

Abstract
Rv3868, a conserved hypothetical protein of the ESAT-6 secretion system of Mycobacterium tuberculosis, is essential for the secretion of at least four virulence factors. Each protein chain is ∼63 kDa and assembles into a hexamer. Limited proteolysis demonstrates that it consists of two domains joined by a linker. The N-terminal domain is a compact, helical domain of ∼30 kDa and apparently functions to regulate the ATPase activity of the C-terminal domain and the oligomerization. The nucleotide binding site is situated in the C-terminal domain, which exhibits ATP-dependent self-association. It is also the oligomerization domain. Dynamic fluorescence quenching studies demonstrate that the domain is proximal to the C terminus in the apoprotein and exhibits a specific movement upon ATP binding. In silico modeling of the domains suggests that Arg-429 of a neighboring subunit forms a part of the binding site upon oligomerization. Mutational analysis of binding site residues demonstrates that the Arg-429 functions as the important “sensor arginine” in AAA-ATPases. Protein NMR experiments involving CFP-10 and activity assays rule out a general chaperone-like function for Rv3868. On the other hand, ATP-dependent “open-close” movements of the individual domains apparently enable it to interact and transfer energy to co-proteins in the ESX-1 pathway.
The ESAT-6 (early secreted antigen 6 kDa) system 1 (ESX-1) is a critical secretion apparatus in Mycobacterium tuberculosis and Mycobacterium marinum, which delivers at least four virulence factors (viz. ESAT-6, CFP-10 (culture filtrate protein-10), EspA, and EspB) to the host macrophages during infection (1-9). ESAT-6 and CFP-10 form a tight 1:1 complex (10), and our earlier work suggests that complex formation confers thermodynamic and biochemical stability (11). The proteins corresponding to this secretion system are encoded by genes of RD1 (region of difference 1) (1, 2, 4) and its surrounding region, together termed extRD1 (extended RD1) (6, 12, 13) in M. tuberculosis and M. marinum. RD1 genes are absent in the Mycobacterium bovis BCG vaccine strain (14-16). A defective ESX-1 secretory apparatus prevents the mutant pathogen from leaving the infected phagocyte and spreading to neighboring cells (17). Further work is necessary to identify the exact functions of the individual ESX-1 genes and proteins.
The proteins encoded by the system can be broadly divided into four groups based on the generated phenotypes upon inactivation of the respective genes (13). Knocking out the pe35 gene (Rv3872) impairs the expression of ESAT-6 (Rv3875) and CFP-10 (Rv3874) virulence factors. Inactivation of Rv3868 (the characterization of which is reported here), Rv3869, Rv3870, Rv3871, and Rv3877 impairs the ability of the pathogen to secrete the virulence factors, although their expression itself was unimpaired. It has been shown that Rv3871, a member of the SpoIIIE/FtsK ATPase family, recognizes a C-terminal signal sequence of CFP-10 (18). Inactivation of a third set of proteins does not impair the RD1-mediated virulence. Inactivation of a fourth group consisting of Rv3865 and Rv3866 attenuated RD1-mediated virulence, although the secretion of ESAT-6 and CFP-10 factors was unimpaired.
Rv3868 is an essential component of the ESX-1 system (13) in M. tuberculosis and in the phylogenetically closely related strain M. marinum. However, its exact role and functions are not characterized. Sequence and phylogeny analysis of Rv3868 shows that it is conserved among a small group of largely hypothetical proteins in mycobacteria (29). Based on yeast two-hybrid and genetic experiments, it was proposed to interact with CFP-10 and also with the PPE-68 protein Rv3873 (19). It has also been hypothesized that it might mediate the formation of the recently observed homodimers (19) and heterodimers in the ESAT-6 and CFP-10 proteins (4, 10), a step that might require chaperone activity. The PPE-68 protein Rv3873 is suggested to be a gating component of the ESX-1 system and regulates the secretion of the ESAT-6·CFP-10 complex (6). Rv3868, on the other hand, is hypothesized to be the chaperone or a source of energy (ATPase activity) required for the export of the factors. Structural and functional characterization of Rv3868 is important to understand its role in ESX-1-mediated secretion and to exploit its potential as a novel drug target.
Here, the protein has been shown to be a hexamer that exhibits ATPase activity. Each chain consists of two distinct domains, and their individual roles have been dissected. Mutational analysis coupled to structural modeling has led to the identification of Arg-429 as the functionally important “sensor arginine” (20). Its mutation abolishes conformational changes in the oligomer and leads to a large reduction in the binding affinity of the substrate nucleotide. Direct interactions that were hypothesized earlier with CFP-10, also as a general chaperone activity, have been ruled out. The picture that emerges is that Rv3868 functions as a novel ATPase with a co-factor-induced “open-close” movement. It most likely interacts with other factors of the ESX-1 machinery to provide energy for the export of the ESAT-6·CFP-10 virulence factors. The detailed characterization of the protein reported here is the first for a protein from the CbxX/CfqX (21) subfamily of AAA-ATPases.
MATERIALS AND METHODS
Phylogenetic Tree and Sequence Analysis—The sequence of Rv3868 was downloaded from the Tuberculist site on the World Wide Web. The multiple sequence alignment and neighbor-joining phylogenetic tree (dendrogram) for the different families of proteins were calculated using the ClustalX package (22). Sequences of proteins from different ATPase families were downloaded from the Swissprot data base.
Cloning, Expression, and Purification of Rv3868, C-terminal Domain, and CFP-10—The full-length Rv3868 gene from M. tuberculosis H37Rv was amplified using the Pfx DNA polymerase (Invitrogen). The C-terminal domain of Rv3868 (amino acids 330-481) (to be called CT-Rv3868) was amplified from the Rv3868 PCR product using the primers detailed in supplemental Table 1. Rv3868 was cloned into pET23a (Novagen) using NdeI and HindIII. CT-Rv3868 was cloned into pET23a using BamHI and HindIII. The Ct-Rv3868 open reading frame of pET23a was mutated by a site-directed mutagenesis kit (Stratagene) using the primers listed in supplemental Table 1. Full-length Rv3868, CT-Rv3868, and mutants of CT-Rv3868 were expressed in BL-21 (DE3) cells (0.5 mm isopropyl 1-thio-β-d-galactopyranoside; OD 0.6; 30 °C). The cells were harvested by centrifugation, resuspended in 40 ml of lysis buffer A (50 mm Tris-HCl, 200 mm NaCl, pH 7.5), and lysed by sonication. Centrifugation at 14,000 rpm was followed by a filtration step using a 0.22-μm filter before loading onto a 5-ml Ni-Hi Trap column equilibrated in buffer A. The column was initially washed with lysis buffer and subsequently with the same buffer containing 40 and 80 mm imidazole, respectively. The proteins were eluted with 15 ml of buffer B containing 200 mm imidazole for Rv3868 and 400 mm imidazole for CT-Rv3868. The samples containing protein were pooled and dialyzed extensively against buffer (50 mm Tris-HCl, 200 mm NaCl, pH 7.5).
For the purification of CFP-10, the plasmid pET28b-cfp10 (11) was grown in M9 medium using (N15) ammonium sulfate as sole nitrogen source and purified as reported earlier (11). The protein was dialyzed against NMR buffer (20 mm NaH2PO4, 50 mm NaCl, 0.1% NaN3, pH 6.5). About 10 mg of protein could be purified per liter of culture.
ATPase Activity Assays—ATPase reactions were carried out in
30 μl of ATPase buffer (25 mm Tris, pH 7.6, 5 mm
MgCl2) at 30 °C for different time periods. Each reaction
mixture contained 0.5 μCi of [γ-32P]ATP. The reaction was
stopped by the addition of 0.5 μl of 10% SDS. 1.0 μl of each reaction
was spotted on a TLC plate. The plate was developed in 0.5 m formic
acid and 0.5 m LiCl and dried at 37 °C. The percentage of ATP
hydrolysis was calculated using the following
formula.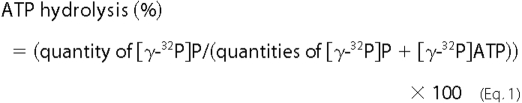
The ATP hydrolysis value was corrected for background by subtracting the value obtained for a reaction mixture containing no protein. Colorimetric assays (23) were performed to determine the ATPase activity of the CT-Rv3868. Except for specified variations, standard ATPase assays were carried out in the assay buffer containing 50 mm Tris-HCl (pH 8.0), 20 mm MgCl2, 1 mm dithiothreitol, 0.5 mm ATP, and 1 μg of protein for 15 min at 37 °C. Briefly, CT-Rv3868 was added to 100 μl of assay buffer; the reaction was carried out at 30 °C for 15 min; and then 200 μl of dye buffer containing 6 mm ammonium heptamolybdate, 120 μm malachite green, 0.06% polyvinyl alcohol, and 4.25% sodium citrate was added. After 20 min of incubation at room temperature, 200 μl from each reaction was transferred to a 96-well plate, and the absorbance at 630 nm was measured. Values from control reactions performed without protein were routinely subtracted from the respective experimental data. The inorganic phosphate released was calculated based on the absorbance standard curve established by KH2PO4 standards. CT-Rv3868 and NT-Rv3868 was purified by affinity chromatography to near homogeneity and used in the assays. All assays were repeated three times, and the average activity is reported. Kinetic parameters, Km, Vmax, and Hill coefficient, were derived using Prism 4.0 (GraphPad Software, Inc.).
Limited Proteolysis and Electrospray Ionization-Mass Spectrometry—2.0 mg/ml protein was subjected to limited proteolysis using trypsin at a protease/protein ratio of 1:50 and 1:100 (w/w) and incubated for different time periods at 30 °C. The protease reaction was stopped by adding phenylmethylsulfonyl fluoride to a final concentration of 1 mm in the reaction mixture, and the samples were analyzed on 12% SDS-PAGE. Digested product was purified by gel filtration chromatography and transferred to a polyvinylidene difluoride membrane for N-terminal sequencing. The electrospray ionization-mass spectrometry analysis was carried out using a MICRO-MASS QUATTRO II mass spectrometer (Micromass, Altricem, UK).
Tryptophan and Tyrosine Fluorescence—Protein concentrations of 0.5 and 1 μm for full-length and purified domains, respectively, were used. Fluorescence spectra were recorded using a PerkinElmer Life Sciences LS 50B instrument with samples placed in a 5-mm path length quartz cell at 25 °C. An excitation wavelength of 285 nm was used, and the spectra were recorded between 300 and 400 nm to monitor tryptophan fluorescence. Tyrosine fluorescence was monitored by using an excitation wavelength of 274 nm.
Analytical Gel Filtration and Dynamic Light Scattering—Gel filtration experiments were carried out using a Superdex 200 HR 10/300 column on an AKTA-FPLC system (GE Healthcare). The column was calibrated using molecular weight standard markers (GE Healthcare). All experiments were carried out using 50 mm Tris, pH 7.5. Other parameters like salt and nucleotide concentrations were varied for the experiments. Typically, 500 μl of the sample was loaded on the column and run at 25 °C at a flow rate of 0.3 ml/min, with detection at 280 nm.
The relative elution volume was calculated as follows,
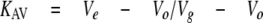 |
(Eq.2) |
where Ve is the elution volume, Vo is the void volume determined by elution of blue dextran 2000 kDa, and Vg is the geometric column volume. For deconvolution of gel filtration peaks, Peakfit (Systat Software, Inc.) software was used for determination of different oligomers in Rv3868.
The dynamic light scattering experiments were carried out on a Zetasizer Nano ZS instrument (Malvern Instruments). Data were acquired at 20 °C over 10 s, repeated 10 times, and averaged. Ten such acquisitions were performed to give 1000 s of data. The in-built software was used to fit the autocorrelation function using the cumulants method and to extract the approximate molecular weight.
Steady-state Nucleotide Binding—Binding of nucleotides to the proteins were determined by monitoring the change in protein fluorescence upon the addition of ligand. Measurements were carried out using a PerkinElmer Life Sciences LS 50B spectrofluorimeter, (excitation 280 nm; emission 330 nm; slit widths 5 nm) for Rv3868 and NT-Rv3868, where tryptophan fluorescence was followed. In the case of the CT-Rv3868, an excitation wavelength of 274 nm was used along with a 5-nm slit width. Tyrosine emission was followed at 304 or 340 nm for lower order and high order oligomeric forms of the domain. Titrations were performed at 25 °C by the addition of ATP to 0.6 ml of 50 mm Tris (pH 7.5), 50 mm NaCl, and 5 mm MgCl2 buffer containing different amounts of proteins. To avoid dilution effects, volume change during the titration was limited to 3% of total volume. Control titrations with buffer alone did not produce any significant change in emission signal. The Kd value was calculated, fitting the data to the Equations 3 and 4. ΔF is the change in emission signal in the presence of ligand (L), and ΔFmax is the maximal change in signal. The corrected data were fitted to the following equations using Prism 4.0 (GraphPad Software).
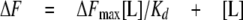 |
(Eq.3) |
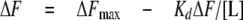 |
(Eq.4) |
The binding stoichiometry of nucleotides and C-terminal domain was determined by plotting the titration data as a mass action plot according to the following equation.
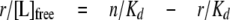 |
(Eq.5) |
The fluorescently labeled ATP analog, N-methylanthraniloyl-ATP (MANT-ATP)3 (Molecular Probes) was used to qualitatively substantiate the binding of the nucleotide to the proteins. All spectra were corrected for the inner filter and dilution effects. Nucleotide binding to CT-Rv3868 was followed by the changes in MANT-ATP emission at 450 nm, (excitation and emission wavelengths). 1 μm MANT-ATP was titrated with increased concentrations of the protein in the experiments.
Stern-Volmer Coefficients—Fluorescence quenching of tryptophan in the presence of increasing concentrations of acrylamide was monitored by following the emission at 340 nm after excitation at 285 nm. Samples were prepared in buffer consisting of 50 mm Tris, pH 7.5, 50 mm NaCl, and 5 mm MgCl2. Aliquots from a 2 m acrylamide stock solution were consecutively added in 5 mm steps to 1 ml of reaction mixture. Experiments were performed in triplets and corrected for dilution effects. Quenching data were plotted as the ratio of florescence in absence of quencher (F0) to the intensity in the presence of quencher (F) against quencher concentration. The resulting data were fit against dynamic parameters according to the Stern-Volmer equation (24).
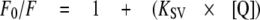 |
(Eq.6) |
KSV is the Stern-Volmer constant for quenching, given by the slope when data are plotted as F0/F versus [Q], where the latter parameter is the concentration of the quencher.
ANS Binding—Titrations were performed to estimate the binding affinities of the proteins to 8-anilino-1-naphthalenesulfonic acid (ANS). Incremental amounts of ANS were added to a series of otherwise identical solutions of protein in buffer (50 mm Tris, pH 7.5, 50 mm NaCl, and 5 mm MgCl2). The excitation wavelengths were set to 370, and the emission was measured from 410 to 600 nm, respectively. For each measurement, the fluorescence intensity was corrected by subtracting the fluorescence of the sample containing only ANS. The data were plotted against the total concentration of ANS. The apparent Kd was estimated by fitting the data to Equation 7,
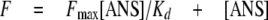 |
(Eq.7) |
where F is the corrected fluorescence intensity, Fmax is the fluorescence intensity upon saturation of the ANS binding sites, [ANS] is the total concentration of ANS, and Kd is the apparent dissociation constant.
Glutaraldehyde Cross-linking—The cross-linking of protein samples was carried out in the presence of 1% glutaraldehyde. CT-Rv3868 was used in the experiments at a concentration of 0.2 mg/ml. The molecular masses of the cross-linked products were determined by 12% SDS-PAGE.
Protein Modeling and NTP Docking—Sequence analysis led to the identification of putative Walker A and B motifs at the C-terminal end. A model of the putative ATP binding site was generated by comparative modeling approaches and corresponds to residues 331-481 of CT-Rv3868 (25). The NT-Rv3868 domain model (residues 18-250) was generated using PHYRE (available on the World Wide Web), following the fold prediction method. The initial models were minimized using the DISCOVER module implemented in Insight II (Accelrys).
The AUTODOCK program was used in the in silico docking studies involving NTPs and CT-Rv3868. Partial charges were assigned using the CVFF force field. The grid maps consisting of 80 × 80 × 80 grid points were centered on the putative ligand-binding site (Walker A motif). The Lamarckian Genetic Algorithm was used for the calculations. Docked complexes were visualized using InsightII (Accelrys) and PyMol (26). The oligomer was modeled by superposing the C-terminal model structure onto the hexameric D2 domain of NSF (Protein Data Bank code 1NSF) (27).
NMR Spectroscopy—For the NMR experiments 15N-labeled CFP10 protein in 20 mm sodium phosphate (pH 6.5), 50 mm NaCl, 0.1% sodium azide, and 5% (v/v) 2H2O was used as reported in earlier experiments by our group (11). The spectra were recorded on a Varian 600-MHz instrument equipped with a triple nuclei inverse probe, at 30 °C. Two-dimensional 15N-1H HSQC spectra were recorded for the 15N-labeled CFP-10 as well as for the 15N-labeled CFP-10-unlabeled Rv3868 protein. The HSQC spectrum for each experiment was acquired with 1024 and 128 complex points in the 1H and 15N dimensions, respectively.
Chaperone-like Activity Assay—The assays were carried out using procedures similar to those described earlier (28) at 43 °C using hen egg white lysozyme (Sigma) and porcine mitochondrial citrate synthase (Sigma) as test substrates.
RESULTS
Sequence and Phylogenetic Analysis—Rv3868 consists of 573 amino acids with molecular mass of ∼63,000 kDa. The protein has been classified as a conserved hypothetical protein in the data bases. Sequence analysis and the construction of a phylogenetic tree using the neighbor-joining method supports that Rv3868 is a member of the CbxX/CfqX subfamily of AAA-ATPases (supplemental Fig. 1). The sister group of CbbX proteins are sporulation factors (29). The related proteins in mycobacteria like Rv3868 have apparently acquired alternate functions.
The Hypothetical Protein Rv3868 of M. tuberculosis Encodes a Hexameric ATPase—At first it was important to probe for the ATPase activity of the protein, if any, in view of the Walker motifs (supplemental Fig. 2) contained in the sequence. It became clear from the initial colorimetric assays that the full-length enzyme is a weak ATPase. The presence of bound nucleotide through the purification process was ruled out by extensive dialysis. The more sensitive radioactive assay involving [γ-32P]ATP as a substrate was therefore used in subsequent experiments, where the release of free phosphate was found to linearly increase over time (Fig. 1A). A Km of 0.8 ± 0.1 μm and Vmax of 139 ± 8.8 fmol/min was derived from a Michaelis-Menten plot (Fig. 1B) following a nonlinear regression analysis using Prism 4.0 (GraphPad Software).
FIGURE 1.

Functional characterization of Rv3868. A, the protein was incubated with [γ-32P]ATP for various time intervals to observe the ATPase activity. The amount of ATP hydrolyzed at each time point is shown as a percentage of the original [γ-32P]ATP before incubation at 30 °C. Each point is the average of the values obtained from three independent experiments. The insets represent the autoradiography profiles taken from a TLC. B, Michaelis-Menten plot of ATP hydrolysis by Rv3868. C, relative NTP hydrolysis (arbitrary units) when [γ-32P]ATP and [γ-32P]GTP were used as substrates of Rv3868. D, the effect of NaCl, casein, and EDTA on ATPase activity. The first bar represents the control experiment. Inset, lanes 1-5 represent the control experiment, the activity in the presence of 0.5 m NaCl, 5 or 10 μg of casein, and 10 mm EDTA, respectively.
Rv3868 was found to be a specific ATPase. The GTPase activity under similar assay conditions was only 20% of that observed with ATP (Fig. 1C). The ATPase assays were also carried out in the presence of casein, DNA, ESAT-6, and CFP-10. Casein and DNA (30, 31) are known to variously stimulate ATPase activity in some AAA-proteins, whereas ESAT-6/CFP-10 have been postulated to interact with Rv3868 (19). No stimulatory or inhibitory effects on the activity were observed in the presence of these factors (data not shown). However, the addition of 0.3 m NaCl or 25 mm EDTA abolished the activity (Fig. 1D). It is possible that NaCl could disrupt the oligomeric associations in the enzyme, and these were investigated subsequently. EDTA apparently chelates out the Mg2+ ions that are necessary for the activity.
Analytical gel filtration experiments show that Rv3868 predominantly exists as a hexamer at protein concentrations up to ∼3 mg/ml and elutes at a molecular mass of ∼380 kDa (Fig. 2A). At high concentrations, the protein forms higher order oligomers with concomitant reduction in the hexamer population (Fig. 2B). Dynamic light scattering experiments further suggest that the higher oligomeric state is a not an aggregate and that the protein exists as a multiple of hexamers (supplemental Fig. 3).
FIGURE 2.

Protein and salt concentration dependence of the oligomerization in Rv3868. A, size exclusion chromatography profile of Rv3868 (∼3 μm) on a Superdex 200HR column in 25 mm Tris, 50 mm NaCl (pH 7.0), at 25 °C. The inset represents the calibration curve of the column. B, Rv3868 at concentrations of ∼7-15 μm (bottom) and >15 μm (second from bottom) was incubated for 1 h at 25 °C before centrifugation and applied onto a Superdex 200HR column in 25 mm Tris buffer. The hexameric form is represented by I, whereas II refers to a high order oligomeric form. The second from top and top lines represent the elution profile in the presence of 500 and 750 mm NaCl, respectively, in the same buffer. Elution volumes corresponding to the Mr of standard proteins used in calibration are indicated by arrows.
The quaternary associations are stabilized by ionic interactions. At an NaCl concentration of ∼0.5 m, the protein is predominantly dimeric. Increasing the concentration to above 0.75 m resulted in the breakage of the dimers to monomers. The addition of ATP did not make any difference to the elution profiles in the experiments (Fig. 2B). Similar effects were observed in the case of guanidinium chloride, a known disrupter of ionic interactions in proteins (data not shown).
Identification of a Stable N-terminal Domain—The vulnerability of a protein for proteolysis depends on parameters such as accessibility, segmental motion, and protrusions. Therefore, limited proteolysis has been effectively used to identify structural domains in proteins, ligand-induced conformational changes, and protein folding/unfolding (32).
The incubation of Rv3868 with trypsin gave rise to mainly two fragments in the SDS-polyacrylamide gels (Fig. 3C). A fragment of molecular mass ∼30 kDa was quite stable under the digestion conditions, whereas the other fragment (∼20 kDa) degraded with time. The former fragment could be purified from the reaction mixture by size exclusion chromatography as a monomer (Fig. 3D) and has a molecular mass of 29.9 kDa, as deduced from electrospray ionization-mass spectrometry. The fragment, unlike the full-length protein, did not exhibit any concentration-dependent self-association. Peptide sequencing established that this fragment/domain occurs in the N terminus of the protein with starting sequence TDRLA (Fig. 3B).
FIGURE 3.

Domain organization and limited proteolysis of Rv3868; purification of NT-Rv3868. A, an estimate of the folded regions in Rv3868 calculated using the program Fold Index. The predicted folded regions are shown in positive values, whereas unstructured regions are shown in negative values and clearly show the demarcation of the protein sequence into two distinct domains. B, a schematic depiction of the predicted domain boundaries and motifs of Rv3868. C, SDS-PAGE analysis of Rv3868 digested with trypsin at a protease to protein ratio of 1:100 (w/w) and 1:50 (w/w). Lanes 2-5 and lanes 6-9 represent the reactions after 5, 10, 15, and 30 min, respectively. D, size exclusion chromatography profile of Rv3868 after trypsin digestion for 30 min (NT-Rv3868). The protein was loaded onto the column in 25 mm Tris, 50 mm NaCl (pH 7.0) at 25 °C. The inset in D depicts the SDS-PAGE analysis of the purified N-terminal domain. Lanes 1-4 represent molecular weight markers, the reaction after 0 min, the reaction after 30 min, and purified protein following size exclusion chromatography, respectively.
Sequence analysis (supplemental Fig. 2) had shown that only the N-terminal stretch contains a tryptophan, whereas the C-terminal stretch does not contain any. We could therefore exploit this fact in later spectroscopy experiments. Subsequent activity assays also revealed that the fragment, as expected, does not possess any ATPase activity observed in the full-length enzyme. The analysis therefore reveals that the N-terminal domain is compact, accounts for approximately half the sequence of the enzyme, and is a monomer, in contrast to the hexameric association observed in the full-length protein.
Identification and Characterization of the ATP-binding Domain—Fold index (33) (Fig. 3A) calculations for Rv3868 suggests that residues between 330 and 481 in the C terminus contain the Walker motifs/ATP binding site and should encode for an ∼18-kDa fragment (Fig. 3A).
The C-terminal domain was accordingly cloned and purified separately. The domain associates predominantly as a dimer in the absence of ATP and forms higher order oligomers in the presence of the nucleotide (ATP), as deduced from gel filtration and glutaraldehyde cross-linking experiments (Fig. 4A). The results suggest that the C-terminal domain is largely responsible for oligomerization.
FIGURE 4.

Purification and functional characterization of CT-Rv3868. A, size exclusion chromatography profiles of CT-Rv3868 in the absence (gray line) and presence of ATP (black line). The column was equilibrated with 25 mm Tris, 50 mm NaCl, pH 7.0, (with or without 2 mm ATP) and at 25 °C. Inset, relative ATPase activity of different forms. B, time dependence of ATPase activity of CT-Rv3868. The reactions were carried out at 30 °C for the indicated time periods (min). C, Michaelis-Menten plot of ATP hydrolysis by CT-Rv3868. The ATPase assays were carried out using 1.0 μg of protein and at the indicated concentrations of ATP. D, concentration dependence of ATP hydrolysis was analyzed using a plot involving specific activity (pmol min-1 mg-1) versus CT-Rv3868 concentration (μg).
Next, the CT-Rv3868 was tested for the ability to hydrolyze ATP using the malachite green assay. A linear release of phosphate was observed during the time course of the assay (Fig. 4B). The activity was found to be maximal between 7.5 and 8.5 pH. The following parameters for the hydrolysis activity were also deduced: Vmax of 141.2 ± 12 with a Km of 73.39 ± 20, Kcat of 2.541 ± 0.23, and Hill coefficient (n) of 1.40 (Fig. 4C). The ATPase activity was found to be severalfold higher compared with the full-length protein. It was also found to be co-operative as suggested from the Hill coefficient.
The cooperativity was also supported by following a plot of ATP hydrolysis versus protein concentration (Fig. 4D). The concave plot indicates that the activity is concentration-dependent. The specific activity increases with an increase in the enzyme concentration until a maximal activity of 50 nmol/min/mg. Concentration-dependent activity is suggestive of cooperative association and has been identified for a number of characterized NTPases, including AAA-ATPases (34).
In Silico Modeling and Docking Studies—The characterization clearly suggests that the CT-Rv3868 is involved in oligomerization. The domain also exhibits homology with the AAA-domain present in other structurally characterized AAA-proteins. We therefore modeled the hexameric association of the protein based on the D2 domain of N-ethylmaleimide-sensitive factor (27) (Protein Data Bank code 1NSF) (Fig. 5A). A detailed examination of the resultant model suggested that the binding pocket is lined by Pro-336, Gly-337, Thr-338, Lys-340, and Arg-429, among other residues. We also carried out in silico docking experiments with different nucleotides, including ATP, ADP, GTP, CTP, and UTP, to examine their respective binding modes. The ATP moiety has the highest affinity for the protein, followed by GTP, and supports the experimental results where the GTPase activity of the protein was found to be only one-third that of ATP (Table 1). The bound ATP lies in a defined area deep within the binding site cleft, and the γ-phosphate is proximal to the side chains of Pro-336, Gly-337, Thr-338, Lys-340, and Arg-429 (Fig. 5B). The latter residue is from the neighboring subunit, and we suspected from the spatial disposition that it might function as an arginine finger/sensor arginine (20) that senses the presence of the nucleotide in the binding site and gives rise to associated mechanochemical outcomes in AAA-ATPases. The modeling results were subsequently substantiated experimentally.
FIGURE 5.

In silico modeling and mutational analysis of Rv3868. A, hexameric association in Rv3868. The nucleotide binding sites occur at the intersubunit interfaces as in other AAA-ATPases. The arrows indicate movement of the N-terminal domain predicted by the dynamic quenching and other experiments. The ATP-binding site is marked by a box. B, close-up of the ATP binding site in Rv3868. The residues corresponding to the Walker A motifs are indicated by cyan space-filled models. Two tyrosine residues (positions 439 and 466) in the close vicinity of the nucleotide are depicted as red sticks. Arg-429, the predicted sensor arginine from the modeling studies, is depicted as a yellow stick and is from a neighboring subunit. C, Michaelis-Menten plots of ATP hydrolysis by CT-Rv3868 (□) and CT-Rv3868P336A (○), CT-Rv3868T338A (•), CT-Rv3868K340A (▵), and CT-Rv3868R429A (*).
TABLE 1.
In silico docking energy and relative activity (percentage) of different nucleotides for CT-Rv3868
| Activity | Free energy | Relative activity |
|---|---|---|
| kcal/mol | % | |
| ATP | −9.06 | 100 |
| GTP | −6.8 | 35 |
| UTP | −5.61 | NDa |
| CTP | −5.92 | ND |
| ADP | −7.52 | 5 |
ND, not determined.
Orientation of Bound Nucleotides—We used the fluorescent ATP analog MANT-ATP, where the fluorophore is attached to the ribose moiety, to probe for the orientation of the nucleotide in the binding site. The binding of this analog close to a hydrophilic pocket causes a decrease in the fluorescence intensity (35, 36). Indeed, a reduction in the fluorescence intensity was observed on titration of the nucleotide analog with increasing amounts of CT-Rv3868 (supplemental Fig. 4). An examination of the docked ATP-CT-Rv3868 complex reveals a hydrophilic pocket near the adenosine moiety (Fig. 5B). Hence, the experiments with the fluorescent ATP analog support the orientation of the nucleotide suggested by the docking experiments.
Mutational Analysis and Identification of Arg-429 as a Sensor Arginine—We generated four mutants of CT-Rv3868 (viz. P336A, T338A, K340A, and R429A) based on the modeling studies to probe for the roles of the residues in ATP binding and hydrolysis (Fig. 5B). The first three mutants correspond to those residues that belong to the same subunit in the nucleotide binding site, whereas the Arg residue is from a symmetry-related subunit of the oliogmer. Thr-338 and Lys-340 lie close to the γ-phosphate in the docked complex. Arg-429 was chosen to examine its role as a probable sensor arginine, whereas the Pro residue was mutated to check for possible structural effects on the binding site architecture.
Table 2 lists the various parameters of the respective mutants. The wild-type protein has a catalytic efficiency of about 577, as suggested by the Kcat/Km ratio. The Hill coefficient of 1.4 is indicative of the positive cooperativity in CT-Rv3868. The P336A mutant does not seem to distort the binding site architecture; the catalytic efficiency as also the Vmax is only marginally reduced in the mutant. The Thr-338 and Lys-340 residues apparently perform different roles in the hydrolysis. Thr-338 contributes to the binding, and its mutation leads to ∼7-fold decrease in the binding of the substrate, as suggested by the Km values. The catalytic efficiency also is reduced ∼10-fold. The reduction in the affinity of ATP in this mutant is also supported by the positive change in the free energy. The K340A mutant does not affect the binding of the substrate, and the Km is relatively unaffected, but there is an ∼35% reduction in the catalytic efficiency as also the Vmax, indicating that this residue has a rigorous role in the catalytic action as opposed to stabilizing the substrate. In the three mutations detailed above, the cooperativity is relatively unaffected.
TABLE 2.
Kinetic parameters of CT-Rv3868 and its mutants
| Vmax | Kcat | Km | Kcat/Km | n (Hill coefficient) | ΔGATPa | |
|---|---|---|---|---|---|---|
| nmol/min/mg | min−1 | μM | M−1 S−1 | kcal/mol | ||
| CT-Rv3868 | 141.2 ± 12 | 2.541 ± 0.23 | 73.39 ± 20 | 577 | 1.40 | 0.0 |
| P336A | 130.9 ± 12 | 2.357 ± 0.23 | 79.97 ± 21 | 491 | 1.37 | 0.096 |
| T338A | 87.33 ± 30 | 1.572 ± 0.5 | 524.3 ± 70 | 49 | 1.34 | 1.47 |
| K340A | 117.2 ± 15 | 2.110 ± 0.28 | 92.29 ± 30 | 380 | 1.31 | 0.256 |
| R429A | 106.4 ± 25 | 1.914 ± 0.5 | 439.8 ± 60 | 72 | 1.15 | 1.244 |
ΔG = -RTln((Kcat/Km)mut)/((kcat /km)wt).
The R429A mutant exhibits a large increase in the Km and a drastic reduction in the catalytic efficiency (Fig. 5C). The positive free energy change also indicates a loss in the binding of the substrate. These parameters are similar to those seen in the T338A mutant. An important difference is that although the Thr mutation did not lead to loss in cooperativity, the R429A mutation almost abolishes the cooperativity, as seen by the Hill coefficient of 1.15. Binding and release of the nucleotide in AAA-ATPases are generally known to lead to changes in the conformation of the oligomer and give rise to cooperative effects. Obviously, the conformational adjustments necessary for the binding of the nucleotide are precluded in the mutant. These properties are consistent with the in silico prediction of Arg-429 as a sensor arginine.
Nucleotide Binding and ATP-dependent Self-association of the Mutants—The loss in the ATP binding affinity of the R429A mutant was further substantiated by the measurement of the dissociation constants using fluorescence spectroscopy (Fig. 6A). We exploited the fact that CT-Rv3868 contains only 4 tyrosine residues, of which two are predicted by modeling to be close to the ATP binding (Fig. 4C). On the other hand, all 8 Trp residues of Rv3868 occur in the N-terminal domain and are quite accessible to the aqueous environment, as delineated by experiments involving the full-length and NT-Rv3868 proteins (supplemental Fig. 5). The affinity of ATP for CT-Rv3868 was found to be 0.27 ± 0.5 mm, whereas it reduces ∼4-fold in the R429A mutant to 1.08 ± 0.02 mm. The stoichiometry calculated through an analysis of the Scatchard plot (Fig. 5A) was found to be ∼1 ATP molecule per CT-Rv3868 chain, as expected.
FIGURE 6.

Steady-state nucleotide binding of CT-Rv3868. A, emission spectra of 1 μm CT-Rv3868 (□) and in the presence of, for example, 0.1 mm (○), 0.2 mm (▵), 0.5 mm (▪), 0.7 mm (▴), and 1.0 mm (+) ATP, respectively. Inset, Scatchard plot of the titration of CT-Rv3868 (Form III) and ATP according to Equation 5 to determine the binding stoichiometry of the complex. B, steady-state binding of ATP to CT-Rv3868 (▪) and CT-Rv3868R429A (▾). Data are plotted as the percentage of fluorescence change versus ATP concentration (mm). C, ATP-induced alterations in the molecular dimension of CT-Rv3868R429A (top) and CT-Rv3868 (bottom). Shown are size exclusion chromatographic profiles for proteins (black) and upon incubation with 2.0 mm ATP (gray) on a Superdex-200 column at pH 7.0 and 25 °C. The columns were run with the same concentration of ATP in which the protein sample was incubated. The samples were incubated for 1 h in ATP before column chromatography.
The above conclusions are supported by ATP-dependent self-association experiments. CT-Rv3868, as mentioned earlier, exhibits ATP-dependent self-association and also shows cooperativity. All mutants except the Arg-429 mutant exhibit ATP-dependent self-association, and also the cooperativity is relatively unaffected. However, the Arg mutant loses the ability to self-associate in the presence of ATP, and the cooperativity is also nearly abolished (Table 2 and Fig. 6C).
N- and C-terminal Domains Exhibit Relative Conformational Changes Linked to Nucleotide Binding—We carried out a dynamic quenching study on the full-length protein and the NT-Rv3868 using acrylamide as a quencher. This moiety, on account of its polar nature, interacts with tryptophan residues, which are exposed or partially buried, and leads to a quenching of the fluorescence. This approach gives insights into relative conformational changes between the domains based on the quenching of the tryptophan fluorescence, as also reported earlier (37). Probing the individual accessibility of each Trp residue rigorously requires the determination of kq, the bimolecular rate constant, kq = KSV × τo, where KSV and τo are the Stern-Volmer constant and fluorescence lifetime, respectively. However, the presence of 8 Trp residues impeded the determination of τo for the individual residues. As is generally accepted, the conformational changes can alternatively be studied by comparing the Stern-Volmer constants rather than the bimolecular rate constants. The Stern-Volmer plots for the NT-Rv3868 and full-length Rv3868 in the presence and absence of ATP are shown in Fig. 7A. The KSV for the N-terminal domain alone is 9.37 ± 0.53 m-1. The KSV corresponding to the full-length protein in the absence of ATP is 5.12 ± 0.54 m-1, whereas it is 4.11 ± 0.96 m-1 in its presence. If the KSV values for the full-length protein and the NT-Rv3868 alone were similar, it would suggest that the two domains in the protein are not in close proximity, since the accessibility of the individual Trp residues is relatively unaffected. However, the present experiments represent a direct evidence for the proximity of the two domains in the protein. A significant reduction was observed in the KSV value for the ATP-bound enzyme compared with the unbound form. This clearly suggests that the two domains move closer to each other from a relatively “open” to a “closed” conformation upon the addition of the nucleotide. From the above results, it is straightforward to visualize that the binding of nucleotide co-factor and its release should be accompanied by a concomitant change in the relative spatial dispositions of the N- and C-terminal domains.
FIGURE 7.

Trp fluorescence quenching studies of Rv3868 and NT-Rv3868 by acrylamide and ATP. The quenching of intrinsic fluorescence of the proteins was followed after the addition of increasing concentrations of acrylamide or ATP. A, fluorescence quenching observed in Rv3868 (•), Rv3868 + ATP (▾), and NT-Rv3868 (▴) proteins upon the addition of acrylamide. The Stern-Volmer quenching constants (KSV) corresponded to plot slopes (Equation 6). B, relative decrease in TRP fluorescence upon the addition of ATP in Rv3868 + ATP + MgCl2 (1), Rv3868 + ATP (2), and NT-Rv3868 + ATP+MgCl2 (3) samples, respectively.
The above results were independently corroborated by following the intrinsic fluorescence of the Trp residues in the presence of ATP. The addition of the nucleotide led to a reduction in the observed Trp fluorescence in the full-length protein. On the other hand, the addition of the nucleotide aliquots to the NT-Rv3868 alone leaves the observed fluorescence relatively undisturbed. Since the Trp residues occur only in the N-terminal segment, which has no nucleotide binding sites, the quenching can only be presumably due to the increased proximity of the two individual domains upon the addition of the nucleotide and corresponding reduction in the accessibility of surface-exposed tryptophan residues (Fig. 7B).
Rv3868 Does Not Interact with CFP-10 and Does Not Exhibit Chaperone-like Activity—Previously, it was suggested that Rv3868 might interact with CFP-10 or ESAT-6 proteins (13, 19). Other groups have suggested a chaperone function for the protein (6, 9, 13, 19). Since a predicted recognition motif is present in the C-terminal segment of CFP-10 (18), NMR studies were undertaken to identify possible interactions with the latter protein. Our own earlier NMR studies have suggested that complex formation confers thermodynamic stability (11). CFP-10 by itself is unstructured, as reported earlier by others and us. The spectra show no change in the presence of unlabeled Rv3868 both in the presence and absence of ATP. The experiments clearly rule out any interactions of CFP-10 and therefore the C-terminal recognition motif with Rv3868 (supplemental Fig. 6).
Possible chaperone-like activities were also probed and ruled out using substrates like hen egg white lysozyme and porcine citrate synthase, where the possible disaggregation of the substrates in the presence of Rv3868 was monitored spectroscopically (supplemental Fig. 7).
In another set of experiments, the presence of hydrophobic patches on the surface of the protein was probed using ANS or bis-ANS binding studies. It is known that substrate polypeptides bind to large hydrophobic patches on the substrate binding domains in related AAA-ATPases like HslU, ClpA, and Hsp with chaperone/protease-like activities (38, 39). Since Rv3868 contains two domains, the N-terminal domain is expected to have hydrophobic patches to bind to substrates if it had a chaperone function. The C-terminal domain, on the other hand, has been shown to be the ATPase domain, which is also involved in oligomerization. The binding data reveal that the N-terminal domain has no hydrophobic patches and is compact, whereas the full-length and ATP binding domains have similar affinity for ANS (supplemental Fig. 8). Altogether, these studies suggest that the enzyme is not likely to have a general chaperone-like function. However, a specific chaperone activity in the presence of as yet unidentified co-factors and/or unknown substrate proteins cannot be ruled out.
DISCUSSION
The present work represents the first detailed characterization of a protein from the CbbX family of proteins. The protein has been shown to be a hexamer, with each chain consisting of two domains. The C-terminal domain is the ATPase and oligomerization domain, whereas the N-terminal domain is compact and has no significant sequence homology to characterized proteins. The full-length protein is a relatively weaker ATPase compared with the CT-Rv3868 alone. Analogous behavior has been observed in some other AAA-ATPases (e.g. E. coli ClpB), where the full-length enzyme hydrolyzed ATP with a lower rate compared with the ATP binding domain alone (30). Often the interactions of the substrate binding domain with target proteins or co-factors stimulate NTPase activity in the respective proteins (e.g. ClpB shows enhanced ATPase activity in the presence of casein). The target protein of Rv3868 is conjectured to be ESAT-6/CFP-10 or Rv3873 based on yeast two-hybrid or genetic experiments (19). Our reported work apparently rules out direct interactions with the ESAT-6/CFP-10 proteins, and their presence also does not stimulate the ATPase activity in the assays. A general chaperone activity was also probed. The protein also does not exhibit large hydrophobic patches on the surface, which is a generally accepted characteristic of a chaperone.
Other groups have identified at least four substrates of the ESX-1 system (viz. ESAT-6, CFP-10, EspA, and EspB), and it is known that disruption of the Rv3868 gene prevents secretion of the substrates, although their expression is not impaired (13, 14). This has led to a suggestion that the protein affects either the translocation or stability of the exported substrates. The lack of a general chaperone activity and absence of interactions with CFP-10 suggests that Rv3868 probably has a role in the translocation of the substrates rather than their stability. This then brings us to the question as to which are the likely interacting partners of Rv3868? One possibility, based on earlier work (13, 19) against the backdrop of the current characterization could be Rv3873, a gating protein. The interactions of Rv3868 with the gating protein would specifically modulate the secretion of the virulence factors, in agreement with the essential role of Rv3868 in secretion, but would not affect the expression of these factors. However, more genetic work is necessary to identify and characterize the interactions of these potential protein partners.
The other conjectured function of Rv3868 is to transfer energy to co-proteins of the ESX-1 system. AAA-ATPases normally translate the conformational changes effected by the ATPase motor to other domains of the protein to effect functional consequences (e.g. HslU (40) undergoes conformational changes upon ATP binding and release to unfold proteins destined for proteolysis). In the case of Rv3868, the two domains are in close proximity upon ATP binding. The release of the nucleotide leads to a distinct relative conformational change between the domains where the N terminus is more accessible to the environment. This open-close movement apparently enables interactions with target proteins and is consistent with the behavior of other characterized AAA-ATPases like HslU or ClpB.
The in silico modeling and docking calculations has helped rationalize the observed activities and also the affinity of the protein for different nucleotides. An exciting outcome of these studies is the identification of Arg-429 as a potential sensor arginine (20). In the model of the monomer alone, the residue is far from the nucleotide binding site. It comes close to the binding site of the neighboring subunit in the oligomer to form a part of the binding site. This residue is known to play a special role by transducing the ATP hydrolysis/binding event into a mechanochemical outcome in AAA-ATPases. However, the catalytic functions in the respective proteins are known to be different, and they play a context-specific role in the ATPases. Although the Thr-338 and Lys-340 mutants affect the binding of the nucleotide to varying degrees, they do not disrupt the observed cooperativity (i.e. the conformational changes in the oligomer that occur upon ATP binding are relatively undisturbed). The Arg-429 mutant abolishes cooperativity and leads to a large reduction in binding of the nucleotide, underscoring its role as a sensor arginine.
In summary, this is the first detailed characterization of the hypothetical protein Rv3868, a critical component of the ESX-1 pathway in M. tuberculosis. The studies suggest a possible molecular mechanism involving co-factor-induced relative conformational changes in the domains through which the protein can interact with other proteins of the pathway. The characterization, molecular modeling, and mutational analysis of Rv3868 set the stage for the identification of novel inhibitors that can disrupt the export of critical tuberculosis virulence factors.
Supplementary Material
This work was supported by Intramural Grant MLP007, and computational support was from the Council of Scientific and Industrial Research, India, Grant CMM0017. This is communication 7469 from the Central Drug Research Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. 1-8.
Footnotes
The abbreviations used are: MANT-ATP, N-methylanthraniloyl-ATP; NT-Rv3868, N-terminal domain of Rv3868; CT-Rv3868, C-terminal ATP-binding domain of Rv3868; ANS, 8-anilino-1-naphthalenesulfonic acid.
References
- 1.Pym, A. S., Brodin, P., Brosch, R., Huerre, M., and Cole, S. T. (2002) Mol. Microbiol. 46 709-717 [DOI] [PubMed] [Google Scholar]
- 2.Hsu, T., Hingley-Wilson, S. M., Chen, B., Chen, M., Dai, A. Z., Morin, P. M., Marks, C. B., Padiyar, J., Goulding, C., Gingery, M., Eisenberg, D., Russell, R. G., Derrick, S. C., Collins, F. M., Morris, S. L., King, C. H., and Jacobs, W. R., Jr. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 12420-12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sassetti, C. M., and Rubin, E. J. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 12989-12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanley, S. A., Raghavan, S., Hwang, W. W., and Cox, J. S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13001-13006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guinn, K. M., Hickey, M. J., Mathur, S. K., Zakel, K. L., Grotzke, J. E., Lewinsohn, D. M., Smith, S., and Sherman, D. R. (2004) Mol. Microbiol. 51 359-370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao, L.-Y., Guo, S., McLaughlin, B., Morisaki, H., Engel, J. N., and Brown, E. J. (2004) Mol. Microbiol. 53 1677-1693 [DOI] [PubMed] [Google Scholar]
- 7.Fortune, S. M., Jaeger, A., Sarracino, D. A., Chase, M. R., Sassetti, C. M., Sherman, D. R., Bloom, B. R., and Rubin, E. J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 10676-10681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu, J., Laine, O., Masciocchi, M., Manoranjan, J., Smith, J., Du, S. J., Edwards, N., Zhu, X., Fenselau, C., and Gao, L.-Y. (2007) Mol. Microbiol. 66 787-800 [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin, B., Chon, J. S., MacGurn, J. A., Carlsson, F., Cheng, T. L., Cox, J. S., and Brown, E. J. (2007) PLoS Pathogens 3 e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renshaw, P. S., Panagiotidou, P., Whelan, A., Gordon, S. V., Hewinson, R. G., Williamson, R. A., and Carr, M. D. (2002) J. Biol. Chem. 277 21598-21603 [DOI] [PubMed] [Google Scholar]
- 11.Meher, A. K., Bal, N. C., Chary, K. V. R., and Arora, A. (2006) FEBS J. 273 1445-1462 [DOI] [PubMed] [Google Scholar]
- 12.Pym, A. S., Brodin, P., Majlessi, L., Brosch, R., Demangel, C., Williams, A., Griffiths, K. E., Marchal, G., Leclerc, C., and Cole, S. T. (2003) Nat. Med. 9 533-539 [DOI] [PubMed] [Google Scholar]
- 13.Brodin, P., Majlessi, L., Marsollier, L., de Jonge, M. I., Bottai, D., Demangel, C., Hinds, J., Neyrolles, O., Butcher, P. D., Leclerc, C., Cole, S. T., and Brosch, R. (2006) Infect. Immun. 74 88-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahairas, G. G., Sabo, P. J., Hickey, M. J., Singh, D. C., and Stover, C. K. (1996) J. Bacteriol. 178 1274-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr, M. A., Wilson, M. A., Gill, W. P., Salamon, H., Schoolnik, G. K., Rane, S., and Small, P. M. (1999) Science 284 1520-1523 [DOI] [PubMed] [Google Scholar]
- 16.Lewis, K., Liao, R., Guinn, K., Hickey, M., Smith, S., Behr, M., and Sherman, D. (2003) J. Infect. Dis. 187 117-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan, T., Lee, W. L., Alexander, D. C., Grinstein, S., and Liu, J. (2006) Cell. Microbiol. 8 1417-1429 [DOI] [PubMed] [Google Scholar]
- 18.Champion, P. A. D., Stanley, S. A., Champion, M. M., Brown, E. J., and Cox, J. S. (2006) Science 313 1632-1636 [DOI] [PubMed] [Google Scholar]
- 19.Teutschbeina, J., Schumannb, G., Möllmannb, U., Grableyc, S., Cole, S. T., and Munder, T. (2006) Microbiol. Res. 10.1016/j.micres.2006.11.016 [DOI] [PubMed]
- 20.Ogura, T., Whiteheart, S. W., and Wilkinson, A. J. (2004) J. Struct. Biol. 146 106-112 [DOI] [PubMed] [Google Scholar]
- 21.Neuwald, A. F., Aravind, L., Spouge, J. L., and Koonin, E. V. (1999) Genome Res. 9 27-43 [PubMed] [Google Scholar]
- 22.Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997) Nucleic Acids Res. 25 4876-4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanzetta, P. A., Alvarez, L. J., Reinach, P. S., and Candia, O. A. (1979) Anal. Biochem. 100 95-97 [DOI] [PubMed] [Google Scholar]
- 24.Lakowicz, J. (1983) Principles of Fluorescence Spectroscopy, pp. 257-301, Plenum Press, New York
- 25.Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29 291-325 [DOI] [PubMed] [Google Scholar]
- 26.DeLano, W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, Palo Alto, CA
- 27.Yu, R. C., Hanson, P. I., Jahn, R., and Brunger, T., (1998) Nat. Struct. Biol. 5 803-811 [DOI] [PubMed] [Google Scholar]
- 28.Ferreira, R. M., de Andrade, L. R., Dutra, M. B., de Souza, M. F., Flosi Paschoalin, V. M., and Silva, J. T. (2006) Protein Expression Purif. 47 384-392 [DOI] [PubMed] [Google Scholar]
- 29.Maier, U.-G., Fraunholz, M., Zauner, S., Penny, S., and Douglas, S. (2000) Mol. Biol. Evol. 17 576-583 [DOI] [PubMed] [Google Scholar]
- 30.Barnett, M. E., Zolkiewska, A., and Zolkiewski, M. (2000) J. Biol. Chem. 275 37565-37571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purkey, R. M., and Ebisuzaki, K. (1977) Eur. J. Biochem. 75 303-310 [DOI] [PubMed] [Google Scholar]
- 32.Hubbard, S. J. (1998) Biochim. Biophys. Acta 1382 191-206 [DOI] [PubMed] [Google Scholar]
- 33.Prilusky, J., Felder, C. E., Zeev-Ben-Mordehai, T., Rydberg, E. H., Man, O., Beckmann, J. S., Silman, I., and Sussman, J. L. (2005) Bioinformatics 21 3435-3438 [DOI] [PubMed] [Google Scholar]
- 34.Pozidis, C., Chalkiadaki, A., Gomez-Serrano, A., Stahlberg, H., Brown, I., Tampakaki, A. P., Lustig, A., Sianidis, G., Politou, A. S., Engel, A., Panopoulos, N. J., Mansfield, J., Pugsley, A. P., Karamanou, S., and Economou, A. (2003) J. Biol. Chem. 278 25816-25824 [DOI] [PubMed] [Google Scholar]
- 35.Nowak, E., Panjikar, S., Morth, J. P., Jordanova, R., Svergun, D. I., and Tucker, P. A. (2006) Structure 14 275-285 [DOI] [PubMed] [Google Scholar]
- 36.Toshiaki, H. (1983) Biochim. Biophys. Acta 742 496-508 [DOI] [PubMed] [Google Scholar]
- 37.Georlette, D., Blaise, V., Bouillenne, F., Damien, B., Thorbjarnardottir, S. H., Depiereux, E., Gerday, C., Uversky, V. N., and Feller, G. (2004) Biophys. J. 86 1089-1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu, X., Zhang, H., Zhang, X., Cao, Y., Jiao, W., Liu, C., Song, Y., Abulimiti, A., and Chang, Z. (2005) J. Biol. Chem. 280 6337-6348 [DOI] [PubMed] [Google Scholar]
- 39.Ramachandran, R., Hartmann, C., Song, H. K., Huber, R., and Bochtler, M. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 7396-7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bochtler, M., Hartmann, C., Song, K., Bourenkov, P., Bartunik, D., and Huber, R. (2000) Nature 403 800-805 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.