

Abstract
The major DNA photoproduct in UV-irradiated Bacillus subtilis spores is the thymine dimer named spore photoproduct (SP, 5-(α-thyminyl)-5,6-dihydrothymine). The SP lesion has been found to be efficiently repaired by SP lyase (SPL) a very specific enzyme that reverses the SP to two intact thymines, at the origin of the great resistance of the spores to UV irradiation. SPL belongs to a superfamily of [4Fe-4S] iron-sulfur enzymes, called “Radical-SAM.” Here, we show that the single substitution of cysteine 141 into alanine, a residue fully conserved in Bacillus species and previously shown to be essential for spore DNA repair in vivo, has a major impact on the outcome of the SPL-dependent repair reaction in vitro. Indeed the modified enzyme catalyzes the almost quantitative conversion of the SP lesion into one thymine and one thymine sulfinic acid derivative. This compound results from the trapping of the allyl-type radical intermediate by dithionite, used as reducing agent in the reaction mixture. Implications of the data reported here regarding the repair mechanism and the role of Cys-141 are discussed.
Because of their chemical reactivity with regard to biological macromolecules (proteins, lipids, DNA, and others), free radicals are responsible for major damages when produced in large amounts within living cells (1). On the other hand, there has been an explosive growth in the number of enzymatic reactions found to proceed by mechanisms involving free radicals as intermediates (2). This chemistry implies that enzymes have evolved delicate mechanisms to generate reaction-initiating primary radicals from stable substrates and to control these highly reactive species within the active sites so that they can undergo subsequent steps in the enzymatic process and do not get inactivated through radical coupling or harmful reactions with the protein itself or with oxygen, for example. This represents a fascinating issue in enzymology, and many mysteries of this chemistry remain to be unraveled.
The spore photoproduct lyase enzyme (SPL)4 studied here is a nice illustration of how a radical chemistry is used for the cleavage of unreactive C–H and C–C bonds to repair DNA and how the control of intermediate radicals can be drastically lost by a single amino acid substitution. During exposure to UV light of bacterial spores, a dormant form produced by some bacteria such as Bacillus and Clostridium species, dimerization of adjacent thymine bases in DNA results in the specific formation of 5-(α-thyminyl)-5,6-dihydrothymine, the so-called spore photoproduct SP (Scheme 1A), a lesion that can block replication and transcription and is thus potentially lethal, mutagenic, and cytotoxic (3,4). Sporulating bacteria are extremely resistant to UV radiation, because they use SPL early in the spore germination cycle, to very efficiently and specifically revert SP to two unmodified thymines (Scheme 1A) (5–7).
SCHEME 1.

A, UV-dependent formation and SPL-catalyzed repair of SP. The wavy line represents the nucleic acid chain. B, proposed chemical mechanism for SP lyase DNA repair (12). AdoMet is S-adenosylmethionine, Ado· is 5′-deoxyadenosyl radical generated from reductive cleavage of AdoMet by the reduced [4Fe-4S]+1 cluster of SPL.
The SPL enzymatic mechanism has been addressed only recently, when pure preparations of SPL became available, and the following data are consistent with a radical mechanism initially proposed by T. P. Begley, on the basis of model compounds (8). First, SPL is a “Radical-SAM (S-adenosyl-methionine)” iron-sulfur enzyme (9). This class of enzymes uses a 5′-deoxyadenosyl radical, produced from reductive cleavage of AdoMet, to initiate the reaction (Scheme 1B) (10, 11). Second, experimental evidence of label transfer to AdoMet from SP in DNA specifically 3H-labeled at C-6 indirectly shows that repair is initiated by abstraction of the C-6 hydrogen atom of SP by the 5′-deoxyadenosyl radical (12) (Scheme 1B). The subsequent steps have been less characterized, but it is likely that C–C bond homolytic cleavage follows to give an allyl-type radical together with a first repaired thymine. The last step of the reaction has been proposed to involve hydrogen atom transfer from 5′-deoxyadenosine (AdoH) to the thymine monomer radical (12). Preliminary experiments have indeed shown some label transfer from [5′-3H]AdoMet into thymine monomers, using a DNA substrate containing the SP lesion, thus supporting the mechanism shown in Scheme 1B (12, 13).
In this report, we provide evidence for the involvement of the allyl-type radical during in vitro B. subtilis SPL-dependent repair reaction. We took advantage of using pure preparations of a dinucleoside monophosphate substrate named SPTpT in the following (Scheme 2), in one hand, and, in another hand, an SPL mutant in which Cys-141, a residue fully conserved in Bacillus species and previously shown to be essential for spore DNA repair in vivo, has been changed into alanine (14). We were indeed intrigued by this observation, because Cys-141 is not a ligand of the [Fe-S] cluster present in the active site, in contrast to cysteines 91, 95, and 98 of the CysXXXCysXXCys motif characteristic of Radical-SAM enzymes. Implications of the data reported here regarding the reaction mechanism and the role of Cys-141 are discussed.
SCHEME 2.

SPTpT formation and repair. SPL is the spore photoproduct lyase enzyme from B. subtilis; DPA is dipicolinic acid.
EXPERIMENTAL PROCEDURES
Materials—Strains: Escherichia coli DH5α was used for routine DNA manipulations. E. coli Tuner (DE3) was used for enzymes overexpression. Enzymes, oligonucleotides, and culture media were purchased from Euromedex (Strasbourg, France). Plasmid DNA purification kit, Qiaprep™, was from Qiagen. DNA sequencing was performed by Genome Express (Grenoble, France).
Cloning and Construction of Mutant C141A SPL-overexpressing Plasmid—The plasmid containing the SplB gene, encoding wild-type N-terminal 6His-tagged SPL (pT7-SPL6H), was obtained as previously described (7). A site-directed mutagenesis was performed to change cysteine 141 into an alanine using QuikChange site-directed mutagenesis kits from Stratagene according to the manufacturer's protocol. The mutant plasmid was entirely sequenced to ensure that no error was introduced during PCR reaction. The plasmid was then named pT7-SPLC141A.
Protein Expression—For both wt SPL and SPL-C141A, expression was conducted in LB medium in the E. coli Tuner (DE3), and proteins were purified under aerobic conditions as previously described (7).
Iron and Sulfide Binding to WT and C141A B. subtilis SPL—[Fe-S] cluster reconstitutions of wt and C141A B. subtilis SPL were carried out under strictly anaerobic conditions in a glove box containing <2 ppm O2 as previously described (7).
Production of SPTpT Substrates—The spore photoproduct of the dinucleoside monophosphate thymidylyl-(3′,5′)thymidine was prepared as previously described (7).
SP-Lyase Activity—To assay SPL activity we followed the same procedure as for Wild-type SPL (7), using 2.5 mm DTT, 1.5 mm dithionite, and 1.5 mm AdoMet. At each time point (0, 15 30, 60, 120, 215, and 280 min) 20 μl and 10 μl of the solution were transferred into Eppendorf tubes, and the reaction was stopped by flash-freezing in liquid nitrogen. Conversion of the spore photoproduct (SPTpT) into the unmodified dinucleoside monophosphate (TpT) and other products in SPL-treated samples was quantified by HPLC coupled to tandem mass spectrometry (HPLC-MS/MS), using the same conditions as for samples treated by wt SPL (7).
HPLC-MS Analysis—HPLC-MS/MS analyses were carried out on an API 3000 tandem mass spectrometer associated to a Series 1100 Agilent chromatography system. An Uptisphere ODB reversed-phase octadecylsilyl silica gel column (3-μm particle size, 150 × 2 mm inner diameter, Interchim, Montluçon, France) was used in all analyses. A gradient of acetonitrile in 2 mm aqueous triethylammonium acetate was used for most analyses, which were carried out in the negative electrospray ionization mode. These conditions were applied for TpT (retention time (Rt) 26.6 min), SPTpT (Rt 19.9 min), the SO2 derivatives of TpT (TpTSO2 Rt 21.7 min and TSO2pT Rt 22.3 min), the hydroxylated derivatives of TpT (TpTOH Rt 25.6 min and TOHpT Rt 24.3 min). Experiments were also carried out in the positive electrospray ionization mode. For that purpose, acetate ammonium was used as the HPLC buffer. Under these conditions, the retention times were the following: TpTSO2 18.3 min, TSO2pT 19.2 min, 5′-deoxyadenosyl (AdoH) 5.1 min, and AdoMet 3.1 min. The chromatograms were recorded either in the MS1 (full spectra without fragmentation), SIM (selected ion monitoring without fragmentation), MS2 (full spectra of fragment arising from a specific parent ion), and MRM (multiple reaction monitoring, specific for transition corresponding to compounds of interest) modes.
SAM Reductase Activity—The cleavage of AdoMet was monitored by HPLC from the formation of AdoH as previously described (15).
Protein Analysis—Protein concentration (by monomer) was determined by the method of Bradford (16). Protein-bound iron (17) and labile sulfide (18) concentrations were determined according to standard procedures.
Preparation and Light-induced Decomposition of Thiophenyl Derivatives of TpT—5-(Thiophenylmethyl)-2′-deoxyuridylyl-(3′,5′)thymidine (TSΦpT) and thymidylyl-(3′,5′)-5-(thiophenylmethyl)-2′-deoxyuridine (TpTSΦ) were prepared as previously described (19). In a first series of experiments, solutions containing 40 μm TpTSΦ or TSΦpT together with 1.5 mm Na2S2O4 were kept overnight in an anaerobic glove box. Samples were then exposed for 5 min to UV-C radiation (254 nm) emitted by a germicidal lamp. A second series of irradiations was performed with aerated solutions of either TpTSΦ or TSΦpT. Samples were then analyzed by HPLC-MS/MS without further treatment. Elution was monitored in the MS2 mode. Parent ions were set at m/z 609 and 561 in the negative mode and 611 in the positive mode.
UV-visible Absorption Spectroscopy—UV-visible absorption spectra were recorded with a Cary 1 Bio (Varian) spectrophotometer.
EPR—Reconstituted SPL-C141A (187 μm, 4.2 iron/protein) were reduced with 2 mm dithionite under anaerobic conditions for 20 min and frozen inside the glove box. X-Band EPR spectra were recorded on a Brucker Instruments ESP 300D spectrometer equipped with an Oxford Instruments ESR 900 flow cryostat (4.2–300 K). Spectra were quantified under non-saturating conditions by double integration against a 1 mm Cu-EDTA standard.
Mössbauer Spectroscopy—For Mössbauer measurements, reconstituted SPL-C141A (9.6 mg and 4.2 iron/protein) was prepared as described above in a final volume of 400 μl. The protein solution was transferred into a Mössbauer cup and frozen in liquid nitrogen. 57Fe-Mössbauer spectra were recorded at zero magnetic field on a spectrometer operating in constant acceleration mode using an Oxford cryostat that allowed temperatures from 1.5 to 300 K and a 57Co source in rhodium. Isomer shifts are reported relative to metallic iron at room temperature.
RESULTS
Cloning, Overexpression, and Purification of SPL C141A Variant—The pT7-SPL-C141A plasmid, obtained as described under “Experimental Procedures,” was used to transform E. coli Tuner (DE3) strain for production of a protein with a His tag at the N-terminal end and an alanine at position 141 in place of a cysteine. A His tag at this extremity is not detrimental to wild-type (wt) SPL enzyme activity (20). The SPL-C141A mutant protein could be obtained in pure form, with traces of protein-bound iron and acid-labile sulfide. Reconstitution of the [Fe-S] cluster yielded a SPL-C141A protein containing ∼3–3.5 iron and 3.5–4 sulfur atoms per monomer, in agreement with the presence of a [4Fe-4S] cluster. The UV-visible spectrum, with two broad bands at 330 and 420 nm, of the reconstituted mutant as well as the EPR spectrum of the dithionite-reduced protein, with a signal characteristic for a S = 1/2 [4Fe-4S]+1 species, were similar to those of wt SPL (supplemental Figs. S1 and S2). The [Fe-S] cluster was further investigated by Mössbauer spectroscopy using a protein sample (277 μm, 4.01 iron/monomer) reconstituted with 57Fe and sulfide as previously described in the case of wt SPL (7). The spectra recorded at 78 K and 4.2 K were dominated by a quadrupole doublet, the parameters of which (δ ∼ 0.44(1) mm.s-1, ΔEQ = 1.07(2) mm.s-1) clearly corresponded to a [4Fe-4S]2+ cluster, accounting for ∼73% of total Fe (supplemental Fig. S3). Extra iron was in the form of high spin ferrous iron, as observed in the case of wt SPL.
In Vitro SPTpT Repair Activity of the Wild-type SPL Enzyme—We assayed SPL activity using the SPTpT dimer, shown in Scheme 2, as a pure substrate. SPTpT is produced by UV irradiation of the unmodified dinucleoside monophosphate TpT. The methylene bridge in SP corresponds to the methyl group of the 3′-end thymine (7). This synthetic compound is identical to that released from DNA extracted from UV-irradiated spores as shown by their identical HPLC retention time and fragmentation mass spectrometry features (7). Use of this substrate facilitated monitoring of the repair reaction, because assay samples could be directly submitted to HPLC-MS/MS analysis at the end of the incubation time, for separation, identification, and quantitation of SPTpT and TpT (Scheme 2). We recently reported that this substrate is totally transformed into TpT under standard assay conditions, using wt SPL (7). For sake of comparison with the SPL-C141A enzyme, a standard highly reproducible wt SPL time-dependent repair experiment is shown in Fig. 1A. The assay mixture contained a pure preparation of reconstituted wt SPL (1 μm) together with 1.5 mm dithionite as a reducing agent, 1.5 mm AdoMet and 10 μm SPTpT in DTT-containing buffer at pH 8 under strict anaerobic conditions. The repair reaction was then assayed at time intervals by HPLC-MS/MS for its content in SPTpT and TpT. Fig. 1A shows that a quantitative conversion of SPTpT to TpT was achieved in <60 min.
FIGURE 1.

A, time-dependent enzymatic repair of SPTpT. SPTpT (10 μm) and SPL (1 μm) in 100 mm Tris-HCl, pH 8, containing 2.5 mm DTT, 1.5 mm dithionite, and 1.5 mm AdoMet anaerobically. SPTpT (•) and TpT (○) were separated by HPLC and quantitated at time intervals. B, time-dependent formation of AdoH (▪ ♦) and TpT (○). Conditions as in A in the presence (▪) or in the absence (♦) of SPTpT. AdoH and TpT are monitored by HPLC. C, time-dependent enzymatic of SPL-C141A on SPTpT. SPTpT (10 μm) and SPL-C141A (1 μm) in 100 mm Tris-HCl, pH 8, containing 2.5 mm DTT, 1.5 mm dithionite, and 1.5 mm AdoMet anaerobically. SPTpT (•), TpT (○), B(▪), and A (▴). SPTpT, TpT, A, and B were separated by HPLC and quantitated at time intervals. D, HPLC chromatogram showing the different reaction products after 15-min incubation. Retention times: SPTpT, 19.9 min; B, 21.7 min; A, 25.6 min; and TpT, 26.6 min.
In parallel, the reaction mixture was assayed for the production of AdoH, the product resulting from AdoMet-reductive cleavage. The results shown in Fig. 1B demonstrate that approximately one equivalent of AdoH with regard to the reaction TpT product was formed during repair. Further production of AdoH still took place after 60-min incubation reflecting the ability of the wt SPL enzyme to reductively cleave AdoMet in the absence of substrate, a property also reported by others (see Fig. 1B) (21,22). These data thus suggest that, at least with SPTpT as a substrate, the SPL enzyme reaction is not catalytic with regard to AdoMet and that AdoH does not serve as a hydrogen atom donor during the final step of the reaction, leading to TpT (see Scheme 1B). This has been confirmed by a labeling experiment, using an AdoMet preparation containing a partially deuterated 5′-methylene group, because no appreciable D incorporation into TpT, during SPTpT repair under standard conditions, could be observed. Buffer used in these in vitro experiments contained DTT, which could function as a hydrogen atom donor.
In Vitro SPTpT Repair Activity of the SPL-C141A Mutant Enzyme—The SPL-C141A enzyme was also competent for AdoMet reductive cleavage in the absence of substrate, under anaerobic conditions showing that the protein interacts with the AdoMet cofactor (data not shown). A time curve for SPL-C141A-dependent conversion of SPTpT, assayed under the conditions used for the wt SPL, is shown in Fig. 1C. The results clearly showed that the SPTpT substrate, corresponding to the peak eluted at 19.9 min (Fig. 1D), was totally consumed after a 30-min reaction but, in contrast to the wt SPL-catalyzed reaction, repaired TpT product (retention time: 26.6 min, Fig. 1D) yield did not exceed 10%. No SPTpT consumption could be detected when protein SPL-C141A or AdoMet was omitted from the reaction mixture (data not shown). This showed that SPTpT was indeed modified by SPL-C141A but not primarily reverted back to TpT with two unmodified thymines.
Samples were then analyzed by HPLC with MS1 monitoring to search for putative incomplete reaction products. Besides SPTpT and TpT, two additional peaks (A, 25.6 min; B, 21.7 min), which increased in intensity during reaction, were observed (Fig. 1, C and D). The molecular weight of the corresponding compounds was found by MS1 to be 562 and 610 for compounds A and B, respectively (Fig. 2A). These values are consistent with an hydroxylated derivative of TpT for compound A and with a SO2 adduct for compound B. Determination of their fragmentation mass spectra (Fig. 2, B and C, see comments below) allowed us to set up a specific and sensitive MRM HPLC-MS/MS method to monitor the time-dependent formation of products A and B upon conversion of SPTpT catalyzed by the SPL-C141A enzyme. The results are those shown in Fig. 1C. At the end of the reaction compound B (90% yield) was by far the major product, and very low amounts of compound A (1–2%) could be detected. In fact, this last product was also observed in tiny amounts in the case of the wt SPL-dependent reaction.
FIGURE 2.

MS1spectra of compounds A and B (panel A) and MS2 features of compound A (OH adduct, panel B) and B (SO2 adduct, panels C and D).
Identification of the Products of the Reaction Catalyzed by the SPL-C141A Enzyme—Further insights into the chemical structure of compounds A and B could be first obtained from the analysis of their MS2 fragmentation mass spectra (Fig. 2). In the negative mode, the fragmentation of unmodified dinucleoside monophosphate compounds is dominated by the loss of the base of the 5′-terminal nucleotide leading to the following fragments: the deprotonated free base and the dinucleoside monophosphate with a single base at the 3′-terminal nucleotide (23). Such a typical fragmentation is illustrated in the supplemental Fig. S4 with uracil/thymine dinucleoside monophosphates dUpT and TpdU. The same fragmentation pattern was unambiguously observed for compound A, with release of unmodified thymine (noted “Thy-H”) and formation of an abasic dinucleoside monophosphate with a single hydroxylated base at the 3′-terminal nucleotide (noted “M-Thy-H”) (Fig. 2B). These data show that compound A is a derivative of TpT with an hydroxyl group at the 3′-end thymine exclusively.
Characterization of compound B was more tedious. Indeed, its fragmentation spectrum recorded in the negative mode (Fig. 2C) was not conclusive, regarding the presence of the SO2 group on the 3′-end or 5′-end thymine: whereas a thymine fragment but no SO2-thymine derivative was observed, detection of a fragment possibly resulting from the loss of ThySO2 could also be explained by the presence of SO2 at the 5′-end base (Fig. 2C). In contrast, fragmentation MS under positive ionization made conclusions possible (Fig. 2D). Under these conditions, a typical fragmentation pattern for dinucleoside monophosphates is the release of the protonated 3′-end nucleotide together with the dehydrated 5′-end nucleoside (see supplemental Fig. S5 for the case of dUpT and TpdU). Fragmentation of product B followed this pattern with evidence for an unmodified dehydrated thymidine at the 5′-end (noted “dThd-H2O+H” in Fig. 2D) and a SO2-modified monophosphate thymidine at the 3′-end (noted “dTMPSO2+H” in Fig. 2D).
These data thus clearly demonstrated that the major product during transformation of SPTpT catalyzed by the mutant enzyme was a dinucleoside monophosphate in which the covalent link between the two bases has been cleaved and that a SO2 group was attached specifically to the base of the 3′-end nucleotide. In the next section we describe experiments aiming at showing, through a chemical approach, that the SO2 moiety comes from dithionite and that SO2 addition occurs at the methyl group of the modified thymine.
A Model Chemistry for the Formation of A and B—We reasoned that formation of B could arise from coupling between an intermediate dinucleoside monophosphate radical and an SO2 dithionite. A good candidate for the former is the carbon-centered allylic radical generated after the homolytic cleavage of the C–C bond, shown in Scheme 1B. To study such a reaction, we generated this allylic radical photochemically in the presence of dithionite and identified the reaction products by HPLC and MS as discussed under “Experimental Procedures.” The precursor was a synthetic dinucleoside monophosphate derivative in which a thiophenyl moiety is covalently attached to the methyl group of the thymine either at the 5′- (named TSΦpT) or the 3′- (named TpTSΦ) end (Scheme 3). This functionality is known to undergo homolytic cleavage of the sulfur-carbon bond during irradiation (19). In the presence of dithionite (1.5 mm) and the absence of oxygen, UV-C photolysis of TSΦpT and TpTSΦ (40 μm) each yielded a compound with an additional mass of 64 consistent with the addition of a SO2 group, as shown by MS (supplemental Figs. S6–S8). However, the two compounds were different in terms of both HPLC retention times (Fig. 3I) and mass spectrometry features (supplemental Figs. S6–S8). Only the reaction product of the TpTSΦ compound irradiated in the presence of dithionite exhibited properties perfectly identical to those of compound B produced during the repair of SPTpT by SPL-C141A. Interestingly, the reaction also produced a very small amount of compound A by an undefined mechanism.
SCHEME 3.

5-(Thiophenylmethyl)uracil dinucleoside monophosphates as precursors of the intermediate allylic radical. When the thiophenyl moiety SΦ is attached on the 5′-end base the compound is named TSΦpT. When SΦ is attached on the 3′-end base the compound is named TpTSΦ. The carbon-centered allylic radical is generated upon UV-C irradiation in the absence of oxygen through the homolytic cleavage of the C–S bond.
FIGURE 3.

HPLC comparison of the reaction products obtained upon repair of SPTpT by SPL-C141A and by photolysis of radical precursor (TpTSΦ and TSΦpT). Panel I, SO2 (*) and OH (**) TpT adducts obtained upon photolysis of TpTSΦ (solid line) and TSΦpT (dotted line), anaerobically in the presence of dithionite. Panel II, SO2 (B) and OH (A) TpT adducts obtained upon incubation of SPTpT with SPL-C141A in the presence of dithionite, anaerobically.
These data then unambiguously established a number of conclusions. First they confirm that compound B had the SO2 adduct at the 3′-end thymine and not at the 5′-end one. Second, they show that the addition occurred at the methyl carbon of the base and that dithionite was the source of SO2.
DISCUSSION
We earlier discovered that the SPTpT dimer (Scheme 2), identical to the dimer released from DNA extracted from UV-irradiated spores, behaves as a substrate of the spore photoproduct lyase (SPL) (7). Now, we show that with SPTpT as a substrate the enzyme reaction is not catalytic with regard to AdoMet and produces the repaired TpT compound and AdoH in stoichiometric amounts. This implies that the product radical intermediate, precursor of TpT, does not abstract a hydrogen atom from AdoH (Scheme 1B). Confirmation is provided from the lack of incorporation of deuterium into TpT when the reaction is carried out in the presence of deuterated AdoMet. The source of H atoms in this reaction has not been determined and will be discussed below. In contrast, in one study using irradiated DNA, SPL-dependent repair was found to be catalytic with regard to AdoMet, thus in agreement with the mechanism shown in Scheme 1B, in which AdoH is the H atom donor. Nevertheless, because SPTpT can be easily produced by UV irradiation of the unmodified dinucleoside monophosphate TpT in significant yields and in pure form and is a SPL substrate easy to monitor, we used it for mechanistic studies rather than an undefined preparation of irradiated DNA containing a mixture of lesions, with the spore photoproduct as a minor one (7). DNA from irradiated bacterial spores is another possible source of substrate but much less easily accessible.
Intrigued by the importance of Cys-141, a conserved cysteine in Bacillus SPLs, for spore survival under UV irradiation (14), we have investigated in detail the properties of the SPL-C141A mutant form and made a number of interesting unexpected observations.
Here, using in vitro experiments, we confirm that this cysteine residue plays an important function in the outcome of the repair reaction. Indeed, whereas SPTpT is still recognized by the mutant enzyme and transformed efficiently, the major product of the reaction is not the TpT dimer. HPLC analysis, mass spectrometry, and chemical model reactions, using photoreactive radical precursors, led us to unambiguously establish that the major product (>90% yield) of the reaction is a modified dinucleoside monophosphate containing one thymine and one thymine with a sulfinate moiety compound B (Scheme 4). We also demonstrate that the sulfinate group: (i) is attached to the methyl carbon of the base; (ii) is exclusively present in the thymine of the 3′-end of the dinucleoside monophosphate; and (iii) is derived from dithionite, the reducing agent present in the in vitro assay mixture. These results are interpreted by the mechanism shown in Scheme 4 for the repair of SPTpT catalyzed by the wt SPL and the mutant SPL-C141A enzyme.
SCHEME 4.

Our proposed mechanism for the repair of SPTpT catalyzed by either wt
SPL or SPL-C141A. After reductive cleavage of AdoMet,
Ado· abstracts one of the H atom at C6 carbon of the
saturated base. The corresponding 6-yl radical is converted to the
carbon-centered allylic radical at the 3′-end base. Whereas wt SPL
allows generation of unmodified thymines, in the case of SPL-C141A, this
allylic radical is trapped by dithionite derivating radical
(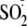 )
leading to compound B (90%).
)
leading to compound B (90%).
Because both enzymes are able to generate products lacking the initial
covalent bond between the bases it is likely that the first steps of the
reaction, during which the cross-link is broken, are the same and that the
substitution of cysteine 141 into alanine is not detrimental to substrate
binding in the active site of the protein and reacting with the cofactor. This
is consistent with the fact that the SPL-C141A enzyme chelates a [4Fe-4S]
cluster with high yield, as shown by Mössbauer spectroscopy, and
catalyzes the reductive cleavage of AdoMet in the absence of substrate, as
does wt SPL (our results and Refs.
21,
22). Thus, in both cases the
reaction starts with the reductive cleavage of AdoMet by the reduced cluster
generating the 5′-deoxyadenosyl radical, which then abstracts one of the
H atoms at the methylene C-6 position. This primary radical undergoes a
β-scission leading to the homolytic cleavage of the C–C bond
linking the two thymines generating an allylic radical. Differences between
the two enzymes appear in the last step of the reaction
(Scheme 4). Our data suggest
that the substitution of Cys-141 into alanine is responsible for a deficient
control/protection of the allylic radical intermediate, which then is
efficiently trapped by the
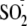 .
radicals generated in solution by dithionite, through a radical-radical
coupling reaction producing the sulfinate compound
(Scheme 4).
.
radicals generated in solution by dithionite, through a radical-radical
coupling reaction producing the sulfinate compound
(Scheme 4).
This mechanism implies two key radical intermediates. The involvement of the first one, derived from H atom abstraction at C-6 methylene by the 5′-deoxyadenosyl radical Ado·, has been demonstrated by DNA labeling experiments, using the wt enzyme, previously reported (12). The involvement of the second one, the allylic radical precursor of the repaired product, is now indirectly proven by the experiments reported here using the SPL-C141A mutant enzyme. The latter in that respect proved remarkable as it indeed unexpectedly allowed an efficient trapping of this second radical by dithionite, which could be easily monitored by HPLC/mass spectrometry.
At this stage it is not known how Cys-141 provides the radical control/protection mechanism, but two possibilities, requiring further investigation, are briefly discussed in the following. The first hypothesis implies Cys-141 as an H atom donor to the intermediate allylic radical. Such a reaction is thermodynamically reasonable, considering that the energy of the S–H bond of a cysteine residue is slightly larger than to that of the C–H bond of an allyl group (25). Substitution of Cys-141 into alanine thus would result in a further stabilization of the intermediate allylic radical, thus facilitating its coupling with highly reactive free radicals in the environment such as those produced by dithionite in the in vitro experiments. To be catalytic, the resulting protein-bound cysteinyl radical generated in SPL after TpT formation needs to be converted back to a normal cysteine. This reaction implies an H atom donor, which has not been identified but might be DTT in vitro. A second hypothesis implies that cysteine 141 participates in the stabilization of a specific structure of the active site, which prevents the intermediate radical from reacting with exogenous free radicals. In this model, the wt enzyme has its active site in a “closed” conformation because of the presence of Cys-141 and the allylic radical does not react with the dithionite-derived radicals present in the in vitro reaction mixture. Instead, it reacts almost exclusively with an H atom donor, by H atom radical abstraction, to generate the final repaired TpT product. In the case of the SPTpT substrate used here, because AdoH is excluded, the nature of the H atom donor is unknown. Considering that with DNA substrate AdoH is the direct H atom donor (Scheme 1B), we consider the second hypothesis as more likely. Obviously a structural characterization of SPL is required to substantiate at the molecular level such a hypothesis.
In conclusion, our data confirm that a single substitution of cysteine-141 into alanine has a major impact on the SPL-dependent repair reaction and are consistent with the high sensitivity of bacterial spores containing a C141A SPL mutant enzyme to irradiation, as previously reported (14). How this amino acid residue contributes to tightly control the outcome of highly energetic and reactive radical intermediates during catalysis remains to be understood. Furthermore, the mutant enzyme studied here has allowed trapping of one of the key catalytically competent free radical intermediates, providing new insights into the mechanism of SP repair by the SPL enzyme.
Supplementary Material
Acknowledgments
We thank Y. Sanakis (Institut of Materials Science, National Center for Scientific Research, Attiki, Greece) for Mössbauer analysis on the SPL-C141A enzyme and P. Frey (Enzyme Institute, University of Madison, Madison, WI) for providing [5′-(2H)2]SAM.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8.
Footnotes
The abbreviations used are: SPL, spore photoproduct lyase; SP, spore photoproduce, 5-(α-thyminyl)-5,6-dihydrothymine; AdoMet, S-adenosylmethionine or S-adenosyl-l-methionine (SAM); AdoH, 5′-deoxyadenosyl; DTT, dithiothreitol; HPLC, high-performance liquid chromatography; MS/MS, tandem mass spectrometry; wt, wild type; Rt, retention time; TpT, dinucleoside monophosphate; TSΦpT, 5-(thiophenylmethyl)-2′-deoxyuridylyl-(3′,5′)thymidine; TpTSΦ, thymidylyl-(3′,5′)-5-(thiophenylmethyl)-2′-deoxyuridine.
References
- 1.Halliwell, B., and Gutteridge, J. (2007) Free Radicals in Biology and Medicine, Oxford University Press, New York, NY
- 2.Frey, P. A., Hegeman, A. D., and Reed, G. H. (2006) Chem. Rev. 106 3302-3316 [DOI] [PubMed] [Google Scholar]
- 3.Donnellan, J. E., Jr., and Setlow, R. B. (1965) Science 149 308-310 [DOI] [PubMed] [Google Scholar]
- 4.Varghese, A. J. (1970) Biochem. Biophys. Res. Commun. 38 484-490 [DOI] [PubMed] [Google Scholar]
- 5.Setlow, P. (1995) Annu. Rev. Microbiol. 49 29-54 [DOI] [PubMed] [Google Scholar]
- 6.Slieman, T. A., Rebeil, R., and Nicholson, W. L. (2000) J. Bacteriol. 182 6412-6417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandor, A., Berteau, O., Douki, T., Gasparutto, D., Sanakis, Y., Ollagnier-de-Choudens, S., Atta, M., and Fontecave, M. (2006) J. Biol. Chem. 281 26922-26931 [DOI] [PubMed] [Google Scholar]
- 8.Mehl, R. A., and Begley, T. P. (1999) Organic Lett. 1 1065-1066 [DOI] [PubMed] [Google Scholar]
- 9.Sofia, H. J., Chen, G., Hetzler, B. G., Reyes-Spindola, J. F., and Miller, N. E. (2001) Nucleic Acids Res. 29 1097-1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontecave, M., Mulliez, E., and Ollagnier-de-Choudens, S. (2001) Curr. Opin. Chem. Biol. 5 506-511 [DOI] [PubMed] [Google Scholar]
- 11.Frey, P. A., Hegeman, A. D., and Ruzicka, F. J. (2008) Crit. Rev. Biochem. Mol. Biol. 43 63-88 [DOI] [PubMed] [Google Scholar]
- 12.Cheek, J., and Broderick, J. B. (2002) J. Am. Chem. Soc. 124 2860-2861 [DOI] [PubMed] [Google Scholar]
- 13.Buis, J. M., Cheek, J., Kalliri, E., and Broderick, J. B. (2006) J. Biol. Chem. 281 25994-26003 [DOI] [PubMed] [Google Scholar]
- 14.Fajardo-Cavazos, P., Rebeil, R., and Nicholson, W. L. (2005) Curr. Microbiol. 51 331-335 [DOI] [PubMed] [Google Scholar]
- 15.Pierrel, F., Douki, T., Fontecave, M., and Atta, M. (2004) J. Biol. Chem. 279 47555-47563 [DOI] [PubMed] [Google Scholar]
- 16.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 17.Fish, W. W. (1988) Methods Enzymol. 158 357-364 [DOI] [PubMed] [Google Scholar]
- 18.Beinert, H. (1983) Anal. Biochem. 131 373-378 [DOI] [PubMed] [Google Scholar]
- 19.Romieu, A., Bellon, S., Gasparutto, D., and Cadet, J. (2000) Organic Lett. 2 1085-1088 [DOI] [PubMed] [Google Scholar]
- 20.Rebeil, R., Sun, Y., Chooback, L., Pedraza-Reyes, M., Kinsland, C., Begley, T. P., and Nicholson, W. L. (1998) J. Bacteriol. 180 4879-4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebeil, R., and Nicholson, W. L. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 9038-9043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pieck, J. C., Hennecke, U., Pierik, A. J., Friedel, M. G., and Carell, T. (2006) J. Biol. Chem. 281 36317-36326 [DOI] [PubMed] [Google Scholar]
- 23.Ni, J., Pomerantz, C., Rozenski, J., Zhang, Y., and McCloskey, J. A. (1996) Anal. Chem. 68 1989-1999 [DOI] [PubMed] [Google Scholar]
- 24.Mees, A., Klar, T., Gnau, P., Hennecke, U., Eker, A. P., Carell, T., and Essen, L. O. (2004) Science 306 1789-1793 [DOI] [PubMed] [Google Scholar]
- 25.Stubbe, J., and van Der Donk, W. A. (1998) Chem. Rev. 98 705-762 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.