

Abstract
Eaf3 is a component of both NuA4 histone acetyltransferase and Rpd3S histone deacetylase complexes in Saccharomyces cerevisiae. It is involved in the regulation of the global pattern of histone acetylation that distinguishes promoters from coding regions. Eaf3 contains a chromo domain at the N terminus that can bind to methylated Lys-36 of histone H3 (H3K36). We report here the crystal structures of the Eaf3 chromo domain in two truncation forms. Unlike the typical HP1 and Polycomb chromo domains, which contain a large groove to bind the modified histone tail, the Eaf3 chromo domain assumes an autoinhibited chromo barrel domain similar to the human MRG15 chromo domain. Compared with other chromo domains, the Eaf3 chromo domain contains a unique 38-residue insertion that folds into two short β-strands and a long flexible loop to flank the β-barrel core. Both isothermal titration calorimetry and surface plasmon resonance studies indicate that the interaction between the Eaf3 chromo domain and the trimethylated H3K36 peptide is relatively weak, with a KD of ∼10-4 m. NMR titration studies demonstrate that the methylated H3K36 peptide is bound to the cleft formed by the C-terminal α-helix and the β-barrel core. Site-directed mutagenesis study and in vitro binding assay results show that the conserved aromatic residues Tyr-23, Tyr-81, Trp-84, and Trp-88, which form a hydrophobic pocket at one end of the β-barrel, are essential for the binding of the methylated H3K36. These results reveal the molecular mechanism of the recognition and binding of the methylated H3K36 by Eaf3 and provide new insights into the functional roles of the Eaf3 chromo domain.
Histone acetyltransferases (HATs)3 and deacetylases (HDACs) are multicomponent protein complexes that modify lysine residues on the N-terminal tails of histones and play important roles in transcriptional activation or repression (1–4). Eaf3 (essential Sas2-related acetyltransferase 1-associated factor 3) is a component of the NuA4 HAT and Rpd3S HDAC complexes in Saccharomyces cerevisiae (5–7). Genetic and biochemical evidence has shown that Eaf3 plays an important role in regulating the genomic profile of histone H3 and H4 acetylation, the loss of which leads to increase of H3 and H4 acetylation at coding regions and decrease at promoters, resulting in an even distribution of histone acetylation across the genome (8). The underlying mechanism by which Eaf3 affects the global pattern of histone acetylation has recently been shown to be related to the N-terminal chromo domain of Eaf3 (5, 7, 9).
Chromo domains are protein modules that are found in many chromatin-related proteins in nucleoprotein complexes, such as HAT and HDAC complexes. They have been shown to be involved in the recognition and binding of Lys-methylated histone tails and nucleic acids and hence play important roles in histone modification and chromatin remodeling that lead to transcriptional activation or repression of a large number of genes (for reviews, see Refs. 10–12). For example, chromo domains of Drosophila chromatin-binding protein HP1 (heterochromatin binding protein 1) and Pc (Polycomb protein) can bind to methylated Lys-9 and Lys-27 of histone H3 (H3K9 and H3K27), respectively (13–18). Human CHD1 (chromo-ATPase/helicase DNA-binding protein 1) double chromo domains can bind to methylated Lys-4 of histone H3 (H3K4) (19). The human MRG15 chromo domain can bind to methylated Lys-36 of histone H3 (H3K36) (20).
The chromo domain of Eaf3 can bind to methylated H3K36 and H3K4 (5, 7, 9). The interaction between the Eaf3 chromo domain and the methylated H3K36 can lead to preferential association of the Rpd3S complex with coding regions, which further mediates preferential histone deacetylation of coding regions. Thus, Eaf3 is ultimately linked to the mechanism by which repressive chromatin structure is restored after transcriptional elongation, because the pattern of H3K36 methylation is determined by the pattern of phosphorylation of the RNA polymerase II C-terminal domain (5, 7, 9). However, the Eaf3 chromo domain and H3K36 methylation do not significantly affect acetylation at promoters, suggesting that Eaf3-dependent effects at promoters and coding regions are mechanistically distinct. Since Eaf3 positively regulates histone acetylation at promoters, it seems likely that this function of Eaf3 might rely on preferential association of the NuA4 HAT complex with promoters through an unknown mechanism (8, 9). No matter how, the existence of Eaf3 in both Rpd3S HDAC and NuA4 HAT complexes, in particular the interaction of the Eaf3 chromo domain with methylated H3K36, provides a vehicle to coordinately or independently regulate the global patterns of histone acetylation at promoters and coding regions throughout the genome. Nevertheless, it is unclear how the Eaf3 chromo domain can recognize and bind to methylated H3K36 and how the Rpd3S and NuA4 complexes can distinguish specific chromatin sites.
Eaf3 belongs to the MRG protein family, whose members are highly conserved from Arabidopsis thaliana to humans (21). Like Eaf3, the other members of the MORF4-related gene (MRG) protein family are also components of HAT and/or HDAC complexes and are involved in histone modification. The human homolog MRG15 is a component of the Tip60 HAT complex (22). It plays a vital role in embryonic development and cell proliferation, and the knock-out mouse shows a decreased level of acetylation in both histone H3 and H4 (23). MRG15 contains a chromo barrel domain at the N terminus, which can bind methylated H3K36 in a way different from that of the HP1/Pc chromo domain (20). Another human homolog, MORF4, which lacks the chromo domain, can induce cellular senescence in immortal cell lines (24). Both MRG15 and MORF4 associate with mSin3A complexes (25). The C. elegans homolog MRG1 is required for establishing the ability of primordial germ cells to initiate mitotic proliferation during postembryonic development (26). The Drosophila homolog MSL3 (male-specific lethal protein 3) is essential for the dosage compensation of X-linked genes in males and is critical for the activation of the HAT activity of dMOF (6, 27–29). Furthermore, male-specific lethal can attract the male-specific lethal complex to open reading frames marked with trimethylated H3K36 in a sequence-independent manner (30). The Saccharomyces pombe homolog Alp13 (altered polarity protein 13) is a component of the Clr6 HDAC complex and affects the histone acetylation level in the fission yeast (31). These results strongly suggest that these MRG proteins might also function through interactions with methylated histones in the HAT and HDAC complexes and participate in the modification and regulation of the histone acetylation pattern.
To understand the molecular basis of the function of the Eaf3 chromo domain and its binding with the methylated histone tail, we determined the crystal structures of the Eaf3 chromo domain in two truncation forms and characterized its interactions with the methylated H3K36 peptides. The Eaf3 chromo domain is more similar to the autoinhibited chromo barrel domain of human MRG15 than the typical HP1 chromo domain. Compared with the other chromo domains, the Eaf3 chromo domain contains a 38-residue insertion that forms part of the extended β-barrel. Isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) analysis results indicate that the Eaf3 chromo domain can bind to methylated H3K36 peptide with a KD of about 10-4 m. NMR titration studies demonstrate that the methylated H3K36 peptide is bound in the cleft formed by the C-terminal α-helix and the β-barrel core. As in the other chromo domain structures, four conserved aromatic residues, Tyr-23, Tyr-81, Trp-84, and Trp-88, form a hydrophobic pocket at one end of the β-barrel core and are essential for the binding of the methylated H3K36, as revealed by site-directed mutagenesis studies and in vitro binding assays. During revision of this paper, a solution structure of the Eaf3 chromo domain (equivalent to the short form Eaf3 chromo domain reported here) with a covalently linked methylated H3K36 peptide was published online (32). This solution structure is very similar to the crystal structures reported here, and their structural results are consistent with our structural and binding data.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification and Peptide Synthesis—Two truncation forms of the Eaf3 chromo domain (corresponding to residues 1–113 (short form) and residues 1–124 (long form), respectively) were amplified using PCR from S. cerevisiae genome DNA. The plasmid pET-22b (+) containing the recombinant Eaf3 chromo domain gene fused with a His6 tag at the C terminus between the NdeI and XhoI sites was expressed in Escherichia coli BL21(DE3) strain. The bacterial cells were grown at 37 °C to A600 of 0.6, and protein expression was induced with 0.2 mm isopropyl-β-d-thiogalactopyranoside at 16 °C for 20 h. The cells were collected by centrifugation at 4,000 × g, suspended in a lysis buffer (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 2 mm β-mercaptoethanol, and 1 mm phenylmethylsulfonyl fluoride), and lysed on ice by sonication. Cell debris was precipitated by centrifugation at 15,000 × g for 30 min, and supernatant was used for protein purification.
The Eaf3 chromo domain was purified by affinity chromatography using an Ni2+-nitrilotriacetic acid superflow column (Qiagen) equilibrated with a binding buffer (50 mm Tris-HCl, pH 8.0, and 500 mm NaCl). The column was washed with the binding buffer plus 30 mm imidazole to remove nonspecific binding proteins. The target protein was eluted with the binding buffer plus 300 mm imidazole. The protein sample was further purified by gel filtration using Superdex G75 26/60 column (Amersham Biosciences) and then concentrated to 20 mg/ml for crystallization and biochemical studies. Mutations of the Eaf3 chromo domain were generated using the QuikChange site-directed mutagenesis kit (Stratagene). Expression and purification of the mutant proteins followed the same procedures as for the wild-type protein.
The yeast methylated H3K36me2/3 (residues 28–44), unmethylated H3K36 (residues 28–44), and methylated H3K9me3 (residues 1–15) peptides were synthesized by HD Biosciences (Shanghai, China).
Isothermal Titration Calorimetry Analysis—Thermodynamic parameters of both short form and long form Eaf3 chromo domain for binding the histone H3 peptides were determined by ITC at 25 °C using a VP-ITC titration calorimeter (Microcal, Northampton, MA) (33). The protein sample was dialyzed against an assay buffer (50 mm Tris-HCl, pH 8.0, and 100 mm NaCl), and the lyophilized peptides were dissolved in the same buffer. The exothermic heat of the reaction was measured by 30 sequential injections of the peptide (1.5 mm), each 10 μl, into 1.41 ml of the protein solution (0.1 mm) with spacing intervals of 150 s. The heat of dilution was obtained by injecting a ligand into the same buffer and was subtracted from the head of the reaction before the fitting process. Binding curves were analyzed by nonlinear least squares fitting of the data using Microcal ORIGIN software.
Surface Plasmon Resonance Binding Analysis—SPR experiments were also performed to measure the binding affinity of both short form and long form Eaf3 chromo domain with the H3K36me3 peptide using ProteOn XPR36 (Bio-Rad) at 25 °C. The Eaf3 chromo domain was first immobilized on a GLC chip at a similar level (∼2000 response units), and the H3K36me3 peptide was used as analytes for binding. The association was monitored over a 90-s period, during which the peptide in the running buffer (PBST, pH 7.4) was flown over the immobilized protein at 100 μl/min. The dissociation was monitored by flowing PBST over the subsequent 600 s. The surface was regenerated with 10 mm NaOH. An irrelevant protein (interleukin-2 antibody) was used as a blank reference. KD values were computed by equilibrium analysis using Bio-Rad ProteOn Manager 2.0.1 software.
In Vitro Binding Assay—In vitro binding assays were carried out to explore the binding site of the short form Eaf3 chromo domain with the H3K36me2 peptide, using the method described previously (20). A His6 tag was fused with both wild-type and mutant Eaf3 chromo domain. 20 μg of the protein was first incubated with 100 μl of 25% Ni2+-nitrilotriacetic acid superflow bead slurry (Qiagen) for 1 h. After removal of the solution, the beads were mixed with 100 μg of the bovine serum albumin protein and 15 μg of the H3K36me2 peptide (protein/peptide molar ratio of 1:3) in 1 ml of a binding buffer (10 mm Na2HPO4, 2.7 mm KCl, 1.8 mm KH2PO4, and 140 mm NaCl, pH 7.4) containing 1 mm phenylmethylsulfonyl fluoride and 2 mm dithiothreitol and incubated at room temperature for 2 h. After extensive washing with the binding buffer, the protein with bound peptide was visualized by peptide dot blot analysis. The Ni2+-nitrilotriacetic acid superflow beads were eluted with 30 μl of a buffer containing 20 mm Tris-HCl (pH 7.4) and 300 mm imidazole. 2.0 μl of the elution sample was spotted onto nitrocellular membrane and probed with anti-H3K36me2 antibody (dilution 1:2000; Upstate Biotechnology). Horseradish peroxidase-conjugated secondary antibody (dilution 1:2000; Chemicon) and ECL-advance Western blotting kit (Amersham Biosciences) were used for exposure detection.
Crystallization, Data Collection, and Structure Determination—Crystallization was performed using the hanging drop vapor diffusion method. Crystals of the short form Eaf3 chromo domain were grown at 4 °C in a drop with an equal volume of the protein solution (20 mg/ml) and the reservoir solution (100 mm MES, pH 6.0, and 30% polyethylene glycol 6000). Crystals of the long form Eaf3 chromo domain were grown at 4 °C in a drop with an equal volume of the protein solution (10 mg/ml) and the reservoir solution (100 mm Bicine, pH 9.0, 2.4 m ammonium sulfate, and 4% acetone). Diffraction data were collected at Photo Factory BL-6A and BL-17A from flash-cooled crystals at 100 K. The diffraction data were processed, integrated, and scaled together with HKL2000 (34). The statistics of the diffraction data are summarized in Table 1.
TABLE 1.
Summary of diffraction data and structure refinement statistics
| Short form | Long form | |
|---|---|---|
| Statistics of diffraction data | ||
| Wavelength (Å) | 1.0000 | 1.0000 |
| Resolution range (Å)a | 50.0–1.80 (1.86–1.80) | 50–2.5 (2.59–2.50) |
| Space group | C2221 | P212121 |
| Cell parameters | ||
| a (Å) | 34.6 | 43.7 |
| b (Å) | 130.3 | 66.5 |
| c (Å) | 56.2 | 98.2 |
| Observed reflections | 78,732 | 66,958 |
| Unique reflections (I/σ(I) > 0) | 12,156 | 10,098 |
| Mosaicity | 0.54 | 0.38 |
| Average redundancy | 6.5 (5.8) | 6.6 (5.5) |
| Average I/σ(I) | 21.6 (3.8) | 112.5 (6.7) |
| Completeness (%) | 99.3 (96.8) | 96.7 (78.3) |
| Rmerge (%)b | 11.0 (24.8) | 7.8 (20.1) |
| Statistics of refinement and model | ||
| No. of reflections (Fo > 0σ(Fo)) | ||
| Working set | 11,531 | 9,537 |
| Free R set | 610 | 521 |
| R factor (%)c | 18.8 | 22.3 |
| Free R factor (%) | 21.7 | 26.7 |
| No. of residues | 97 | 216 |
| No. of water molecules | 123 | 123 |
| No. of MES molecules | 1 | 0 |
| Average B factor (Å2) | ||
| All atoms | 26.4 | 45.4 |
| Water | 38.7 | 46.5 |
| MES | 36.9 | |
| Root mean square bond lengths (Å) | 0.015 | 0.006 |
| Root mean square bond angles (degrees) | 1.57 | 1.04 |
| Ramachandran plot (%) | ||
| Most favored regions | 95.2 | 94.1 |
| Allowed regions | 4.8 | 5.9 |
Numbers in parentheses refer to the highest resolution shell
Rmerge = ΣhklΣi|Ii(hkl)i – 〈I(hkl)〉|/ΣhklΣiIi(hkl)
R factor = Σhkl||Fo| – |Fc||/Σhkl|Fo
The structure of the short form Eaf3 chromo domain was solved by the molecular replacement method using the program CNS (35) with the MRG15 chromo domain as the search model (Protein Data Bank code 2F5K) (20). The program ARP/WARP was used to build an initial model, followed by manual building with the programs O (36) and Coot (37). The structure of the long form Eaf3 chromo domain was solved by molecular replacement with the short form Eaf3 chromo domain as the search model. Structure refinement was carried out using the programs CNS and REFMAC5 (38). The statistics of the structure refinement and the quality of the final structure model are summarized in Table 1.
NMR Spectroscopy Analysis—All NMR data were collected on a Varian Unity Inova 600 MHz spectrometer at 298 K. The samples used in the experiments contain 0.5–1.0 mm 15N/15N,13C-labeled protein in a buffer consisting of 20 mm phosphate, pH 6.6, and 0.1 m NaCl with 10% D2O. Sequential assignments of the Eaf3 chromo domain were accomplished using three-dimensional sensitivity-enhanced HNCACB and CBCA-(CO)NH experiments. All of the spectra were processed with the NMRPipe software (39) and analyzed with the Sparky software (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco).
Two-dimensional 1H-15N HSQC spectra were used to monitor the chemical shift changes of the Eaf3 chromo domain upon the binding of the H3K36me3 peptide. A series of 1H-15N HSQC spectra were recorded with the use of increasing concentrations of the peptide until the molar ratio of protein to peptide reached 1:10. For analysis of the chemical shift perturbations of 1H and 15N backbone resonance, the average chemical shift changes were calculated using the following formula.
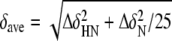 |
(Eq. 1) |
A significant chemical shift change was defined as a value larger than the average plus the S.D. The residues with significant chemical shift change are thought to be involved in interaction with the peptide.
RESULTS AND DISCUSSION
Structure of the Eaf3 Chromo Domain—The structure of the short form Eaf3 chromo domain (residues 1–113) was solved by molecular replacement, and the structure refinement converged to an R factor of 18.8% and a free R factor of 21.7% at 1.8 Å resolution (Table 1). This crystal form belongs to space group C2221 and contains one Eaf3 chromo domain molecule in the asymmetric unit. In the final model, the N-terminal six residues (numbered 1–6) and residues 44–53 were disordered. The Eaf3 chromo domain is composed of an extended, twisted β-barrel and a C-terminal α-helix (Fig. 1A). A number of hydrophobic residues (including Phe-8, Tyr-77, Ile-79, Val-92, and Ile-97) form a hydrophobic core to stabilize the overall scaffold. The C-terminal α-helix flanks one side of the β-barrel and is in the opposite direction from that of the HP1/Pc chromo domains. Residues Ile-105, Met-107, and Leu-111 of the α-helix have hydrophobic interactions with the side chains of Leu-15 and Phe-17 of β1, Leu-21 and Met-22 of β2, and Arg-98 of β5. The side chain Nδ2 of Asn-109 forms a hydrogen bond with the main chain carbonyl of Ala-99. These interactions dictate the orientation of the C-terminal α-helix, which is important for the binding of histone peptide (see below).
FIGURE 1.

Structure of the Eaf3 chromo domain. A, overall structure and electrostatic surface of the short form Eaf3 chromo domain (residues 1–113). B, overall structure and electrostatic surface of the long form Eaf3 chromo domain (residues 1–124). The C-terminal part forms a stable α-helix in both structures. The Eaf3 chromo domain contains a 38-residue insertion (shown in red), which, together with the β-barrel core, forms a long, deep surface groove. C, sequence comparison of the chromo domain from yeast Eaf3, human MRG15, and Drosophila HP1. Strictly conserved residues are highlighted in shaded red boxes, and conserved residues are highlighted in open red boxes. The secondary structure of the Eaf3 chromo domain is placed above the alignment. D, structural comparison of the Eaf3 chromo domain (cyan, insertion in red) with the MRG15 (gray, Protein Data Bank code 2F5K) and HP1 (purple, Protein Data Bank code 1KNA) chromo domains. Residues forming the hydrophobic pocket are shown with side chains. The bound peptide in the HP1 chromo domain complex is shown in magenta.
Recently, the NMR solution structure of the MRG15 chromo domain was released by the Protein Data Bank (code 2EFI), in which the C-terminal region forms two short α-helices connected by three residues rather than one long α-helix, as seen in the crystal structure, and the C-terminal short α-helix makes a sharp bend and folds onto the β-barrel core. To investigate the conformational flexibility of the C-terminal region of the Eaf3 chromo domain, we also determined the crystal structure of the long form Eaf3 chromo domain (residues 1–124), whose C terminus is equivalent to that seen in both the crystal and solution structures of the MRG15 chromo domain, and the structure refinement converged to an R factor of 22.3% and a free R factor of 26.7% at 2.5 Å resolution (Table 1). This crystal form belongs to space group P212121 and contains two Eaf3 chromo domain molecules in the asymmetric unit. The structure of the long form Eaf3 chromo domain shows no substantial difference compared with the short form Eaf3 chromo domain (a root mean square deviation of 0.4 Å for all Cα atoms), and the C-terminal region forms a long α-helix, as seen in the crystal structure of the MRG15 chromo domain (Fig. 1B). In addition, the average B-factor of the C-terminal α-helix in both the Eaf3 and MRG15 chromo domains is comparable with that of the whole protein (22.1 Å2 versus 26.1 Å2 for the short form Eaf3 chromo domain, 47.4 Å2 versus 45.4 Å2 for the long form Eaf3 chromo domain, and 53.6 Å2 versus 52.6 Å2 for the MRG15 chromo domain). These results suggest that the C-terminal region of the Eaf3 and MRG15 chromo domain should form a stable long α-helix. Nevertheless, we cannot rule out the effect of crystal packing, because in all structures of the Eaf3 and MRG15 chromo domains, a few residues of the C-terminal α-helix are involved in interactions with symmetry-related molecules. Thus, in the structural analysis and discussion below, we will not distinguish the short form and long form Eaf3 chromo domain unless otherwise specified.
Sequence comparison shows that the Eaf3 chromo domain shares a high sequence homology with the MRG15 chromo domain (30 of 71 residues identical) (Fig. 1C). Structural comparison indicates that the Eaf3 chromo domain assumes an autoinhibited chromo barrel domain structure, which is more similar to the MRG15 chromo domain (a root mean square deviation of 0.78 Å) than the typical HP1 chromo domain (a root mean square deviation of 1.49 Å) (Fig. 1D). Specifically, the Eaf3 chromo domain contains an extra β-strand (β1), which occupies the binding groove of the histone peptide seen in the HP1/Pc chromo domains complexed with the histone peptides (13–18). Interestingly, compared with the other chromo domains, the Eaf3 chromo domain contains a 38-residue insertion, which folds into two short β-strands (β3 and β4) and a long loop linking strand β4 back to strand β5 (Fig. 1C). As a result, the β-barrel in the Eaf3 chromo domain comprises seven β-strands (β1–β7), forming a twisted β-sheet, which can be divided into two parts; strands β1-β2 and β5-β7 form one part of the β-barrel, and strands β2-β4 and β5-β6 form the other part of the β-barrel. At the junction of the two parts, there is a long and deep groove, which could be a potential binding site for other proteins (Fig. 1, A and B).
Like the other chromo domains, the Eaf3 chromo domain contains a highly hydrophobic pocket at one end of the β-barrel formed by three conserved aromatic residues Tyr-23, Tyr-81, and Trp-84 (equivalent to Tyr-26, Tyr-46, and Trp-49 of the MRG15 chromo domain; Tyr-21, Trp-42, and Phe-45 of the mouse HP1 chromo domain; or Tyr-26, Trp-47, and Trp-50 of the Drosophila Pc chromo domain, respectively) which could be the potential binding site for a methylated lysine of histone H3 (Fig. 1D). In addition, Trp-88 of the Eaf3 chromo domain (equivalent to Trp-53 of the MRG15 chromo domain) also forms part of the pocket. As in the structures of the HP1 and Pc chromo domains, this pocket is shallow in the absence of a bound methylated H3 peptide.
It is noteworthy that in the short form Eaf3 crystal structure, there is a positive electron density above the hydrophobic pocket, which could be fitted very well with a MES molecule that exists in the crystallization solution. The morpholino ring of the putative MES molecule has extensive hydrophobic interactions with the side chains of Tyr-23, Trp-84, and Trp-88 (about 3.4–4.2 Å), and its sulfonic group forms a hydrogen bond with the main chain amide of Lys-85 (2.8 Å) (Fig. S1). However, the biological implication of the bound MES is unclear.
Characterization of the Eaf3 Chromo Domain Binding to Histone Tail Peptides—To understand the specificity of the Eaf3 chromo domain binding to methylated histone tails, we carried out ITC and SPR experiments with a number of synthetic histone tail peptides. The ITC results show that the short form Eaf3 chromo domain can bind with the H3K36me3/2 peptides and very weakly with the H3K4me3/2 peptides but not with the unmethylated H3K36 or H3K9me3 peptide (Fig. 2A). The binding with the H3K36me3 peptide (dissociation constant KD = 0.18 ± 0.09 mm) is tighter than the H3K36me2 peptide. The binding affinity of the long form Eaf3 chromo domain for the H3K36me3 peptide (KD = 0.37 ± 0.04 mm) is comparable with that of the short form, indicating that truncation of the C-terminal 11 residues does not significantly affect the binding. These results were further confirmed by the SPR experiments which show the KD of 0.21 ± 0.02 and 0.38 ± 0.01 mm for the short form and long form Eaf3 chromo domain, respectively (Fig. 2, B and C).
FIGURE 2.

Characterization of the Eaf3 chromo domain binding to histone H3 peptides. A, characterization of binding of the Eaf3 chromo domain with various histone H3 peptides using isothermal titration calorimetry: H3K36me3 peptide (a); H3K36me2 peptide (b); H3K36 peptide (c); H3K4me3 peptide (d); H3K4me2 peptide (e); H3K9me3 peptide (f). The upper panels show the raw data for injections of the peptides into the Eaf3 chromo domain, and the lower panels show the integrated heats of the injections. B, binding of the short form Eaf3 chromo domain with the H3K36me3 peptide measured by ITC and SPR. C, binding of the long form Eaf3 chromo domain with the H3K36me3 peptide measured by ITC and SPR. RU, response units. D, binding of the wild-type and mutant Eaf3 chromo domain with the H3K36me2 peptide using in vitro binding assay. Negative control 1 (N1) shows that the H3K36me2 peptide does not have nonspecific binding with the nickel beads; negative control 2 (N2) shows that the Eaf3 chromo domain does not have nonspecific reaction with the anti-H3K36me2 antibody; positive control (P) shows the specific binding between the H3K36me2 peptide and the anti-H3K36me2 antibody.
The Eaf3 Chromo Domain Uses a Hydrophobic Cleft to Bind the Methylated H3K36—The previous cell biological data (9) and our biochemical data have shown that the Eaf3 chromo domain can bind with methylated H3K36 and weakly with methylated H3K4. In the structures of the Eaf3 chromo domain, the histone peptide binding groove seen in the HP1/Pc chromo domains is pre-occupied by an extra β-strand (β1), suggesting that the binding mode of the Eaf3 chromo domain with the histone tail has to be different from that of the HP1/Pc chromo domains. Due to the weak binding of the Eaf3 chromo domain with the methylated H3K36 peptides, we were unsuccessful in obtaining crystal of the complexes. Since NMR chemical shift perturbation is sensitive to molecular interaction, we thus used the NMR chemical shift perturbation method to map the binding site of the Eaf3 chromo domain with the H3K36me3 peptide. Fig. 3A shows superposition of the two-dimensional 15N-1H HSQC spectra of the short form Eaf3 chromo domain in unliganded form and in complex with the H3K36me3 peptide. Fig. 3B shows the chemical shift changes of the Eaf3 chromo domain residues versus residue number before and after titration with the H3K36me3 peptide. We also performed the titration experiment of the long form Eaf3 chromo domain with the H3K36me3 peptide. Consistent with the results from the short form Eaf3 chromo domain, the C-terminal 11 residues of the long form do not show significant chemical shift changes (data not shown). This indicates that the C-terminal part does not participate in the interaction, which is in agreement with the results of the ITC and SPR experiments.
FIGURE 3.

NMR titrations of the Eaf3 chromo domain with the H3K36me3 peptide. A, superposition of 1H-15N HSQC spectra of the 15N-labeled Eaf3 chromo domain in unliganded form (red) and in complex with the H3K36me3 peptide at a molar ratio of 1:2 (orange), 1:3 (green), 1:4 (blue), 1:5 (purple), 1:6 (gray), 1:7 (pink), 1:8 (gold), and 1:10 (magenta). B, average chemical shift changes versus residue number of the 15N-labeled Eaf3 chromo domain. Solid line, average value of chemical shift changes; dashed line, sum of the average value and S.D. The blank column means that the residue cannot be assigned. C, molecular surface representation of the Eaf3 chromo domain (short form) showing the potential binding site for the methylated H3K36 peptide. Residues exhibiting a chemical shift change larger than the average value are colored orange, and residues exhibiting chemical shift change larger than the average plus S.D. are colored red.
The titration results show that most of the residues of the Eaf3 chromo domain exhibiting significant chemical shift changes are clustered into three patches that are located on the surface cleft formed by the β-barrel core and the C-terminal α-helix (Fig. 3, B and C). Specifically, residues Tyr-23, Tyr-81, Trp-84, and Trp-88 in the hydrophobic pocket and several residues surrounding them form the first patch. Consistently, the results of the in vitro pull-down assays showed that this pocket is a potential binding site for the methylated Lys-36. The second patch consists of a number of residues that are located in the β1-β2 turn. It is possible that this turn might undergo a substantial conformational change upon the binding of the peptide. The third patch comprises several residues (positions 110–113) of the C-terminal α-helix, suggesting that the C-terminal α-helix is also involved in the binding with the peptide. On the other hand, none of the residues in the insertion region of the Eaf3 chromo domain shows significant chemical shift change, further confirming that the insertion region is not involved in the binding of the methylated H3K36. Taking the structural and biochemical data together, we conclude that the hydrophobic cleft formed by the β-barrel core and the C-terminal α-helix is the potential binding site for the methylated H3K36 peptide. Recently, Xu et al. (32) reported a solution structure of the Eaf3 chromo domain (equivalent to the short form described here) with a covalently linked methylated H3K36 peptide. The solution structure is very similar to our crystal structures. Consistent with our results, the C-terminally linked H3K36 peptide indeed lies in the cleft formed by the β-barrel core and the C-terminal α-helix.
We also performed the titration experiment of the Eaf3 chromo domain with the H3K4me3 peptide. The chemical shift perturbation pattern is quite similar to that from the H3K36me3 peptide titration experiment (data not shown). However, the magnitudes of the chemical shift changes in the H3K4me3 peptide titration are smaller than those in the H3K36me3 peptide titration. This is in agreement with the results of the in vitro binding assays showing that the binding of the Eaf3 chromo domain with the H3K4me3 peptide is weaker than that with the H3K36me3 peptide. Similar results were obtained from the titration experiment of the MRG15 chromo domain with the H3K36me3 peptide (data not shown). Since the MRG15 chromo domain also assumes the chromo barrel domain structure with a previously occupied histone peptide binding groove (20), it is likely that it may recognize and bind the methylated H3K36 in a manner similar to that of the Eaf3 chromo domain.
A Conserved Hydrophobic Pocket of the Eaf3 Chromo Domain Is Essential for the Binding of the Methylated H3K36—The typical HP1/Pc chromo domains contain a hydrophobic pocket formed by three aromatic residues (Tyr-21, Trp-42, and Phe-45 in mouse HP1 or Tyr-26, Trp-47, and Trp-45 in Drosophila Pc) at one end of the β-barrel to accommodate the methylated lysine (13–18). Structural analysis of the Eaf3 chromo domain also reveals a shallow hydrophobic pocket formed by the conserved aromatic residues Tyr-23, Tyr-81, and Trp-84. Besides, residue Trp-88 also forms part of the pocket. Our NMR titration studies show that Tyr-81, Trp-84, and Trp-88 exhibit significant chemical shift changes and Tyr-23 displays a moderate chemical shift change when titrated with the H3K36me3 peptide. These data suggest that these conserved aromatic residues are involved in the interaction with the methylated H3K36. To further confirm the functional role of these residues and identify possibly other residues involved in the binding of the methylated H3K36, we carried out an in vitro binding assay of both wild-type and mutant Eaf3 chromo domains containing point mutations in the hydrophobic pocket and other regions with the H3K36me2 peptides. Compared with the wild-type protein, mutations Y23A, W84A, and W88A in the hydrophobic pocket significantly impaired and mutation Y81A completely abolished the binding of the protein with the peptide, indicating that, like the other chromo domains, the hydrophobic pocket is essential for the binding of the methylated H3K36 and probably is the binding site for the methylated Lys-36 (Fig. 2D). We also selected residue His-18 of the β1-β2 loop for mutagenesis study, and the result shows that mutation H18A does not significantly affect the binding of the H3K36me3 peptide (data not shown), indicating that His-18 and, possibly, the whole β1-β2 loop are not directly involved in or are not essential for the binding. These results are also consistent with the recently reported solution structure of the Eaf3 chromo domain, in which the four aromatic residues (Tyr-23, Tyr-81, Trp-84, and Trp-88) adopt different side chain conformations, thus creating a deep pocket to accommodate the methylated Lys-36 of the peptide, whereas His-18 is not essential for the binding (32).
The previous cell biological and biochemical studies have shown that loss of Eaf3 in yeast results in increased histone acetylation of the transcribed region of certain genes, and the interaction of the Eaf3 chromo domain with the methylated H3K36 is essential for association of Rpd3S with coding regions, which further mediates preferential histone deacetylation of coding regions (7). Mutations of the conserved residues Tyr-81 and Trp-84 to Ala disrupted the association, and the mutants behaved very similarly to the eaf3Δ deletion strain (7). As a negative control, the mutant bearing the double mutation R96A/I97A in Eaf3, both mutations of which are not located in the peptide binding cleft and thus not involved in the binding of the methylated H3K36, showed an acetylation level similar to that of the wild-type strain. These data further indicate that the binding of Eaf3 to the methylated H3K36 is crucial for its biological function, and the conserved residues forming the hydrophobic pocket are essential for the binding, consistent with our structural and in vitro binding data.
Compared with the other chromo domains, the Eaf3 chromo domain contains a 38-residue insertion that, together with the β-barrel core, forms a long and deep groove that could be a potential binding site for other proteins (see above). However, a series of mutations in this insertion region do not affect the binding of the Eaf3 chromo domain with the methylated H3K36 peptide (data not shown), indicating that the insertion region is not involved in the binding of the methylated histone.
In comparison, the binding affinity of the Eaf3 chromo domain with the methylated H3K36 peptides is 10–100-fold weaker than that of the HP1/Pc chromo domains with the methylated H3K9 and H3K27 peptides (14, 15, 17) and that of the human CHD1 double chromo domain with the methylated H3K4 peptides (19). Because the methylated H3K36 peptide is shown to bind in a hydrophobic surface cleft formed by the β-barrel core and the C-terminal α-helix instead of the surface groove, as seen in the HP1/Pc chromo domains, it is not surprising to see a weak binding of the methylated H3K36 peptide with the Eaf3 chromo domain. So far, a number of protein modules have been identified to recognize and bind specifically with modified histones, including the chromo domain, bromo domain, Tudor domain, PHD finger, and PWWP domain, etc. However, in many cases, the binding affinity of these domains with the modified histones is not strong enough so that one module domain-histone interaction is not sufficient to recruit a given complex, and therefore it is proposed that multivalent recognition and binding of several module domains on one or more histone tails of the same, adjacent, or discontinuous nucleosomes may play a significant role in chromatin functions (40). Multimodule domains may bind their substrates cooperatively to determine the overall affinity and specificity with specific chromatin region. For example, the A. thaliana DNA methyltransferase CMT3 contains two copies of the chromo domain that together bind only to the combination of H3K9me3 and H3K27me3 in the same peptide but not to the individual modification (41). Similarly, the single bromo domain can only bind weakly to the acetylated histone peptides (KD of 0.1–0.35 mm), whereas the double bromo domains can bind to the doubly acetylated histone peptides with much higher binding affinity (KD of 1–20 μm) (42, 43). Thus, it seems that the weak binding of the Eaf3 chromo domain to the methylated H3K36 may be not sufficient to recruit the NuA4 or Rdp3S complexes to specific chromatin site. Participation of other module domain(s) may be required to provide extra binding affinity and specificity to exert biological function(s). The argument is supported by recent studies showing that the Rco1 subunit in the Rpd3S complex can greatly enhance Rpd3S interaction with nucleosomes and that the PHD finger of Rco1 is responsible for this effect (44) and that the Rco1 PHD finger is able to interact with histone H3 peptides irrespective of lysine methylation (45). It is possible that the Eaf3 chromo domain provides specificity for H3K36-methylated nucleosomes and that the Rco1 PHD finger enhances the overall affinity of Rpd3S for nucleosomes. The binding of the Rco1 PHD finger to nucleosomes may anchor Rpd3S in a specific configuration which allows the Eaf3 chromo domain to recognize H3K36 methylation with greater affinity. A similar scenario may also happen for the function of Eaf3 in the NuA4 complex.
Supplementary Material
Acknowledgments
We thank the staff members at Photon Factory, Japan, for support in diffraction data collection and other members of our groups for helpful discussion.
The atomic coordinates and structure factors (codes 3E9F and 3E9G) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by Ministry of Science and Technology of China Grants 2006CB806501, 2006AA02Z112, 2006AA02A313, 2007CB914302, and 2007CB914304; National Natural Science Foundation of China Grants 30570379, 30570352, 30623002, and 30730028; Chinese Academy of Sciences Grants KSCX2-YW-R-107 and KSCX2-YW-R-104; and Science and Technology Commission of Shanghai Municipality Grants 07XD14032 and 07DZ22009. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: HAT, histone acetyltransferase; HDAC, histone deacetylase; MES, 2-(N-morpholino)ethanesulfonic acid; ITC, isothermal titration calorimetry; SPR, surface plasmon resonance; Bicine, N,N-bis(2-hydroxyethyl)glycine.
References
- 1.Clayton, A. L., Hazzalin, C. A., and Mahadevan, L. C. (2006) Mol. Cell 23 289-296 [DOI] [PubMed] [Google Scholar]
- 2.Grunstein, M. (1997) Nature 389 349-352 [DOI] [PubMed] [Google Scholar]
- 3.Roth, S. Y., Denu, J. M., and Allis, C. D. (2001) Annu. Rev. Biochem. 70 81-120 [DOI] [PubMed] [Google Scholar]
- 4.Struhl, K. (1998) Genes Dev. 12 599-606 [DOI] [PubMed] [Google Scholar]
- 5.Carrozza, M. J., Li, B., Florens, L., Suganuma, T., Swanson, S. K., Lee, K. K., Shia, W. J., Anderson, S., Yates, J., Washburn, M. P., and Workman, J. L. (2005) Cell 123 581-592 [DOI] [PubMed] [Google Scholar]
- 6.Eisen, A., Utley, R. T., Nourani, A., Allard, S., Schmidt, P., Lane, W. S., Lucchesi, J. C., and Cote, J. (2001) J. Biol. Chem. 276 3484-3491 [DOI] [PubMed] [Google Scholar]
- 7.Keogh, M. C., Kurdistani, S. K., Morris, S. A., Ahn, S. H., Podolny, V., Collins, S. R., Schuldiner, M., Chin, K., Punna, T., Thompson, N. J., Boone, C., Emili, A., Weissman, J. S., Hughes, T. R., Strahl, B. D., Grunstein, M., Greenblatt, J. F., Buratowski, S., and Krogan, N. J. (2005) Cell 123 593-605 [DOI] [PubMed] [Google Scholar]
- 8.Reid, J. L., Moqtaderi, Z., and Struhl, K. (2004) Mol. Cell Biol. 24 757-764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi, A. A., and Struhl, K. (2005) Mol. Cell 20 971-978 [DOI] [PubMed] [Google Scholar]
- 10.Berger, S. L. (2007) Nature 447 407-412 [DOI] [PubMed] [Google Scholar]
- 11.Brehm, A., Tufteland, K. R., Aasland, R., and Becker, P. B. (2004) BioEssays 26 133-140 [DOI] [PubMed] [Google Scholar]
- 12.Daniel, J. A., Pray-Grant, M. G., and Grant, P. A. (2005) Cell Cycle 4 919-926 [DOI] [PubMed] [Google Scholar]
- 13.Bannister, A. J., Zegerman, P., Partridge, J. F., Miska, E. A., Thomas, J. O., Allshire, R. C., and Kouzarides, T. (2001) Nature 410 120-124 [DOI] [PubMed] [Google Scholar]
- 14.Fischle, W., Wang, Y., Jacobs, S. A., Kim, Y., Allis, C. D., and Khorasanizadeh, S. (2003) Genes Dev. 17 1870-1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs, S. A., and Khorasanizadeh, S. (2002) Science 295 2080-2083 [DOI] [PubMed] [Google Scholar]
- 16.Lachner, M., O'Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. (2001) Nature 410 116-120 [DOI] [PubMed] [Google Scholar]
- 17.Min, J., Zhang, Y., and Xu, R. M. (2003) Genes Dev. 17 1823-1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen, P. R., Nietlispach, D., Mott, H. R., Callaghan, J., Bannister, A., Kouzarides, T., Murzin, A. G., Murzina, N. V., and Laue, E. D. (2002) Nature 416 103-107 [DOI] [PubMed] [Google Scholar]
- 19.Flanagan, J. F., Mi, L. Z., Chruszcz, M., Cymborowski, M., Clines, K. L., Kim, Y., Minor, W., Rastinejad, F., and Khorasanizadeh, S. (2005) Nature 438 1181-1185 [DOI] [PubMed] [Google Scholar]
- 20.Zhang, P., Du, J., Sun, B., Dong, X., Xu, G., Zhou, J., Huang, Q., Liu, Q., Hao, Q., and Ding, J. (2006) Nucleic Acids Res. 34 6621-6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertram, M. J., and Pereira-Smith, O. M. (2001) Gene (Amst.) 266 111-121 [DOI] [PubMed] [Google Scholar]
- 22.Doyon, Y., Selleck, W., Lane, W. S., Tan, S., and Cote, J. (2004) Mol. Cell Biol. 24 1884-1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tominaga, K., Kirtane, B., Jackson, J. G., Ikeno, Y., Ikeda, T., Hawks, C., Smith, J. R., Matzuk, M. M., and Pereira-Smith, O. M. (2005) Mol. Cell Biol. 25 2924-2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertram, M. J., Berube, N. G., Hang-Swanson, X., Ran, Q., Leung, J. K., Bryce, S., Spurgers, K., Bick, R. J., Baldini, A., Ning, Y., Clark, L. J., Parkinson, E. K., Barrett, J. C., Smith, J. R., and Pereira-Smith, O. M. (1999) Mol. Cell Biol. 19 1479-1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yochum, G. S., and Ayer, D. E. (2002) Mol. Cell Biol. 22 7868-7876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujita, M., Takasaki, T., Nakajima, N., Kawano, T., Shimura, Y., and Sakamoto, H. (2002) Mech. Dev. 114 61-69 [DOI] [PubMed] [Google Scholar]
- 27.Morales, V., Regnard, C., Izzo, A., Vetter, I., and Becker, P. B. (2005) Mol. Cell Biol. 25 5947-5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morales, V., Straub, T., Neumann, M. F., Mengus, G., Akhtar, A., and Becker, P. B. (2004) EMBO J. 23 2258-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith, E. R., Pannuti, A., Gu, W., Steurnagel, A., Cook, R. G., Allis, C. D., and Lucchesi, J. C. (2000) Mol. Cell Biol. 20 312-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larschan, E., Alekseyenko, A. A., Gortchakov, A. A., Peng, S., Li, B., Yang, P., Workman, J. L., Park, P. J., and Kuroda, M. I. (2007) Mol. Cell 28 121-133 [DOI] [PubMed] [Google Scholar]
- 31.Nakayama, J., Xiao, G., Noma, K., Malikzay, A., Bjerling, P., Ekwall, K., Kobayashi, R., and Grewal, S. I. (2003) EMBO J. 22 2776-2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu, C., Cui, G., Botuyan, M. V., and Mer, G. (2008) Structure 16 1740-1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiseman, T., Williston, S., Brandts, J. F., and Lin, L. N. (1989) Anal. Biochem. 179 131-137 [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 35.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D 54 905-921 [DOI] [PubMed] [Google Scholar]
- 36.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 37.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 38.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D 53 240-255 [DOI] [PubMed] [Google Scholar]
- 39.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 40.Ruthenburg, A. J., Li, H., Patel, D. J., and Allis, C. D. (2007) Nat. Rev. Mol. Cell Biol. 8 983-994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindroth, A. M., Shultis, D., Jasencakova, Z., Fuchs, J., Johnson, L., Schubert, D., Patnaik, D., Pradhan, S., Goodrich, J., Schubert, I., Jenuwein, T., Khorasanizadeh, S., and Jacobsen, S. E. (2004) EMBO J. 23 4286-4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K., and Zhou, M. M. (1999) Nature 399 491-496 [DOI] [PubMed] [Google Scholar]
- 43.Mujtaba, S., He, Y., Zeng, L., Yan, S., Plotnikova, O., Sachchidanand, Sanchez, R., Zeleznik-Le, N. J., Ronai, Z., and Zhou, M. M. (2004) Mol. Cell 13 251-263 [DOI] [PubMed] [Google Scholar]
- 44.Li, B., Gogol, M., Carey, M., Lee, D., Seidel, C., and Workman, J. L. (2007) Science 316 1050-1054 [DOI] [PubMed] [Google Scholar]
- 45.Shi, X., Kachirskaia, I., Walter, K. L., Kuo, J. H., Lake, A., Davrazou, F., Chan, S. M., Martin, D. G., Fingerman, I. M., Briggs, S. D., Howe, L., Utz, P. J., Kutateladze, T. G., Lugovskoy, A. A., Bedford, M. T., and Gozani, O. (2007) J. Biol. Chem. 282 2450-2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.