

Abstract
Nitrile hydratases (NHases) have an unusual iron or cobalt catalytic center with two oxidized cysteine ligands, cysteine-sulfinic acid and cysteine-sulfenic acid, catalyzing the hydration of nitriles to amides. Recently, we found that the NHase of Rhodococcus erythropolis N771 exhibited an additional catalytic activity, converting tert-butylisonitrile (tBuNC) to tert-butylamine. Taking advantage of the slow reactivity of tBuNC and the photoreactivity of nitrosylated NHase, we present the first structural evidence for the catalytic mechanism of NHase with time-resolved x-ray crystallography. By monitoring the reaction with attenuated total reflectance-Fourier transform infrared spectroscopy, the product from the isonitrile carbon was identified as a CO molecule. Crystals of nitrosylated inactive NHase were soaked with tBuNC. The catalytic reaction was initiated by photo-induced denitrosylation and stopped by flash cooling. tBuNC was first trapped at the hydrophobic pocket above the iron center and then coordinated to the iron ion at 120 min. At 440 min, the electron density of tBuNC was significantly altered, and a new electron density was observed near the isonitrile carbon as well as the sulfenate oxygen of αCys114. These results demonstrate that the substrate was coordinated to the iron and then attacked by a solvent molecule activated by αCys114-SOH.
Nitrile hydratases (NHases)2 catalyze the hydration of nitriles to the corresponding amides and are used as catalysts in the production of acrylamide, making them one of the most important industrial enzymes (1, 2). NHases contain a nonheme Fe3+ or non-corrin Co3+ catalytic center. Iron-type NHases show unique photoreactivity; the enzyme is inactivated by nitrosylation in the dark and immediately reactivated by photo-induced denitrosylation (3-5). The protein structure is highly conserved among all known NHases (6-9) as well as a related enzyme, thiocyanate hydrolase (10). The metal site is also conserved, with a distorted octahedral geometry. All ligand residues are involved in a strictly conserved motif of the α subunit, Cys1-Xaa-Leu-Cys2-Ser-Cys3, where two amide nitrogens of Ser and Cys3 and three Cys sulfurs are coordinated to the metal (6). Cys2 and Cys3 are post-translationally modified to cysteine-sulfinic acid and cysteine-sulfenic acid, respectively (7), which probably take deprotonated forms at the metal site (11). The sixth ligand site is occupied by a solvent molecule (8) or by a NO molecule in nitrosylated iron-type NHase (7).
Several reaction mechanisms have been proposed based on the protein structures (1, 6). First, nitriles directly bind to the metal to facilitate the nucleophilic attack of a water molecule on the nitrile carbon. In the other mechanisms, a water molecule activated by the metal directly or indirectly attacks nitriles trapped near the metal. In all cases, the metal is suspected to function as a Lewis acid. By reconstituting iron-type NHase from recombinant unmodified subunits, we demonstrated that the post-translational modifications of its cysteine ligands are essential for its catalytic activity (12). We also found that specific oxidation of the cysteine sulfenic acid ligand to cysteine sulfinic acid resulted in irreversible inactivation (13). Kovacs and co-workers (14) studied the ligand exchange reaction in the low spin Co3+-containing NHase model complexes and concluded that the trans-thiolate sulfur played an important role in promoting the ligand exchange at the sixth site. Later, by using a sulfenate-ligated iron complex, they showed that protonation/deprotonation states of the sulfenate oxygen were modulated by the unmodified Cys thiolate ligand (15). An N3S2-type model complex, [Co(PyPS)(H2 O)]-, slowly hydrolyzes nitriles (18 turnovers in 4 h) (16). Interestingly, the hydration activity was enhanced by the mono-oxygenation of one of two sulfur ligands (17). Heinrich et al. (18) demonstrated that Na[Co(l-N2SOSO)(tBuNC)2] exhibited the nitrile hydration activity but that (Me4N)[Co(l-N2SO2SO2)(tBuNC)2] did not. These results indicate that the oxidized cysteine ligands, especially the cysteine sulfenic acid ligand, play an important role in the catalysis. Recently, theoretical calculations, including density functional calculations (19, 20) as well as molecular dynamics simulations (21), have been applied to the mechanisms described above. However, the detailed mechanism remains unclear because of a lack of direct information on the reaction intermediates.
We recently found that an iron-type NHase from Rhodococcus erythropolis N771 (ReNHase) catalyzes the conversion of isonitriles to the corresponding amines (22). Although the other product derived from the isonitrile carbon was not identified, the kinetic analyses revealed that the Km for tBuNC was comparable with that for methacrylonitrile, whereas kcat (1.8 × 10-2 s-1) was 1.8 × 105 times smaller. In this study, taking advantage of the slow reactivity of tBuNC as well as the photoreactivity of nitrosylated inactive ReNHase (3, 4), we obtained structural evidence on the reaction mechanism by studying the time course of the tBuNC catalysis with x-ray crystallography. Based on the results, we propose a reaction mechanism in which the sulfenate group of αCys114-SO- plays a key role in the catalysis.
EXPERIMENTAL PROCEDURES
Materials—Nitrile hydratase from R. erythropolis N771 (ReNHase) was inactivated by endogenous NO molecules in living cells in the dark (4, 23). ReNHase was purified in the nitrosylated form in the dark as described previously (23). The purified nitrosylated ReNHase was stored in 50 mm Tris-HCl, pH 7.5, at -80 °C in the dark at a concentration of 20 mg/ml. The concentration of the nitrosylated NHase was determined by measuring the absorbance at 280 nm (ε280 = 1.7 ml mg-1 cm-1). tBuNC was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). All the other reagents used in this study were of the highest grade available.
ATR-FTIR Measurements—The nitrosylated ReNHase (70 mg/ml) in 50 mm sodium phosphate, pH 7.5, was loaded on the surface of a three-reflection silicon prism (3 mm in diameter) in the ATR accessory (DuraSamplIR II, Smiths Detection, Danbury, CT) and dried under nitrogen gas flow. Subsequently, 1.5 μl of water (H216O or H218 O) was added to the sample. The sample space was sealed with a CaF2 plate and a greased Teflon spacer (0.7 mm in thickness). The substrate, 0.5 μl of neat tBuNC, was also enclosed in this sealed space as a drop on the CaF2 plate, to be supplied to the ReNHase solution as a vapor. The sample was stabilized at room temperature in the dark for 4 h.
FTIR spectra were measured on a Bruker IFS-66/S spectro-photometer equipped with an MCT detector (D313-L). All of the spectra were recorded at 4 cm-1 resolution. A single-beam spectrum was recorded for 100 s before illumination, and ten spectra (100 s scans) were successively recorded after 10 s of illumination by continuous white light from a halogen lamp (Hoya-Schott HL150; ∼60 milliwatt cm-2 at the sample).
Light-induced difference spectra were calculated by subtracting the dark spectra from each spectrum after illumination. The base-line distortion was corrected by subtracting the corresponding spectra measured in the same manner but without illumination.
For CO detection, 6 μl of hemoglobin (50 mg/ml) in 50 mm Tris-HCl, pH 7.5, was lightly dried on a silicon ATR prism, and 6 μl of the nitrosylated ReNHase sample (70 mg/ml) in Tris-HCl, pH 7.5, was placed beside the hemoglobin. The sample space was sealed with a CaF2 plate, on which 0.5 μl of tBuNC was placed, and a greased Teflon spacer (Fig. 1). The ReNHase sample was photoactivated by white light illumination for 1 min. FTIR spectra with 10-s scans were recorded at 0, 20, 30, and 60 min after illumination.
FIGURE 1.

The sample unit of the ATR-FTIR measurement for CO detection using hemoglobin. Hemoglobin was loaded on a silicon ATR prism, and an NHase solution and tBuNC were separately placed in a sealed space. tBuNC was supplied to the NHase solution as a vapor, and the reaction was initiated by photoactivation of NHase.
Crystallization of ReNHase—Crystals of the nitrosylated ReNHase were grown using the vapor diffusion hanging drop method at 20 °C. Two microliters of the nitrosylated ReNHase (20 mg/ml protein in 50 mm Tris-HCl, pH 7.5) was mixed with an equal amount of the precipitant solution (20% polyethylene glycol 8000, 0.10 m Tris-HCl, pH 7.5, 0.30 m MgCl2) and equilibrated against 0.40 ml of precipitant solution. Crystals with dimensions of approximately 0.4 × 0.3 × 0.3 mm3 grew within a day in the dark at 20 °C. When crystals of the nitrosylated NHase were dissolved in 50 mm Tris-HCl, pH 7.5, the enzyme solution exhibited trace amounts of methacrylonitrile hydration activity in the dark, but it had a specific activity of 7.3 × 102 units/mg after light-induced denitrosylation (10,000 l×) with a cold light illumination system (LG-PS2; Olympus, Tokyo, Japan) for 15 min.
Preparation of the ReNHase Crystals without or with tBuNC—Crystals of the nitrosylated ReNHase were first vapor-soaked with cryoprotectant solution (30% polyethylene glycol 8000, 0.10 m Tris-HCl, pH 7.5, 0.60 m MgCl2) for 1 day by being swapped in mother liquor. They were then vapor-soaked for a day with mother liquor solution containing tBuNC at a final concentration of 0.10 m. After being mounted, ReNHases in the crystals were activated by light-induced denitrosylation (10,000 l×) with a cold light illumination system (LG-PS2; Olympus), and the reaction proceeded for 18, 120, 340, and 440 min at 20 °C. At each elapsed time, the reaction was terminated by flash cooling with N2 gas at 95 K.
X-ray Data Collections, Structure Determinations, and Refinements—Diffraction data were collected using a Quantum 315 CCD detector (Area Detector Systems Corporation, Poway, CA) at the beamline BL-5A (λ = 1.000 Å) of the Photon Factory (Tsukuba, Japan) at 95 K. Each data set was indexed, merged, and scaled with the HKL2000 program suite (24). The ReNHase crystals belonged to the C2 space group. One heterodimer of α and β subunits populated the asymmetric unit. Molecular replacement was performed with MOLREP (25) in the CCP4 program suite (26) using the structure of the nitrosylated ReNHase in the P21212 space group (Protein Data Bank code 2ahj) (7) as the initial coordinates. The obtained models were improved by iterative cycles of crystallographic refinement using REFMAC5 (27) and manual model rebuilding using Coot (28). The models were cross-validated by the SigmaA-weighted electron density maps (29) calculated with both 2mFobs - DFcalc and mFobs - DFcalc coefficients. The refinements were performed using a maximum likelihood target with bulk solvent corrections. During the structure refinement, ∼5% of the amplitude data were set aside to monitor the progress of refinement using the Rfree factor. Solvent water molecules were gradually introduced if the peaks that were contoured at more than 4.0 σ in the mFobs - DFcalc electron density were in the range of a hydrogen bond. tert-Butyl groups of tBuNC were fit on the resultant difference electron density map by handling, and their coordinate data were then refined using REFMAC5 (27). All of the structural figures were generated using PyMol.
RESULTS AND DISCUSSION
Identification of the Product from the Isonitrile Carbon by ATR-FTIR Measurements—To identify all products except for the amine, the reaction was monitored using ATR-FTIR. tBuNC was added as a vapor to nitrosylated NHase, and the enzyme was activated by light-induced denitrosylation. Several prominent positive peaks, all arising from tert-butylamine (tBuNH2), increased their intensities as the reaction proceeded, whereas no signals from other origins were detected (Fig. 2). Supposing that the other product possessing a carbon atom was CO, which escaped from the solution as a gas, we attempted CO detection by trapping using hemoglobin. Hemoglobin was located on a silicon ATR crystal, and nitrosylated ReNHase solution and tBuNC were separately placed in a sealed space (Fig. 1). A CO molecule-bound hemoglobin was monitored by ATR-FTIR after light activation of ReNHase (Fig. 3). The CO stretching signal of CO-hemoglobin was observed at 1953 cm-1, and its intensity increased with the reaction time. The CO peak appeared at a downshifted frequency of 1908 cm-1 when the ReNHase reaction was performed in an H218O buffer, confirming that CO was produced by the ReNHase-consuming water. Thus, we concluded that ReNHase hydrolyzed tBuNC to produce tBuNH2 and CO (tBuNC + H2O → tBuNH2 + CO).
FIGURE 2.

ATR-FTIR difference spectra showing product formation by the NHase reaction with tBuNC. FTIR spectra of H216O(A) and H218O(B) solutions including NHase and tBuNC were recorded before and 100 (purple), 300 (blue), 500 (cyan), 700 (green), and 900 (red) s after illumination of ReNHase, and difference spectra were calculated relative to before illumination values. C, the spectrum of tBuNH2 in an aqueous solution (in a protonated tBuNH3+ form) after subtraction of water absorption is presented for comparison.
FIGURE 3.

The CO stretching region of the ATR-FTIR spectra of hemoglobin. Hemoglobin was loaded on an Si ATR prism, and NHase in an H216O(A) or H218O(B) buffer and tBuNC were separately placed in a sealed space (Fig. 2). The spectra at 0 (blue), 20 (cyan), 30 (green), and 60 (red) min after photoactivation of NHase were recorded.
Time-resolved X-ray Crystallography of the Reaction of ReNHase with
tBuNC—Crystals of nitrosylated ReNHase were soaked with
tBuNC, and the reaction was started by light-induced denitrosylation
at 293 K. At 18, 120, 340, and 440 min, the reaction was stopped by flash
cooling at 95 K, at which point the crystal structure was determined. Details
of data collection and refinement statistics are summarized in
Table 1. Unfortunately, we
could not collect data from the crystals that were incubated longer because
those crystals were damaged. The overall structure at each elapsed time was
essentially unchanged except for the pocket above the Fe3+ center
(Fig. 4).
αCys112-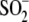 (αCSD112) and αCys114-SO- (αCSO114)
modifications were clearly observed in all of the structures determined.
(αCSD112) and αCys114-SO- (αCSO114)
modifications were clearly observed in all of the structures determined.
TABLE 1.
Data collection and refinement statistics
| Nitrosylated NHasea | 0 mina,b | 18 mina | 120 mina | 340 mina | 440 mina | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | C2 | C2 | C2 | C2 | C2 | C2 |
| Cell dimensions | ||||||
| a (Å) | 114.7 | 114.0 | 114.0 | 114.1 | 113.9 | 114.0 |
| b (Å) | 60.5 | 60.0 | 60.0 | 60.1 | 60.2 | 60.2 |
| c (Å) | 81.9 | 81.7 | 81.7 | 81.7 | 81.4 | 81.5 |
| α, β, γ (°) | 125.0 | 125.1 | 125.1 | 125.1 | 125.1 | 125.1 |
| Wavelength (Å) | 1.00000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| Resolution (Å) | 50.0-1.30 | 50.0-1.48 | 50.0-1.48 | 50.0-1.39 | 50.0-1.59 | 50.0-1.49 |
| Resolution of highest resolution shell (Å) | 1.35-1.30 | 1.53-1.48 | 1.53-1.48 | 1.44-1.39 | 1.65-1.59 | 1.54-1.49 |
| Rmerge | 0.051 | 0.035 | 0.038 | 0.038 | 0.036 | 0.042 |
| Rmerge of highest resolution shell | 0.293 | 0.118 | 0.108 | 0.271 | 0.208 | 0.216 |
| I/σI | 24.4 | 26.8 | 26.1 | 24.1 | 27.9 | 24.4 |
| I/σI of highest resolution shell | 3.49 | 10.5 | 10.8 | 3.71 | 4.04 | 3.97 |
| Completeness (%) | 98.8 | 98.3 | 97.0 | 98.0 | 97.3 | 93.8 |
| Completeness of highest resolution shell (%) | 97.2 | 93.6 | 93.1 | 90.2 | 85.3 | 75.9 |
| Redundancy | 3.7 | 2.1 | 2.0 | 1.8 | 1.8 | 1.9 |
| Refinement | ||||||
| Resolution (Å) | 7.96-1.30 | 8.00-1.48 | 8.00-1.48 | 7.98-1.39 | 7.99-1.59 | 8.00-1.49 |
| No. reflections | 105,634 | 69,876 | 68,831 | 84,855 | 55,536 | 65,146 |
| Rwork/Rfree | 16.9/18.7 | 16.8/19.4 | 16.8/19.0 | 17.7/20.1 | 15.8/18.5 | 15.9/18.3 |
| No. atoms | ||||||
| Protein | 3,288 | 3,252 | 3,255 | 3,222 | 3,185 | 3,185 |
| Ligand/ion | 4 | 5 | 5 | 5 | 12 | 13 |
| Water | 756 | 663 | 643 | 601 | 538 | 545 |
| B-factors | ||||||
| Protein | 11.7 | 13.4 | 14.1 | 14.5 | 11.2 | 11.7 |
| Ligand/ion | 18.7 | 16.6 | 20.9 | 29.8 | 18.6 | 20.4 |
| Water | 28.8 | 28.3 | 29.1 | 20.7 | 27.2 | 28.6 |
| Root mean square deviations | ||||||
| Bond length (Å) | 0.007 | 0.008 | 0.008 | 0.007 | 0.009 | 0.007 |
| Bond angles (°) | 1.151 | 1.198 | 1.188 | 1.184 | 1.184 | 1.160 |
| Ramachandran plot | ||||||
| Most favored (%) | 98.5 | 98.0 | 98.2 | 98.5 | 98.2 | 98.2 |
| Additionally allowed region (%) | 1.5 | 2.0 | 1.8 | 1.5 | 1.8 | 1.8 |
One crystal was used to collect the data of each complex.
“0 min” represents nitrosylated NHase soaked with tBuNC.
FIGURE 4.

Structures around the non-heme Fe3+ center
of ReNHase. Fo - Fc
electron densities (3.0 σ contour as green and -3.0 σ
contour as red) superimposed on the refined structures. NO and
tBuNC molecules and Sγ of βMet40 were excluded
from the calculation. A, nitrosylated NHase before soaking;
B, nitrosylated NHase with tBuNC; C-E, NHase with
tBuNC after light illumination for 18 (C), 120 (D),
and 440 min (E). F, Fo - Fc
electron density (3.0 σ contour as green and -3.0σ
contour as red) and 2Fo - Fc
electron density (1.0σ contour as gray) superimposed on the
refined structure at 440 min. tBuNH2 was included in the
calculation. alphaCSD112 and alphaCSO114 indicate
αCys112-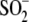 and
αCys114-SO-, respectively. Yellow, blue, red,
green, and brown spheres represent carbon, nitrogen, oxygen,
sulfur, and iron atoms, respectively.
and
αCys114-SO-, respectively. Yellow, blue, red,
green, and brown spheres represent carbon, nitrogen, oxygen,
sulfur, and iron atoms, respectively.
Before soaking with tBuNC, an NO molecule was observed at a distance of 2.1 Å from the Fe3+ (Fig. 4A). The Fe-N(NO) distance is 0.6 Å longer than observed in the previous structure (Protein Data Bank code 2ahj) (7). In the previous structure, the NO was likely to be pushed toward Fe3+ by 1,4-dioxane, the co-precipitant used (7). After soaking with tBuNC, the electron density of tBuNC was clearly observed in the pocket (Fig. 4B) with the tert-butyl group facing the NO molecule coordinated to the Fe3+. Because of the limited space in the hydrophobic pocket, the bulky tert-butyl group must face the iron in its nitrosylated state. In addition to the original conformation (conformer A), Sγ of βMet40 took another conformation (conformer B) with occupancies of A:B = 0.25:0.75. Movement of Sγ of βMet40 to conformer B is likely due to the occupation of the hydrophobic pocket by tBuNC. We hypothesize that conformer B is less stable because of steric hindrance between Sγ and the amide oxygen of βMet40 (Fig. 5).
FIGURE 5.

The steric hindrance at Sγ of βMet40 caused by tBuNC. The refined structure around βMet40 in the nitrosylated NHase without (A) and with (B) tBuNC. Yellow, blue, red, and green spheres represent carbon, nitrogen, oxygen, and sulfur atoms, respectively. The black and red dashed lines indicate the distances between Sγ of βMet40 and the isonitrile carbon and between Sγ of βMet40 and the amide oxygen of βMet40.
At 18 min, electron densities of NO and tBuNC, especially that of the isonitrile group, were attenuated (Fig. 4C). Sγ of βMet40 remained disordered, but the occupancy of conformer A increased to 0.55. At 120 min, the NO disappeared, and a tBuNC molecule was coordinated to Fe3+ with an Fe-C(-NC) length of 2.1 Å (Fig. 4D). βMet40 took conformer A again. The rotation of the tBuNC molecule could be driven by the recovery of βMet40 to conformer A.
The Fo - Fc electron density at 340 (supplemental Fig. S1) and 440 min (Fig. 4E) were very similar to one another but distinct from those observed at 120 min. In both structures, the Fo - Fc electron density corresponding to the tert-butyl group was moved ∼1.0 Å away from the iron, and an extra electron density was observed near the isonitrile carbon as well as the sulfenate oxygen of αCys114. When the products, tBuNH2 and CO, were included in the calculation of the electron density at 440 min, the refined model of tBuNH2 was well fit on the 2Fo - Fc electron density, but that of CO was not (supplemental Fig. S2). In addition, two positive electron densities were observed near the CO molecule in the Fo - Fc electron density. Alternatively, we calculated the electron density at 440 min by assuming the presence of only tBuNH2. As shown in Fig. 4F, tBuNH2 was well fit on the 2Fo - Fc electron density, and two positive electron densities were observed above the iron ion and near Oδ of the sulfenate group, in the Fo - Fc electron density. We assigned the positive densities as the carbon of the isonitrile group and the solvent water molecule (named as H2Oa), respectively (Fig. 4F). All distances of Fe-C(-NC), C(-NC)-N(-NC), C(-NC)-O(H2Oa), N(-NC)-O(H2Oa), and O(H2Oa)-O(-SO) converged at less than 2.2 Å (Table 2). The O(H2Oa)-O(-SO) distance cannot be explained. These atoms may be disordered because the occupancies of H2Oa and Oδ of αCys114-SO- converged on 0.50. Interestingly, a positive difference density was observed below Sγ of αCys114-SO- in the 2Fo - Fc electron density map after coordination of tBuNC (Fig. 4, D-F). The distance between the density and Sγ of αCys114-SO- is 1.4 Å, and the angle Oδ(αCys114-SO-) - Sγ(αCys114-SO-) - the density was 133°. The positive density may represent an alternative position of Oδ of αCys114-SO-.
TABLE 2.
Selected bond lengths for the complex of nitrosylated NHase with tBuNC after light irradiation for 440 min
| Bond lengths | |
|---|---|
| Å | |
| Fe-C(-NC) | 2.1 |
| C(-NC)-N(-NC) | 1.4 |
| C(-NC)-O(H2Oa) | 2.3 |
| N(-NC)-O(H2Oa) | 1.8 |
| O(SO)-O(H2Oa) | 1.6 |
Proposed Catalytic Mechanisms of NHase—Based on the results,
we propose the following catalytic mechanism: the tBuNC substrate
binds the metal directly, and then a water molecule, activated by Oδ of
αCys114-SO-, makes a nucleophilic attack on the
isonitrile carbon to produce tBuNH2 and CO
(Fig. 6A). Considering
the similarity between isonitriles and nitriles, nitrile hydration is likely
to proceed in a similar manner (Fig.
6B). When a nitrile coordinates to the metal, the nitrile
carbon is attacked by a water molecule, activated by Oδ of
αCys114-SO-. The low kcat
value for isonitrile may be due to limited accessibility of the activated
water molecule because of steric hindrance by Oδ of
αCys114-SO-. Nitrile coordination to the
Fe3+ was suggested by electron spin resonance measurements
(30). Involvement of
αCys114-SO- in the catalytic reaction had been
suggested by our previous studies using the inhibitor, 2-cyano-2-propyl
hydroperoxide (13),
specifically oxidizing αCys114-SO- to
Cys-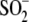 , and the site-directed mutant
NHases (31). Yano et
al. (32) have extensively
studied the N2S2 (tBuNC)2-type
Co3+model complexes with different sulfur oxidation states and
concluded that sulfur oxidations promoted the Lewis acidity of the
Co3+ center and that only the sulfenyl oxygen exhibited a
nucleophilic character. Theoretical calculation studies have indicated that
Oδ of αCys114-SO- could be a catalytic base
when nitrile coordination was assumed
(19). These antecedent studies
support the mechanism of the substrate coordinated to the iron being attacked
by water activated by αCys114-SO-. Recently,
involvement of the Ser ligand (αSer112; corresponding to
αSer113 of ReNHase) and of the vicinal Tyr and Trp
residues (βTyr68 and βTrp72; corresponding to
βTyr72 and βTyr76 of ReNHase) in the
catalytic mechanism was suggested by temperature- and pH-dependent kinetic
studies of the Co-type NHase from Pseudonocardia thermophila JCM 3095
(33). However, the
corresponding residues of ReNHase were unchanged during our
investigations (Fig. 4). Our
findings represent the first structural evidence of reaction intermediates in
NHase catalysis. The present results demonstrate a reaction mechanism in which
the sulfenate group of αCys114-SO- plays a key
role in the catalysis. Cysteine oxidation has been found to play important
roles in various proteins
(34). The present work reveals
a novel role of cysteine sulfenic acid as a catalytic base.
, and the site-directed mutant
NHases (31). Yano et
al. (32) have extensively
studied the N2S2 (tBuNC)2-type
Co3+model complexes with different sulfur oxidation states and
concluded that sulfur oxidations promoted the Lewis acidity of the
Co3+ center and that only the sulfenyl oxygen exhibited a
nucleophilic character. Theoretical calculation studies have indicated that
Oδ of αCys114-SO- could be a catalytic base
when nitrile coordination was assumed
(19). These antecedent studies
support the mechanism of the substrate coordinated to the iron being attacked
by water activated by αCys114-SO-. Recently,
involvement of the Ser ligand (αSer112; corresponding to
αSer113 of ReNHase) and of the vicinal Tyr and Trp
residues (βTyr68 and βTrp72; corresponding to
βTyr72 and βTyr76 of ReNHase) in the
catalytic mechanism was suggested by temperature- and pH-dependent kinetic
studies of the Co-type NHase from Pseudonocardia thermophila JCM 3095
(33). However, the
corresponding residues of ReNHase were unchanged during our
investigations (Fig. 4). Our
findings represent the first structural evidence of reaction intermediates in
NHase catalysis. The present results demonstrate a reaction mechanism in which
the sulfenate group of αCys114-SO- plays a key
role in the catalysis. Cysteine oxidation has been found to play important
roles in various proteins
(34). The present work reveals
a novel role of cysteine sulfenic acid as a catalytic base.
FIGURE 6.

Proposed catalytic mechanisms of NHase. A, isonitrile hydrolysis. B, nitrile hydration.
Supplementary Material
Acknowledgments
We are grateful to beamline assistants of the Photon Factory for data collection at beamline BL-5A. We thank Drs. K. Noguchi and A. Ohtaki (Tokyo University of Agriculture and Technology), Prof. N. Kamiya (Osaka City University), and Dr. M. Nojiri (Osaka University) for useful advice and discussion on the structural determination and analyses of the intermediate structure of NHase. We also thank Prof. K. Nagasawa and Mr. M. Tera (Tokyo University of Agriculture and Technology) and Prof. T. Ozawa (Nagoya Institute of Technology) for fruitful discussion on the catalytic mechanism.
The atomic coordinates and structure factors (code 2ZPB, 2ZPE, 2ZPF, 2ZPG, 2ZPH, and 2ZPI) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported in part by a Grant-in-aid for scientific research from the “Future nano materials” section of the “21st Century Center of Excellence” Project, Grant-in-Aid for Scientific Research (B) KAKENHI 19350080 (to M. O.), Grant-in-Aid for Creative Scientific Research 17GS0314 (to T. N.), Grant-in-Aid from the Japanese Society for the Promotion of Science Fellows 19252 (to H. S.), and a grant from the National Project on Protein Structural and Functional Analyses from the Ministry of Education, Science, Sports and Culture of Japan (to M. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: NHase, nitrile hydratase; ReNHase, nitrile hydratase from R. erythropolis N771; tBuNC, tert-butylisonitrile; tBuNH2, tert-butylamine; ATR-FTIR, attenuated total reflectance-Fourier transform infrared.
References
- 1.Kobayashi, M., and Shimizu, S. (1998) Nat. Biotechnol. 16 733-736 [DOI] [PubMed] [Google Scholar]
- 2.Endo, I., Nojiri, M., Tsujimura, M., Nakasako, M., Nagashima, S., Yohda, M., and Odaka, M. (2001) J. Inorg. Biochem. 83, 247-253 [DOI] [PubMed] [Google Scholar]
- 3.Noguchi, T., Hoshino, M., Tsujimura, M., Odaka, M., Inoue, Y., and Endo, I. (1996) Biochemistry 35 16777-16781 [DOI] [PubMed] [Google Scholar]
- 4.Odaka, M., Fujii, K., Hoshino, M., Noguchi, T., Tsujimura, M., Nagashima, S., Yohda, M., Nagamune, T., Inoue, Y., and Endo, I. (1997) J. Am. Chem. Soc. 119 3785-3791 [Google Scholar]
- 5.Bonnet, D., Artaud, I., Moali, C., Petre, D., and Mansuy, D. (1997) FEBS Lett. 409 216-220 [DOI] [PubMed] [Google Scholar]
- 6.Huang, W., Jia, J., Cummings, J., Nelson, M., Schneider, G., and Lindqvist, Y. (1997) Structure 5 691-699 [DOI] [PubMed] [Google Scholar]
- 7.Nagashima, S., Nakasako, M., Dohmae, N., Tsujimura, M., Takio, K., Odaka, M., Yohda, M., Kamiya, N., and Endo, I. (1998) Nat. Struct. Biol. 5 347-351 [DOI] [PubMed] [Google Scholar]
- 8.Miyanaga, A., Fushinobu, S., Ito, K., and Wakagi, T. (2001) Biochem. Biophys. Res. Commun. 288 1169-1174 [DOI] [PubMed] [Google Scholar]
- 9.Hourai, S., Miki, M., Takashima, Y., Mitsuda, S., and Yanagi, K. (2003) Biochem. Biophys. Res. Commun. 312 340-345 [DOI] [PubMed] [Google Scholar]
- 10.Arakawa, T., Kawano, Y., Kataoka, S., Katayama, Y., Kamiya, N., Yohda, M., and Odaka, M. (2007) J. Mol. Biol. 366 1497-1509 [DOI] [PubMed] [Google Scholar]
- 11.Noguchi, T., Nojiri, M., Takei, K., Odaka, M., and Kamiya, N. (2003) Biochemistry 42 11642-11650 [DOI] [PubMed] [Google Scholar]
- 12.Murakami, T., Nojiri, M., Nakayama, H., Odaka, M., Yohda, M., Dohmae, N., Takio, K., Nagamune, T., and Endo, I. (2000) Protein Sci. 9 1024-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsujimura, M., Odaka, M., Nakayama, H., Dohmae, N., Koshino, H., Asami, T., Hoshino, M., Takio, K., Yoshida, S., Maeda, M., and Endo, I. (2003) J. Am. Chem. Soc. 125 11532-11538 [DOI] [PubMed] [Google Scholar]
- 14.Shearer, J., Kung, I. Y., Lovell, S., Kaminsky, W., and Kovacs, J. A. (2001) J. Am. Chem. Soc. 123 463-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lugo-Mas, P., Dey, A., Xu, L. K., Davin, S. D., Benedict, J., Kaminsky, W., Hodgson, K. O., Hedman, B., Solomon, E. I., and Kovacs, J. A. (2006) J. Am. Chem., Soc. 128 11211-11221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noverson, J. C., Olmstead, M. M., and Mascharak, P. K. (1999) J. Am. Chem. Soc. 121 3553-3554 [Google Scholar]
- 17.Tyler, L. A., Noverson, J. C., Olmstead, M. M., and Mascharak, P. K. (2003) Inorg. Chem. 42 5751-5761 [DOI] [PubMed] [Google Scholar]
- 18.Heinrich, L., Mary-Verla, A., Li, Y., Vassermann, J., and Chottard, J. C. (2001) Eur. J. Inorg. Chem. 9 2203-2206 [Google Scholar]
- 19.Hopmann, K. H., Guo, J. D., and Himo, F. (2007) Inorg. Chem. 46 4850-4856 [DOI] [PubMed] [Google Scholar]
- 20.Hopmann, K. H., and Himo, F. (2008) Eur. J. Inorg. Chem., 2008 1406-1412 [Google Scholar]
- 21.Kubiak, K., and Nowak, W. (2008) Biophys. J. (2008) 94 3824-3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taniguchi, K., Murata, K., Murakami, Y., Takahashi, S., Nakamura, T., Hashimoto, K., Koshino, K., Dohmae, N., Yohda, M., Hirose, T., Maeda, M., and Odaka, M. (2008) J. Bioeng. Biosci. 106 174-179 [DOI] [PubMed] [Google Scholar]
- 23.Tsujimura, M., Odaka, M., Nagashima, S., Yohda, M., and Endo, I. (1996) J. Biochem. (Tokyo) 119 407-413 [DOI] [PubMed] [Google Scholar]
- 24.Otowinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 25.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1024 [Google Scholar]
- 26.CCP4 (Collaborative Computational Project, Number 4) (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 27.Murshudov, G. N., Vargin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 28.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 29.Read, R. J. (1986) Acta Crystallogr. Sect. A 42 140-149 [Google Scholar]
- 30.Sugiura, Y., Kuwahara, J., Nagasawa, T., and Yamada, H. (1987) J. Am. Chem. Soc. 109 5848-5850 [Google Scholar]
- 31.Takarada, H., Kawano, Y., Hashimoto, K., Nakayama, H., Ueda, S., Yohda, M., Kamiya, N., Dohmae, N., Maeda, M., and Odaka, M. (2006) Biosci. Biotechnol. Biochem. 70 881-889 [DOI] [PubMed] [Google Scholar]
- 32.Yano, T., Wasada-Tsutsui, Y., Arii, H., Yamaguchi, S., Funahashi, Y., Ozawa, T., and Masuda, H. (2007) Inorg. Chem. 46 10345-10353 [DOI] [PubMed] [Google Scholar]
- 33.Mitra, S., and Holz, R. C. (2007) J. Biol. Chem. 282 7397-7404 [DOI] [PubMed] [Google Scholar]
- 34.Claiborne, A., Mallett, T. C., Yeh, J. I., Luba, J., and Parsonage, D. (2001) Adv. Protein Chem. 58 215-276 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.