

Abstract
Clustered DNA damages—two or more closely spaced damages (strand breaks, abasic sites, or oxidized bases) on opposing strands—are suspects as critical lesions producing lethal and mutagenic effects of ionizing radiation. However, as a result of the lack of methods for measuring damage clusters induced by ionizing radiation in genomic DNA, neither the frequencies of their production by physiological doses of radiation, nor their repairability, nor their biological effects are known. On the basis of methods that we developed for quantitating damages in large DNAs, we have devised and validated a way of measuring ionizing radiation-induced clustered lesions in genomic DNA, including DNA from human cells. DNA is treated with an endonuclease that induces a single-strand cleavage at an oxidized base or abasic site. If there are two closely spaced damages on opposing strands, such cleavage will reduce the size of the DNA on a nondenaturing gel. We show that ionizing radiation does induce clustered DNA damages containing abasic sites, oxidized purines, or oxidized pyrimidines. Further, the frequency of each of these cluster classes is comparable to that of frank double-strand breaks; among all complex damages induced by ionizing radiation, double-strand breaks are only about 20%, with other clustered damage constituting some 80%. We also show that even low doses (0.1–1 Gy) of high linear energy transfer ionizing radiation induce clustered damages in human cells.
Ionizing radiation may produce cancer, death, and loss of neural function in humans and animals, and it may induce killing, mutation, and chromosomal aberrations in cells (1). Humans are exposed to low doses of radiation—during air travel, from radon in homes, during space travel, or in areas of low-level contamination, including former sites of nuclear weapon production—and can encounter much higher radiation doses in contaminated areas such as Chernobyl or during radiotherapy (1–5). However, ionizing radiation induces a plethora of types of DNA damages (6), and the identity of specific lesions responsible for the biological effects of radiation remains uncertain. Understanding the long-term effects of low and high doses of ionizing radiation on living organisms requires identification of critical radiation-induced DNA lesions, measurement of their repairability, and determination of the consequences of misrepaired or unrepaired persistent lesions.
Lethal and mutagenic effects of ionizing radiation result principally from incompletely or incorrectly repaired DNA lesions (7, 8). Ionizing radiation induces high levels of isolated DNA lesions, including SSBs, damaged bases, and abasic sites, located at a distance from other damages (6). Such isolated damages are generally repaired efficiently, and their repair may be increased by priming ionizing radiation doses (9).
Ionizing radiation also induces closely spaced lesions, including double-strand breaks (DSBs)—two or more SSBs on opposing strands within about 10–20 bp (10, 11)—and has been postulated to produce other clustered damages (12), composed of other lesions on both DNA strands. Although such clustered damages have been postulated to be significant lesions that induce biological damage (7, 8, 13, 14), it has not been possible to evaluate their biological impact by measuring their induction and repair. Model system studies of oligonucleotides with defined lesions at specific relative spacings on opposing strands indicate that clusters may comprise nonrepairable, highly repair-resistant, or premutagenic damages (15–17). Whether low doses of ionizing radiation produce significant levels of clustered DNA damages in cells—and, if so, their compositions and frequencies—is not known.
We reasoned that clustered lesions could be identified by treating irradiated DNA with lesion-specific enzymes—endonucleases making single-strand nicks at oxidized bases or abasic sites: at each cluster site such an enzyme produces two closely spaced single-strand nicks on opposing strands, resulting in a DSB. We could then determine the frequency of the induced DSBs (each resulting from a damage cluster) by quantitative nondenaturing agarose gel electrophoresis to determine the mobility distribution of the DNA (18–20) and hence the number-average molecular length (Ln) of the resulting DNA population (21). To test our hypothesis, we determined whether γ-irradiation of DNA in solution induced levels of clustered lesions that could be measured by this approach. We show in this paper that ionizing radiation induces clustered DNA lesions at significant levels in DNA irradiated in solution. Furthermore, we show that clustered lesions are induced and can be detected by these methods in human cells exposed to 0.1- to 1-Gy doses of high linear energy transfer (LET) radiation, 1-GeV Fe26+ particles, doses that result in high cell survival.
Methods
Irradiation and Enzyme Treatment of DNA in Solution.
Bacteriophage T7 DNA in 20 mM potassium phosphate buffer, pH 7.4, or in 10 mM Tris⋅HCl, pH 8.0, was irradiated in plastic tubes with 137Cs γ rays (0.16–1.6 Gy/min) or 50-kVp x rays (5 Gy/min). Samples were equilibrated with 70 mM Hepes–KOH, pH 7.6/100 mM KCl/1 mM EDTA/1 mM DTT/50 ng/μl BSA. They were then treated with saturating levels (for reaction mixtures containing 500 ng of T7 DNA, from 0 to 100 Gy, Fpg protein, 60 ng; Nth protein, 120 ng; and Nfo protein, 156 ng) of a lesion-specific enzyme (Table 1) or without endonuclease for determination of frank DSBs. After digestion was complete, traces of these enzymes were removed by addition of proteinase K and EDTA to final concentrations of 1.33 mg/ml and 0.1 M, respectively, and incubation at 37°C overnight. A neutral stop mixture (0.125% bromphenol blue/0.5% sodium lauryl sulfate in 50% glycerol) was then added to ensure dissociation of any persistent DNA–protein complexes.
Table 1.
Enzymes for identifying clustered and isolated damages and their substrates
| Enzyme | Principal lesion class recognized | Lesions recognized* |
|---|---|---|
| Escherichia coli formamidopyrimidine-DNA glycosylase (Fpg protein) | Oxidized purines | FaPy, 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine FaPy-adenine, FaPy-guanine, C8-oxoguanine, some abasic sites, C8-oxoadenine, and to a lesser extent, other modified purines (45–49) |
| E. coli Nth protein (endonuclease III) | Oxidized pyrimidines | Thymine residues damaged by ring saturation, fragmentation, or ring contraction, including 5,6-dihydrothymine, thymine glycol, urea, 5-hydroxy-5-methyl hydantoin, DNA damaged at guanine sites, and some abasic sites (48–51) |
| E. coli Nfo protein (endonuclease IV) | Abasic sites | Several types of abasic sites, including oxidized abasic sites, abasic sites modified with alkoxyamines, and DNA containing urea residues (34, 52) |
5-Hydroxycyctosine and 5-hydroxy-2′-deoxyuridine are substrates for Fpg protein and Nth protein, but neither is formed at significant levels during aerobic irradiation.
Damage Cluster Measurement.
Samples were electrophoresed along with molecular length standards (DNA from bacteriophages T4 and λ and a HindIII digest of λ DNA) in 0.4% agarose, in Tris–acetate buffer, pH 8, using static field electrophoresis (30 V, 6°C, with buffer recirculation) for cluster frequencies more than ≈30 sites per megabase pair (Mbp) or unidirectional pulsed-field electrophoresis (22) (15 V/cm; 0.3-s pulse, 10-s interpulse, 16 hr, 10°C with buffer recirculation) for lower cluster frequencies. Gels were stained with ethidium bromide (1 μg/ml) and destained, and a quantitative electronic image was obtained by using a charge-coupled device-based system (20). A DNA dispersion curve (migration position on the gel vs. molecular length) was constructed from the length standard DNA lanes. DNA lane profiles (fluorescence from ethidium bromide bound to DNA vs. migration distance) of the experimental samples were determined and their Ln values were calculated. From these Ln values, the frequencies of DSBs, φDSB, was calculated from the equation (21):
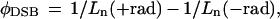 |
1 |
where 1/Ln(+rad) and 1/Ln(−rad) are the reciprocals of the Lns of irradiated and unirradiated samples. The frequencies of other cluster sites, φC, (determined by treatment of samples subjected to dose D, then subdivided; part was digested with Nfo protein, Fpg protein, or Nth protein while the companion part was incubated without enzyme), were calculated from the equation
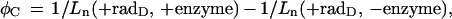 |
2 |
where 1/Ln(+rad, +enzyme) and 1/Ln(+rad, −enzyme) are reciprocals of Lns of sample pairs with and without enzyme, respectively.
Clustered Damage Measurement in DNA from Irradiated Human Cells.
Human monocytes (ATCC CRL 9855) were grown on Iscove's modified Dulbecco's medium (GIBCO/BRL) containing 10% fetal bovine serum (HyClone) without antibiotics, and were ascertained to be free of mycoplasma by periodic testing (Bionique, Saranac Lake, NY). Cells in medium (1 × 106 cells per 0.25 ml in small vials, chilled on ice), were exposed to low doses (0–0.70 Gy, 0.1–1 Gy/min) of 1-GeV/atomic mass unit (amu) Fe26+ ions (LET, 149 keV/amu) from the Brookhaven Alternating Gradient Synchrotron (23). Radiation doses were monitored by ionization chambers and beam uniformity, by a scintillator/charge-coupled device camera system. Immediately after irradiation, cells were harvested by immersion of the vial into liquid N2 and stored in liquid N2 until processed. Each vial of frozen cells was thawed rapidly, EDTA was added to 83 mM final concentration, and the cell suspension was mixed with an equal volume of 2% agarose (InCert; FMC), and formed into plugs. The solidified plugs were treated with lysis solution [10 mM Tris⋅HCl/20 mM NaCl/0.1 M EDTA, pH 8.3 (lysis buffer) containing 1 mg/ml proteinase K (Boehringer Mannheim) and 0.2% N-lauroylsarcosine] for at least 96 hr with daily changes of lysis solution. Digested plugs were rinsed with TE (10 mM Tris⋅HCl/1 mM EDTA, pH 8) to remove detergent, treated with 10 vol of TE containing 40 μg/ml phenylmethylsulfonyl fluoride (PMSF), and then rinsed twice (30 min each) with TE, stored in L buffer (10 mM Tris⋅HCl, pH 8.0/20 mM NaCl/0.1 M EDTA). DNA was digested with NotI for damage analysis (24); digestion conditions were according to the supplier's instructions (New England Biolabs). The effectiveness of NotI cleavage of DNA isolated by this method was determined by Southern blotting using a 1.7-kb ClaI–EcoRI probe to the third exon of the c-myc gene. Human DNA, along with molecular length standards, was dispersed on a 1% PFGE (Amresco; Solon OH) or FastLane (FMC) agarose transverse alternating field electrophoretic gel (25) (TAFE; 30 min at 90 V with a 4-s switching time, then 16 hr at 190 V with a 60-s switching time; electrophoresis buffer, 10 mM Tris acetate/0.5 mM EDTA, pH 8). The human DNA showed a single hybridizing band of 70 kb, the expected length of this fragment.
For glycosylase treatment, duplicate plugs from each experimental treatment were transferred to 70 mM Hepes⋅KOH/100 mM KCl/1 mM EDTA, pH 7.6, then to the same buffer containing 1 mM DTT and 0.1 mg/ml BSA. Plugs were then treated with sufficient homogeneous, lesion-specific enzyme (for one plug containing 500 ng of DNA, 1.2 μg of Nth protein) to cleave at all substrate sites. To remove traces of these enzymes, plugs were treated with proteinase K (1 mg/ml), rinsed, and equilibrated into electrophoresis buffer (10 mM Tris/87 mM acetic acid/0.5 mM EDTA free acid, pH 8). Samples were electrophoresed along with molecular length standard DNAs (Saccharomyces cerevisiae chromosomes, λ ladders) using neutral TAFE (25). Gels were stained with ethidium bromide and destained, and an electronic image was obtained. A DNA dispersion curve was determined by using a spline-fitting procedure, the Lns of the experimental DNAs were calculated, and cluster lesion frequencies (φC) in the 1.3-Mbp DNA populations were computed according to Eq. 2 (19).
Results and Discussion
Fig. 1 shows the principles of identification of clustered damages by this method. In A, DNA is either unirradiated or exposed to ionizing radiation. In B, the left oval shows that unirradiated DNA remains the same size, whereas the right oval shows that radiation induces DSBs and clustered damages (closely opposed damages on opposing strands—either an oxidized base opposite a SSB or two closely apposed oxidized bases). C (block 1 vs. block 2) shows that treatment with a lesion-specific enzyme does not affect the size of the unirradiated DNA. Block 3 of C shows that, in the absence of lesion-specific enzyme treatment, the size of the irradiated DNA is decreased by DSBs but not by clustered damages. Block 4 of C shows that treatment with the lesion-specific enzyme induces a de novo DSB at the site of each clustered damage. D is a schematic representation of a neutral pH agarose gel. Comparison of lanes 1 and 2 (containing DNA from blocks 1 and 2 of C) shows that the lesion-specific enzyme does not affect the size distribution of the unirradiated DNA. Lane 3 (DNA from block 3 of C) shows that radiation-induced DSBs reduce the size of the DNA, and lane 4 (DNA from block 4 of C) shows that action of the lesion-specific enzyme results in a de novo DSB at the site of each clustered damage, further reducing the size of the DNA.
Figure 1.

Determination of clustered lesions. (A) DNA remains unirradiated (Left) or is exposed to ionizing radiation (Right). (B) Unirradiated DNA is unchanged in size (left oval), whereas radiation produces DSBs and clustered lesions containing damaged bases or abasic sites at approximately equal frequencies (right oval). (C) Treatment of unirradiated DNA with a lesion-specific enzyme has little or no effect on the size of the DNA molecules (block 2 relative to block 1). Irradiated DNA contains both DSBs (which reduce the size of the DNA) and clustered lesions (which do not reduce DNA size) (block 3); however, lesion-specific enzyme treatment of irradiated DNA (through release of damaged bases and AP endonuclease action) generates de novo DSBs at cluster sites (block 4). (D) DNA molecules from experimental samples, along with molecular length standard DNAs (ML), are dispersed according to double-strand molecular length by agarose gel electrophoresis under neutral conditions. Ionizing radiation also produces isolated DNA damages (not shown) that lesion-specific enzyme treatment converts to SSBs but not to DSBs.
Fig. 2 shows a representative neutral agarose gel for damage cluster analysis. Lanes 1–10 contain T7 DNA exposed to 0–50 Gy of 50-kVp x rays, and 11–20, T7 DNA exposed to 0–10 Gy of 137Cs γ rays. The samples are paired at each dose, with the first of each pair not treated with enzyme, and the second, treated with Fpg protein. Inspection of lanes 1, 3, 5, 7, and 9 clearly shows that x irradiation alone induces DSBs; likewise, lanes 11, 13, 15, 17, and 19 show induction of DSBs by γ rays. Treatment of the unirradiated DNA with Fpg protein (lanes 2 and 12) indicates the presence of very few, if any, clustered sites in unirradiated isolated T7 DNA. However, inspection of DNAs treated with Fpg protein after irradiation—for x rays, lanes 4, 6, 8, and 10, and for γ rays, lanes 14, 16, 18, and 20—clearly shows that this enzyme induces de novo DSBs, compared with those induced by the radiation alone. These de novo DSBs represent sites of clustered damage revealed by Fpg protein treatment.
Figure 2.

Electronic image of a neutral unidirectional pulsed-field agarose gel for damage cluster analysis. Lanes 1–10 contain T7 DNA exposed to 0–50 Gy of 50-kVp x rays, and lanes 11–20, T7 DNA exposed to 0–10 Gy of 137Cs γ rays. The samples are paired, with the first of each pair not treated with enzyme, and the second, treated with Fpg protein. M lanes contain molecular length standards (λ DNA and a HindIII digest of λ DNA); unirradiated T7 DNA was also used as a length standard in calculating the DNA dispersion function.
Fig. 3 shows quantitative data resulting from such an analysis. Irradiation of isolated DNA with 137Cs γ rays produces at least four classes of clustered damages. Fig. 3A depicts a frank DSB, defined functionally as two or more SSBs on opposite strands within ≈10–20 bp (10, 11), and the yields of DSBs induced by irradiation of T7 DNA in solution under our conditions. Upon electrophoresis in nondenaturing conditions, DSBs decrease the average length of a population of DNA molecules; such changes in average molecular length can be quantitated by number-average length analysis (21). Data are presented on a log–log plot to display both the full range of cluster yields and doses for analysis of mechanism(s) of cluster production.
Figure 3.

Schematic representation of the configuration of clustered lesion classes in DNA and dose responses for their induction in isolated T7 DNA by exposure to γ rays. (A) DSBs. (B) Abasic clusters (abasic site opposite SSB, upper diagram, or abasic site, lower diagram) converted to a DSB by Nfo protein digestion. (C) Oxidized purine clusters (oxidized purine opposite SSB, upper diagram, or oxidized purine, lower diagram) converted to a DSB by treatment with Fpg protein. (D) Oxidized pyrimidine clusters (oxidized pyrimidine opposite SSB, upper diagram, or oxidized pyrimidine, lower diagram) converted to a DSB by treatment with Nth protein. In B–D, levels of radiation-induced DSB are subtracted. Clusters of oxidized purine opposite oxidized pyrimidine, and of multiple lesions on opposing strands are not considered. Symbols: small, results of 2–10 individual gels; large, averages; A–C, least-squares lines fit on a log–log plot of the averages of the site frequencies as a function of the doses; D, least-squares line for 0- to 5-Gy data.
In the absence of further treatment and under neutral conditions, none of the other clustered lesions (Fig. 3 B–D) result in a decrease of the length of double-stranded DNA molecules. Although the frequencies of individual lesions composing the clusters may be measured by treating DNA with a lesion-specific endonuclease, followed by electrophoresis under denaturing conditions (see, e.g., refs. 26–28), this measurement yields the total lesion frequency, regardless of its configuration as an isolated or clustered damage.
To determine cluster frequencies, we combined lesion-specific DNA cleavage with subsequent analysis by quantitative neutral agarose gel electrophoresis and electronic imaging. Fig. 3B shows that abasic clusters—a damage cluster containing at least one abasic site—are produced by γ-irradiation (1–100 Gy) of isolated T7 DNA, and can be measured by treatment with E. coli Nfo protein, which cleaves the phosphodiester backbone principally (Table 1) at abasic sites. Similarly, clustered lesions containing at least one Fpg protein site (principally oxidized purines) (Fig. 3C) are converted to frank DSBs by digestion with Fpg protein, which has glycosylase activity and nicks at apurinic sites by β-lyase activity. Likewise, radiation-induced clusters containing at least one Nth protein site [principally oxidized pyrimidines (Table 1), Fig. 3D] are revealed by treatment with that enzyme. Clusters whose recognition requires cleavage by two glycosylases were not measured routinely (e.g., an oxidized purine opposite an oxidized pyrimidine, with no other sites cleavable by these enzymes or SSBs nearby).
We used nonradioscavenging conditions (dilute DNA in phosphate buffer) to maximize damage yields and decrease the irradiation times at our 137Cs γ source for the largest doses from hours to minutes. The range of ionizing radiation-induced lesions depends strikingly on the level of scavengers, with SSBs varying as much as 500-fold with high to low scavenger concentrations (29). Measurement of SSBs in the DNA samples shown in Fig. 3 allows us to assess the level of scavengers in our DNA solutions and to compare with the results of others. Milligan et al. (29) obtained 66 SSBs per Mb per Gy in the absence of scavengers, and ≈0.1 SSB per Mb per Gy in highly radioprotective solution. Prise et al. (30) obtained 0.34 SSB per Mb per Gy by irradiating DNA in Tris, indicating the radioprotective nature of this solution. Brake (31) obtained 55–67 total and 30 frank breaks for DNA irradiated in phosphate buffer, and Chen and Sutherland (32) obtained 36 frank breaks under similar conditions. In phosphate buffer, we obtained 57 total breaks (and calculated 32 frank breaks), in good agreement with previous data. Similarly, the yields of DSBs depend strongly on the scavenger levels; the yields of Prise et al. of 0.015 DSB per Mbp per Gy reflect their highly radioprotective conditions, and ours of 2.4 DSBs per Mbp per Gy, the result of the low level of scavengers in our solutions. Similar DSB yields (2.1 DSBs per Mbp per Gy) were obtained by Chen and Sutherland (32). We also measured DSBs and other clustered damages in T7 DNA irradiated with γ rays under moderately radioprotective solution (10 mM Tris, pH 8) and obtained yields of 0.07, 0.03, 0.03, and 0.08 site per Mbp per Gy for DSBs, oxidized pyrimidine clusters, abasic clusters, and oxidized purine clusters, respectively (data not shown), indicating much lower yields of all damages in the presence of scavengers.
The observation that the log–log plots in Fig. 3 of the DSB frequency vs. the radiation dose fall close to straight lines implies that the frequency of breaks is proportional to some power of the dose and hence can be described by the equation:
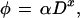 |
3 |
where φ is the frequency of DSBs induced either by irradiation alone (Eq. 1) or by irradiation followed by enzymatic treatment (Eq. 2), D is the radiation dose, α is a proportionality constant, and x is an exponent indicating the degree of the power law relationship. Experimentally, x is determined from the slope of the log–log plot from the equation:
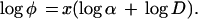 |
4 |
The experimental result that x is close to unity for all the results presented in Fig. 3 (DSBs, 1.00; abasic clusters, 1.05; Fpg sites/oxidized purine clusters, 1.05; Nth protein sites/oxidized pyrimidine clusters, initial slope 0.94) indicates a direct proportionality between cluster lesion frequency and dose—i.e., φ = αD and thus each cluster we measure results from a single radiation track. The relative frequencies of various cluster sites per unit dose, α, are characterized on the log–log plots of Fig. 3 by the vertical displacement of the straight lines (see below).
In addition to frank strand breaks and damaged bases, ionizing radiation also produces at least three kinds of abasic sites, with only about 10% of the sites resulting from oxidation of deoxyribose being “regular” abasic sites (the same as those produced by DNA glycosylases). The rest are oxidized abasic sites, both 4′ and (less likely) 2′, with a minor fraction of 1′ sites (33). Because Nfo protein cleaves oxidized abasic sites efficiently, and regular abasic sites fairly well (34), it is likely that the abasic clusters we measure contain regular as well as oxidized abasic sites. Since Fpg protein is reported to cleave oxidized abasic sites poorly (34), the clusters revealed by Fpg cleavage probably contain largely oxidized purine sites, with a minority of radiation-induced abasic sites. The Nth protein cleaves regular abasic sites well, but seems to cleave oxidized abasic sites poorly (34); thus the Nth-site clusters probably contain principally oxidized pyrimidine sites and ionizing radiation-induced regular abasic sites. Comparison of the cluster yields in Fig. 3 at a constant dose (e.g., 1 Gy) shows that the frequencies of the different cluster classes produced by γ-irradiation of DNA in phosphate buffer vary: normalized to the frequency of DSBs as 1, the ratios are 2 oxidized purine clusters to 1.5 abasic clusters to 0.56 oxidized pyrimidine clusters.
Because the Nth and Fpg proteins have multiple substrates (see Table 1), clusters revealed by treatment with these enzymes will include all substrate lesions in clusters that are recognized and DNA nicked under our conditions. In addition to the simple configurations shown, clustered lesions (including DSBs) may include other nearby damages. In model systems, clusters containing very closely spaced opposing damages are cleaved poorly or not at all by lesion-recognizing enzymes, and the identity of the constituent partners and opposing bases can determine susceptibility to cleavage (15–17). Additionally, in nondenaturing electrophoresis, opposing lesions separated by more than ≈10–20 bp would not be expected to produce a DSB and hence under our conditions would not be detected as a cluster. Thus, the cluster levels we measure may underestimate the total frequencies of multiple lesion sites that produce biological effects ascribed to clustered lesions. We also investigated whether lower energy photons produce clustered lesions. We found that 50 kVp x rays induced all four cluster classes in isolated DNA (See Fig. 2), apparently producing somewhat different ratios of frequencies of the classes (data not shown) than are produced by γ rays.
Fig. 3 shows that irradiation of DNA in solution induces clustered damages. However, because the internal cellular milieu contains radical scavengers, ionizing radiation induces lower damage yields in intact cells than in isolated DNA (35). However, the spatial condensation of DNA within cells compared with isolated DNA in solution might increase the production of clustered lesions. We asked whether measurable levels of damage clusters were formed in cells irradiated with low doses of high LET radiation. Fig. 4 shows that irradiation of human cells with 0–0.70 Gy of 1-GeV per nucleon Fe26+ ions produces approximately comparable levels of DSBs and oxidized pyrimidine clusters. Thus, these data indicate that such clusters are formed at significant levels in human cells, even at low doses corresponding to cell survival >90%. Preliminary results indicate that low LET radiation (137Cs γ rays and 50-kVp x rays) also produce at least one cluster class (oxidized purine clusters, recognized by Fpg cleavage) at yields comparable to DSBs (data not shown).
Figure 4.

Induction of DSBs and oxidized pyrimidine clusters in human cells by low doses of 1 GeV/atomic mass unit Fe26+ nuclei, normalized to the average frequency obtained for DSBs at 0.35 Gy (taken as 100%). Symbols: small, individual data (● and ♦, DSBs; ▴ and ▾, net Nth protein (oxidized pyrimidine) clusters; large, averages (○, DSBs; □, net oxidized pyrimidine clusters). Least-squares lines are shown.
Closely spaced abasic sites in DNA have also been detected by a fluorescence energy transfer method (36), but the limit of sensitivity of ≈1 per 17,000 bp would correspond to high radiation doses (≈400 Gy, cf. Fig. 3). Irradiation of plasmid DNA with 900–10,000 Gy of neutrons produced short DNA fragments consistent with clustered DSBs (37). Thermal denaturation and S1 nuclease analysis of γ-irradiated λ DNA suggested production of bulky lesions (38). S1 nuclease and gamma endonuclease treatment of λ DNA irradiated with 2,000–8,000 Gy of γ rays suggested the close proximity of unpaired DNA regions and base damages (39), and S1 analysis of human cells exposed to 100 Gy of γ rays indicated closely spaced damages, probably nicks and gaps (40). At these high doses, clusters could include sites resulting from multiple independent radiation hits. Lam and Reynolds (41) used Micrococcus luteus endonuclease treatment of mammalian DNA to show that ultraviolet radiation apparently produces clustered pyrimidine dimers at extremely low frequencies at physiological doses.
Clustered lesions are expected to constitute poorly repairable lesions that could produce mutations, induce inaccurate transcription, or constitute persistent lesions even in cells exposed to low levels of radiation. The mechanisms of repair of lesion clusters in cells are not yet clear, although studies with small oligonucleotides suggest possible pathways (15, 16, 42). In the cell, clusters may be subject to the same rejoining signals and pathways as are frank DSBs. However, if one member of a cluster were repaired independently of opposing lesions, the same mechanisms that deal with isolated damages might prevail (43, 44). Further, the identity of the individual lesion in the cluster may dictate the repair path and susceptibility to repair by that path, and thus the severity of biological consequences of the cluster.
Our results show that clustered damages involving abasic sites and oxidized bases are induced in isolated DNA by ionizing radiation, both photons and particle radiation, and that such clusters constitute about 80% of the total complex damages, with DSBs being only about 20%. We show that high LET radiation produces oxidized pyrimidine clusters in human cells. Our approach also provides a foundation for correlating clustered lesions induced by ionizing radiation in cells, tissues, or organisms with biological effects including survival, mutation, and oncogenesis.
Acknowledgments
We thank Dr. B. Demple (Harvard School of Public Health) for the endonuclease IV and Drs. J. Sutherland, B. Demple, and R. Setlow for comments on the manuscript. This work was supported by the Office of Biological and Environmental Research of the U.S. Department of Energy, the Space Radiation Health Program of the U.S. National Aeronautics and Space Administration, and by the Centre National de la Recherche Scientifique and Electricité de France, Contrat Radioprotection.
Abbreviations
- DSB
double-strand break
- LET
linear energy transfer
- Mb and Mbp
megabase and megabase pair (one million bases or base pairs), respectively
- SSB
single-strand break
References
- 1.Bissell M J, Warner H W, Berget S M, Fry R J M, Hanawalt P C, Kastan M, Kornberg A, Lutze-Mann L, Souza K A, Ullrich R, et al. Modeling Human Risk: Cell and Molecular Biology in Context. Univ. of California, Berkeley: Lawrence Berkeley National Laboratory; 1997. [Google Scholar]
- 2.Yang, T. C., George, K., Johnson, A. S., Durante, M. & Fedorendo, B. S. (1997) Radiat. Res.148 (5 Suppl.), S17–S23. [PubMed]
- 3.Tucker J D, Tawn E J, Holdsworth D, Morris S, Langlois R, Ramsey M J, Kato P, Boice J D, Jr, Tarone R E, Jensen R H. Radiat Res. 1997;148:216–226. [PubMed] [Google Scholar]
- 4.Bigbee W L, Jensen R H, Veidebaum T, Tekkel M, Rahu M, Strengrevics A, Auvinen A, Hakulinen T, Servomaa K, Obrams G I, Boice J D., Jr Radiat Res. 1997;147:215–224. [PubMed] [Google Scholar]
- 5.Fry R J, Grosovsky A, Hanawalt P C, Jostes R F, Little J B, Morgan W F, Oleinick N L, Ullrich R L. Radiat Res. 1998;150:695–705. [PubMed] [Google Scholar]
- 6.Wallace, S. S. (1998) Radiat. Res.150 (5 Suppl.), S60–S79. [PubMed]
- 7.Ward J F. Radiat Res. 1985;104:S103–S111. [PubMed] [Google Scholar]
- 8.Ward J F. Int J Radiat Biol. 1994;66:427–432. doi: 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- 9.Le X C, Xing J Z, Lee J, Leadon S A, Weinfeld M. Science. 1998;280:1066–1069. doi: 10.1126/science.280.5366.1066. [DOI] [PubMed] [Google Scholar]
- 10.Van Der Schans G P. Int J Radiat Biol. 1978;33:105–120. doi: 10.1080/09553007814550011. [DOI] [PubMed] [Google Scholar]
- 11.Olive P L. Radiat Res. 1998;150,(Suppl.):S42–S51. [PubMed] [Google Scholar]
- 12.Ward J F. Radiat Res. 1981;86:185–195. [PubMed] [Google Scholar]
- 13.Goodhead D T. Int J Rad Biol. 1994;65:7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 14.Ward J F. Radiat Res. 1995;142:362–368. [PubMed] [Google Scholar]
- 15.Chaudhry M A, Weinfeld M. J Biol Chem. 1997;272:15650–15655. doi: 10.1074/jbc.272.25.15650. [DOI] [PubMed] [Google Scholar]
- 16.Chaudhry M A, Weinfeld M. J Mol Biol. 1995;249:914–922. doi: 10.1006/jmbi.1995.0348. [DOI] [PubMed] [Google Scholar]
- 17.Harrison L, Hatahet Z, Purmal A A, Wallace S S. Nucleic Acids Res. 1998;26:932–941. doi: 10.1093/nar/26.4.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutherland B M, Bennett P V, Sutherland J C. In: Methods in Molecular Biology: DNA Repair Protocols. Henderson D, editor. Totowa NJ: Humana; 1999. pp. 183–202. [Google Scholar]
- 19.Sutherland B M, Bennett P V, Sutherland J C. Anal Biochem. 1996;239:53–60. doi: 10.1006/abio.1996.0290. [DOI] [PubMed] [Google Scholar]
- 20.Sutherland J C, Lin B, Monteleone D C, Mugavero J, Sutherland B M, Trunk J. Anal Biochem. 1987;163:446–457. doi: 10.1016/0003-2697(87)90247-8. [DOI] [PubMed] [Google Scholar]
- 21.Freeman S E, Blackett A D, Monteleone D C, Setlow R B, Sutherland B M, Sutherland J C. Anal Biochem. 1986;158:119–129. doi: 10.1016/0003-2697(86)90599-3. [DOI] [PubMed] [Google Scholar]
- 22.Sutherland J C, Monteleone D C, Mugavero J H, Trunk J. Anal Biochem. 1987;162:511–520. doi: 10.1016/0003-2697(87)90427-1. [DOI] [PubMed] [Google Scholar]
- 23.Zeitlin C, Heilbronn L, Miller J. Radiat Res. 1998;149:560–569. [PubMed] [Google Scholar]
- 24.Lobrich M, Ikpeme S, Kiefer J. Radiat Res. 1994;138:186–192. [PubMed] [Google Scholar]
- 25.Gardiner K, Laas W, Patterson D. Somatic Cell Mol Genet. 1986;12:185–195. doi: 10.1007/BF01560665. [DOI] [PubMed] [Google Scholar]
- 26.Setlow R B, Carrier W L. Nat New Biol. 1973;241:170–172. doi: 10.1038/newbio241170a0. [DOI] [PubMed] [Google Scholar]
- 27.Paterson M C, Setlow R B. Proc Natl Acad Sci USA. 1972;69:2927–2931. doi: 10.1073/pnas.69.10.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pouget J-P, Ravanat J-L, Douki T, Richard M-J, Cadet J. Int J Radiat Biol. 1999;75:51–58. doi: 10.1080/095530099140807. [DOI] [PubMed] [Google Scholar]
- 29.Milligan J R, Aguilera J A, Nguyen T-T D, Ward J F, Kow Y W, Cunningham R P. Radiat Res. 1999;151:334–342. [PubMed] [Google Scholar]
- 30.Prise K M, Pullar C H, Michael B D. Carcinogenesis. 1999;20:905–909. doi: 10.1093/carcin/20.5.905. [DOI] [PubMed] [Google Scholar]
- 31.Brake R J. Ph.D. dissertation. Knoxville: Univ. Tennessee; 1979. [Google Scholar]
- 32.Chen C Z, Sutherland J C. Anal Biochem. 1989;10:318–326. [Google Scholar]
- 33.von Sonntag C. The Chemical Basis of Radiation Biology. London: Taylor and Francis; 1987. [Google Scholar]
- 34.Haring M, Rudiger H, Demple B, Boiteux S, Epe B. Nucleic Acids Res. 1994;22:2010–2015. doi: 10.1093/nar/22.11.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oleinick N L, Balasubramaniam U, Xue L, Chiu S. Int J Radiat Biol. 1994;66:523–529. doi: 10.1080/09553009414551561. [DOI] [PubMed] [Google Scholar]
- 36.Makrigiorgos G M, Chakrabarti S, Mahmood A. Int J Radiat Biol. 1998;74:99–109. doi: 10.1080/095530098141762. [DOI] [PubMed] [Google Scholar]
- 37.Pang D, Berman B L, Chasovskikh S, Rodgers J E, Dritschilo A. Radiat Res. 1998;150:612–618. [PubMed] [Google Scholar]
- 38.Martin-Bertram H, Rumpf E, Winkler C. Radiat Environ Biophys. 1983;21:305–307. doi: 10.1007/BF01341467. [DOI] [PubMed] [Google Scholar]
- 39.Kohfeldt E, Bertram H, Hagen U. Radiat Environ Biophys. 1988;27:123–132. doi: 10.1007/BF01214602. [DOI] [PubMed] [Google Scholar]
- 40.Legault J, Tremblay A, Ramotar D, Mirault M-E. Mol Cell Biol. 1997;17:5437–5452. doi: 10.1128/mcb.17.9.5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam L H, Reynolds R J. Mutat Res. 1986;166:187–198. doi: 10.1016/0167-8817(86)90017-9. [DOI] [PubMed] [Google Scholar]
- 42.Harrison L, Hatahet Z, Purmal A A, Wallace S S. Nucleic Acids Res. 1998;26:932–941. doi: 10.1093/nar/26.4.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frosina G, Fortini P, Rossi O, Carrozino F, Raspaglio G, Cox I S, Lane D P, Abbondandolo A, Dogliotti E. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 44.Demple B, Johnson A, Fung D. Proc Natl Acad Sci USA. 1986;83:7731–7735. doi: 10.1073/pnas.83.20.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boiteux S, O'Conner T R, Lederer F, Gouvette A, Laval J. J Biol Chem. 1990;265:3916–3922. [PubMed] [Google Scholar]
- 46.Boiteux S, Gajewski E, Laval J, Dizdaroglu M. Biochemistry. 1992;31:106–110. doi: 10.1021/bi00116a016. [DOI] [PubMed] [Google Scholar]
- 47.Tchou J, Kasai H, Shibutani S, Chung M-H, Laval J, Grollman A P, Nishimura S. Proc Natl Acad Sci USA. 1991;88:4690–4694. doi: 10.1073/pnas.88.11.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hatahet A, Kow W Y, Purmal A A, Cunningham R P, Wallace S S. J Biol Chem. 1994;269:18814–18820. [PubMed] [Google Scholar]
- 49.Jurado J, Saparbaev M, Matray T J, Greenberg M M, Laval J. Biochemistry. 1998;37:7757–7763. doi: 10.1021/bi972982z. [DOI] [PubMed] [Google Scholar]
- 50.Asahara H, Wistort P M, Bank J F, Bakerian R H, Cunningham R P. Biochemistry. 1989;28:4444–4449. doi: 10.1021/bi00436a048. [DOI] [PubMed] [Google Scholar]
- 51.Dizdaroglu M, Laval J, Boiteux S. Biochemistry. 1993;32:12105–12111. doi: 10.1021/bi00096a022. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, Kim E Y, Demple B. J Biol Chem. 1998;273:28837–28844. doi: 10.1074/jbc.273.44.28837. [DOI] [PubMed] [Google Scholar]