EGFP chromophore to the axis of rotation for γ, ε, and c10 supports this assumption of EGFP oriented parallel to the membrane plane and to the c ring. In addition, a hypothetical orientation of EGFP perpendicular to the membrane would place a large fraction of the protein inside the lipid membrane; this seems unlikely.
Electron microscopic images of the mitochondrial F0F1 (50) support the localization of a. There an additional protein mass was found in an asymmetric position to the right of the mitochondrial subunits comprising the peripheral static connection between F1 and F0. However, the mitochondrial enzyme consists of several additional small subunits in the membrane portion of F0. Similarly the proton-translocating subunit of the chloroplast ATP synthase had been placed asymmetrically near the peripheral stalk (51).
What might be a structural or functional advantage for subunit a located in this position with respect to b2? During ATP synthesis the rotation of the ring of c subunits is expected to occur clockwise when viewed from the periplasmic side of the membrane. This will force the adjacent a subunit to move relatively into the opposite direction. With subunit a placed to the right side of b, the forced movement will push a against b, or vice versa, the b subunits will prevent a to counter-rotate. In the case of ATP hydrolysis, the counterclockwise rotation of γ and ε induces an opposing torque on F1 and the peripheral stalk that is transmitted to the b2-a interface (52). Accordingly the b dimer is again pushed against the a subunit. It seems to be likely that the stator subunits not only hold the non-rotating F1 subunits mechanically in place but are arranged in the F0 part so that the force that is imposed on the a-b2 interface as the two rotary motors work against one another acts to push the membrane domains of these subunits together rather than pulling them apart. Thus, their relationship is stabilized rather than destabilized as rotational forces are strengthened.
Supplementary Material
Acknowledgments
We thank P. Gräber (University of Freiburg, Germany) for support of F0F1-ATP synthase preparation and catalytic activity measurements, Becker & Hickl for the loan of the SPC152 TCSPC electronics, and Cobolt for the loan of the solid state laser Jive for the single molecule anisotropy measurements.
This work was supported in part by Deutsche Forschungsgemeinschaft Grant BO 1891/8-1 (to M. B.) and by Canadian Institutes of Health Research Grant MT-10237 (to S. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S4.
Footnotes
The abbreviations used are: FRET, fluorescence resonance energy transfer; EGFP, enhanced green fluorescent protein; P, proximity factor; TCSPC, time-correlated single photon counting; L, low FRET efficiency; M, medium FRET efficiency; H, high FRET efficiency.
References
- 1.Boyer, P. D. (1997) Annu. Rev. Biochem. 66 717-749 [DOI] [PubMed] [Google Scholar]
- 2.Weber, J., and Senior, A. E. (1997) Biochim. Biophys. Acta 1319 19-58 [DOI] [PubMed] [Google Scholar]
- 3.Angevine, C. M., Herold, K. A., and Fillingame, R. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13179-13183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottcher, B., Bertsche, I., Reuter, R., and Graber, P. (2000) J. Mol. Biol. 296 449-457 [DOI] [PubMed] [Google Scholar]
- 5.Meier, T., Polzer, P., Diederichs, K., Welte, W., and Dimroth, P. (2005) Science 308 659-662 [DOI] [PubMed] [Google Scholar]
- 6.Dmitriev, O., Jones, P. C., Jiang, W., and Fillingame, R. H. (1999) J. Biol. Chem. 274 15598-15604 [DOI] [PubMed] [Google Scholar]
- 7.Del Rizzo, P. A., Bi, Y., Dunn, S. D., and Shilton, B. H. (2002) Biochemistry 41 6875-6884 [DOI] [PubMed] [Google Scholar]
- 8.Dickson, V. K., Silvester, J. A., Fearnley, I. M., Leslie, A. G., and Walker, J. E. (2006) EMBO J. 25 2911-2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumamoto, C. A., and Simoni, R. D. (1986) J. Biol. Chem. 261 10037-10042 [PubMed] [Google Scholar]
- 10.Kumamoto, C. A., and Simoni, R. D. (1987) J. Biol. Chem. 262 3060-3064 [PubMed] [Google Scholar]
- 11.Zimmermann, B., Diez, M., Zarrabi, N., Graber, P., and Borsch, M. (2005) EMBO J. 24 2053-2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster, T. (1948) Ann. Phys. 2 55-75 [Google Scholar]
- 13.Stryer, L., and Haugland, R. P. (1967) Proc. Natl. Acad. Sci. U. S. A. 58 719-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ha, T., Ting, A. Y., Liang, J., Chemla, D. S., Schultz, P. G., and Weiss, S. (1999) Biophys. J. 76 A386-A386 [Google Scholar]
- 15.Deniz, A. A., Laurence, T. A., Dahan, M., Chemla, D. S., Schultz, P. G., and Weiss, S. (2001) Annu. Rev. Phys. Chem. 52 233-253 [DOI] [PubMed] [Google Scholar]
- 16.Myong, S., Stevens, B. C., and Ha, T. (2006) Structure (Lond.) 14 633-643 [DOI] [PubMed] [Google Scholar]
- 17.Antonik, M., Felekyan, S., Gaiduk, A., and Seidel, C. A. (2006) J. Phys. Chem. B 110 6970-6978 [DOI] [PubMed] [Google Scholar]
- 18.Andrecka, J., Lewis, R., Bruckner, F., Lehmann, E., Cramer, P., and Michaelis, J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 135-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo, P. H., Ketchum, C. J., and Nakamoto, R. K. (1998) FEBS Lett. 426 217-220 [DOI] [PubMed] [Google Scholar]
- 20.Valiyaveetil, F. I., and Fillingame, R. H. (1998) J. Biol. Chem. 273 16241-16247 [DOI] [PubMed] [Google Scholar]
- 21.McLachlin, D. T., Coveny, A. M., Clark, S. M., and Dunn, S. D. (2000) J. Biol. Chem. 275 17571-17577 [DOI] [PubMed] [Google Scholar]
- 22.Gogol, E. P., Lucken, U., Bork, T., and Capaldi, R. A. (1989) Biochemistry 28 4709-4716 [DOI] [PubMed] [Google Scholar]
- 23.Aggeler, R., and Capaldi, R. A. (1992) J. Biol. Chem. 267 21355-21359 [PubMed] [Google Scholar]
- 24.Aggeler, R., Ogilvie, I., and Capaldi, R. A. (1997) J. Biol. Chem. 272 19621-19624 [DOI] [PubMed] [Google Scholar]
- 25.Borsch, M., Diez, M., Zimmermann, B., Reuter, R., and Graber, P. (2002) FEBS Lett. 527 147-152 [DOI] [PubMed] [Google Scholar]
- 26.Fischer, S., and Graber, P. (1999) FEBS Lett. 457 327-332 [DOI] [PubMed] [Google Scholar]
- 27.Ding, K., Alemdaroglu, F. E., Borsch, M., Berger, R., and Herrmann, A. (2007) Angew. Chem. Int. Ed. Engl. 46 1172-1175 [DOI] [PubMed] [Google Scholar]
- 28.Alemdaroglu, F. E., Wang, J., Borsch, M., Berger, R., and Herrmann, A. (2008) Angew. Chem. Int. Ed. Engl. 47 974-976 [DOI] [PubMed] [Google Scholar]
- 29.Heitkamp, T., Kalinowski, R., Bottcher, B., Borsch, M., Altendorf, K., and Greie, J. C. (2008) Biochemistry 47 3564-3575 [DOI] [PubMed] [Google Scholar]
- 30.Zarrabi, N., Duser, M. G., Ernst, S., Reuter, R., Glick, G. D., Dunn, S. D., Wrachtrup, J., and Borsch, M. (2007) Proc. Soc. Photo-Opt. Instrum. 6771 67710F [Google Scholar]
- 31.Peneva, K., Mihov, G., Herrmann, A., Zarrabi, N., Borsch, M., Duncan, T. M., and Mullen, K. (2008) J. Am. Chem. Soc. 130 5398-5399 [DOI] [PubMed] [Google Scholar]
- 32.Zarrabi, N., Zimmermann, B., Diez, M., Graber, P., Wrachtrup, J., and Borsch, M. (2005) Proc. Soc. Photo-Opt. Instrum. 5699 175-188 [Google Scholar]
- 33.Diez, M., Zimmermann, B., Borsch, M., Konig, M., Schweinberger, E., Steigmiller, S., Reuter, R., Felekyan, S., Kudryavtsev, V., Seidel, C. A., and Graber, P. (2004) Nat. Struct. Mol. Biol. 11 135-141 [DOI] [PubMed] [Google Scholar]
- 34.McKinney, S. A., Joo, C., and Ha, T. (2006) Biophys. J. 91 1941-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsien, R. Y. (1998) Annu. Rev. Biochem. 67 509-544 [DOI] [PubMed] [Google Scholar]
- 36.Duser, M. G., Zarrabi, N., Bi, Y., Zimmermann, B., Dunn, S. D., and Borsch, M. (2006) Proc. Soc. Photo-Opt. Instrum. 6092 60920H [Google Scholar]
- 37.Cotlet, M., Goodwin, P. M., Waldo, G. S., and Werner, J. H. (2006) Chemphyschem 7 250-260 [DOI] [PubMed] [Google Scholar]
- 38.Zimmermann, B., Diez, M., Borsch, M., and Graber, P. (2006) Biochim. Biophys. Acta 1757 311-319 [DOI] [PubMed] [Google Scholar]
- 39.Yasuda, R., Noji, H., Yoshida, M., Kinosita, K., Jr., and Itoh, H. (2001) Nature 410 898-904 [DOI] [PubMed] [Google Scholar]
- 40.Panchuk-Voloshina, N., Haugland, R. P., Bishop-Stewart, J., Bhalgat, M. K., Millard, P. J., Mao, F., Leung, W. Y., and Haugland, R. P. (1999) J. Histochem. Cytochem. 47 1179-1188 [DOI] [PubMed] [Google Scholar]
- 41.Rodgers, A. J., and Wilce, M. C. (2000) Nat. Struct. Biol. 7 1051-1054 [DOI] [PubMed] [Google Scholar]
- 42.Fillingame, R. H., Jiang, W., and Dmitriev, O. Y. (2000) J. Exp. Biol. 203 9-17 [DOI] [PubMed] [Google Scholar]
- 43.Stalz, W. D., Greie, J. C., Deckers-Hebestreit, G., and Altendorf, K. (2003) J. Biol. Chem. 278 27068-27071 [DOI] [PubMed] [Google Scholar]
- 44.Krebstakies, T., Zimmermann, B., Graber, P., Altendorf, K., Borsch, M., and Greie, J. C. (2005) J. Biol. Chem. 280 33338-33345 [DOI] [PubMed] [Google Scholar]
- 45.Nakanishi-Matsui, M., Kashiwagi, S., Hosokawa, H., Cipriano, D. J., Dunn, S. D., Wada, Y., and Futai, M. (2006) J. Biol. Chem. 281 4126-4131 [DOI] [PubMed] [Google Scholar]
- 46.Zarrabi, N., Duser, M. G., Reuter, R., Dunn, S. D., Wrachtrup, J., and Borsch, M. (2007) Proc. Soc. Photo-Opt. Instrum. 6444 64440E [Google Scholar]
- 47.Ormo, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R. Y., and Remington, S. J. (1996) Science 273 1392-1395 [DOI] [PubMed] [Google Scholar]
- 48.Rubinstein, J., and Walker, J. (2002) J. Mol. Biol. 321 613-619 [DOI] [PubMed] [Google Scholar]
- 49.Dmitriev, O. Y., Freedman, K. H., Hermolin, J., and Fillingame, R. H. (2008) Biochim. Biophys. Acta 1777 227-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubinstein, J. L., Walker, J. E., and Henderson, R. (2003) EMBO J. 22 6182-6192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mellwig, C., and Bottcher, B. (2003) J. Biol. Chem. 278 18544-18549 [DOI] [PubMed] [Google Scholar]
- 52.Panke, O., and Rumberg, B. (1999) Biochim. Biophys. Acta 1412 118-128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.