

Abstract
A library of photocrosslinkable poly(β-amino ester)s (PBAEs) was recently synthesized to expand the number of degradable polymers that can be screened and developed for a variety of biological applications. In this work, the influence of variations in macromer chemistry and macromer molecular weight (MMW) on network reaction behavior, overall bulk properties, and cell interactions were investigated. The MMW was controlled through alterations in the initial diacrylate to amine ratio (≥1) during synthesis and decreased with an increase in this ratio. Lower MMWs reacted more quickly and to higher double bond conversions than higher MMWs, potentially due to the higher concentration of reactive groups. Additionally, the lower MMWs led to networks with higher compressive and tensile moduli that degraded slower than networks formed from higher MMWs due to an increase in the crosslinking density and decrease in the number of degradable units per crosslink. The adhesion and spreading of osteoblast-like cells on polymer films was found to be dependent on both the macromer chemistry and the MMW. In general, the number of cells was similar on networks formed from a range of MMWs, but the spreading was dramatically influenced by MMW (higher spreading with lower MMWs). These results illustrate further diversity in photocrosslinkable PBAE properties and that the chemistry and macromer structure must be carefully selected for the desired application.
Keywords: polymers, degradable, photopolymerization, molecular weight, poly(β-amino ester), tissue engineering
INTRODUCTION
Injectable biomaterials have been used for many years for applications such as bone cements and for filling dental caries.1 In recent years, injectable materials and specifically, photocrosslinkable polymers, have been developed for a variety of clinical applications; including adhesion prevention,2 tissue engineering,3-10 and drug and gene delivery.11 Photopolymerization can be advantageous over other polymerization techniques for many of these applications because they occur rapidly at physiological temperature (37°C) and in biological environments without the use of solvents, which permits network formation in vivo.3 Additionally, the polymerization is temporally and spatially controlled, which is not afforded by thermal initiation and allows for better control over the polymerization reaction (e.g., for minimizing exotherms)1,12 and for the fabrication of materials with complex structures (e.g., for tissue engineering scaffolds and microfluidic devices).13-15
For many of these intended applications, the overall mechanical properties of the biomaterial and the ability of the material to degrade (either enzymatically or hydrolytically) in a controlled fashion are extremely important. For instance, degradation is important in tissue engineering scaffolds to prevent the necessity of a secondary surgery for implant removal and may aid in healing if the degradation rate is matched to cell penetration and tissue formation. In drug delivery, the controlled release of the payload is typically controlled by properties such as degradation and diffusion. Mechanics are an import design criteria for many applications if the material is to bear the load of the tissue,16 to control the interaction of cells with the material,17-19 and may be correlated to properties like swelling which subsequently controls encapsulated cell viability.20
Numerous photopolymerizable and degradable biomaterials have been developed to date. These include materials like poly(propylene fumarate)s (PPF),21 photocrosslinkable polyanhydrides,22 poly(ethylene glycol)s,18,23-25 and crosslinkable polysaccharides.4,8,10 Typically, the syntheses of these polymers involve several reaction and purification steps. These lengthy and often tedious synthesis procedures limit the potential variations in polymer chemistry and structure that can be developed and rapidly tested for various applications. This is particularly important since the degradation, mechanical, and cellular interaction properties of these degradable polymer systems have proven difficult to predict based on chemistry alone; leading to long iterative processes for material development.
To expedite the development of novel photocrosslinkable polymers, conjugate addition polymerizations have been used to create large libraries of poly(β-amino ester) (PBAE) macromers by combining commercially available primary and secondary amines with diacrylates.26,27 This procedure is relatively simple and does not require protection/deprotection schemes, does not generate byproducts that must be removed through further purification steps, and is tolerant of most functional groups. Similarly, through precise control over reagent concentration and selection of appropriate precursor molecules, a large library of PBAE macromers with acrylate end groups (ratio of acrylate to amine > 1) was synthesized that form degradable networks using photoinitiated polymerizations.28 This macromer library was produced with only a diacrylate to amine ratio of 1.2, so variations in molecular weight were not investigated. Networks formed from this macromer library were screened for degradation and initial mechanical properties and showed that a wide range of degradation behavior and mechanical properties are possible from the library. Now, application-specific design criteria can be used to identify candidate polymers from this library that can be further developed for a variety of applications (e.g., tissue engineering, drug delivery, microfluidics).
The overall aim of this study is to utilize the advantages of this synthesis process to further expand the library by investigating the control over MMW during synthesis and examine how this influences network reaction behavior, bulk properties, and degradation. Specifically, three macromers from the original library were selected and utilized for this fundamental study of macromer structure/function relationships. Additionally, for the first time, the interaction of cells with these novel materials was investigated. This work represents another step towards the development of advanced polymers for a variety of biological applications, and specifically as scaffolding for tissue engineering.
MATERIALS AND METHODS
Macromer synthesis and characterization
Acrylate terminated PBAEs were synthesized in parallel by the step-growth polymerization of commercially available primary amines and diacrylates.28 The diacrylates and amines (Figure 1) included triethylene glycol diacrylate (J) and 1,6-hexanediol diacrylate (C) (Scientific Polymer Products, Ontario, NY); 1,6-hexanediol ethoxylate diacrylate (E), 1-amino-2-methylpropane (6), and 1-(3-aminopropyl)imidazole (12) (Sigma, St Louis, MO); and 3-methoxypropylamine (1) (TCI America, Portland, OR). These liquid reagents were mixed at molar ratios of 1:1, 1.05:1, 1.1:1, 1.2:1, and 1.4:1 of diacrylate to amine (J6, C12 or E1) in glass scintillation vials at 90°C overnight while stirring (700 rpm, Telesystem HP15/RM, Variomag USA, Daytona Beach, FL). The sample notation is consistent with our previous report on the development of the initial photocrosslinkable macromer library.28 The viscous liquid macromers were immediately used for testing, polymerized into networks, or stored at 4°C until use.
Figure 1.
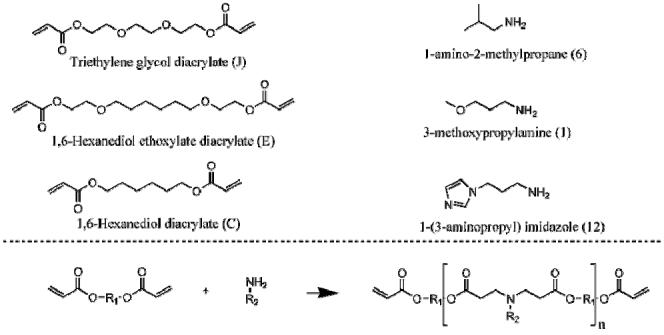
Chemical structures of diacrylates (J, E, C) and amines (6, 1, 12) used in the synthesis of acrylate terminated poly(β-amino ester)s. These macromers can be crosslinked into degradable networks using a free-radical photoinitiated polymerization.
The macromer chemical structures were analyzed with proton nuclear magnetic resonance (1H-NMR, 360 MHz, DMX360, Bruker, Madison, WI) and Fourier transform infrared (FTIR, Nicolet 6700, Thermo Electron, Waltham, MA) spectroscopy. The 1H-NMR spectra were taken for the initial diacrylates and amines and compared to the spectra for the synthesized macromers in each of the diacrylate to amine ratios. The MMW was calculated using ratios of the areas of acrylate peaks (ppms) to the peaks in the macromer backbone. For FTIR analysis, a drop of the liquid macromer was placed on a zinc selenium crystal and spectra were obtained in attenuated total reflectance (ATR) FTIR mode. The ATR-FTIR spectra were then analyzed to monitor variations in the areas of the acrylate peaks (~1630 cm-1) normalized to the carbonyl peak (~1730cm-1) as the ratio of diacrylate:amine was altered.
Photopolymerization and network characterization
The photoinitiator 2,2-dimethoxy-2-phenyl acetophenone (DMPA, Sigma) was added to the liquid macromers at a final concentration of 0.5% (w/w) by addition of a 10% (w/v) solution of DMPA in methylene chloride and solvent was removed in a vacuum desiccator overnight. The polymerization behavior was monitored using an ATR-FTIR system with a zinc selenium crystal. A drop of the macromer/initiator solution was placed directly on the horizontal crystal, covered with a glass cover slip, and exposed from above to ultraviolet light (~1.3 W/cm2 at tip of light guide, distance of 24 cm, 365 nm, Omnicure Series 1000, Exfo, Quebec, Canada). The change in area of the double bond peak (~1630 cm-1) was used to monitor double-bond conversion in real-time. Values were normalized to the area of the carbonyl peak (~1730 cm-1) and converted to double bond conversion using the initial peak areas.
For mechanical testing, the macromer/initatior solution was placed between glass slides with either a 1 mm (tensile and degradation samples) or 2 mm (compression samples) spacer and polymerized with exposure to ~10 mW/cm2 ultraviolet light (365nm, Blak-Ray B-100 AP, Ultraviolet Products, Upland, CA) for 5 minutes. The polymers were then cut into 25 mm × 5 mm slabs for tensile testing and 5 mm disks were punched for compression testing. Tensile testing was performed on a materials testing machine (Microtester 5848, Instron, Norwood, MA) with a constant strain rate of 0.1% per second until macroscopic failure. The elastic modulus was then calculated as the slope of the linear portion of the stress versus strain plot. Unconfined compression testing was completed on a custom made mechanical testing device designed as described in Soltz et al.29 Samples were creep tested under a 2 gram load until equilibrium and then stressed to 10% strain at 1μm/sec and allowed to relax. The compressive moduli were calculated as the slope of the stress versus strain plots between 5 and 10% strain.
The soluble (sol) fraction of the various networks was determined by placing polymer discs in methylene chloride overnight. This allowed unreacted macromer to swell from the network. After drying, the sol fraction was calculated as the percent of initial mass lost during overnight swelling (only one cycle). Negligible mass loss was observed with subsequent swelling and drying cycles indicating that the sol fraction was removed during the first cycle.
Polymer degradation
Samples were polymerized as described above and 5 mm × 5 mm squares were cut from the 1 mm thick slabs and weighed (initial mass). Samples were placed in histology cassettes and degraded in phosphate buffered saline (PBS) at 37°C (pH 7.4) on an orbital shaker. Samples were removed after 1, 2, 4, 7, 10, 13, and 17 weeks of degradation, dried in an oven, and weighed (final mass). The overall mass loss was calculated from the recorded initial and final mass values. Mechanical properties with degradation were determined through tensile and compressive analysis of samples as described above after 2, 4, and 8 weeks of degradation.
Cell interaction studies
To prepare films for cell interaction studies, the macromer/initiator solutions were dissolved in ethanol at a 1:2 (w/v) ratio and pipetted (30 μl) into 24-well plates. The ethanol was allowed to evaporate off overnight to leave a thin film of the macromer and initiator. The plates were placed in a transparent chamber being purged with nitrogen and polymerized for 10 minutes with ultraviolet light (Blak Ray). To sterilize the films, the plates were placed under a germicidal lamp in a laminar flow hood for 30 minutes. Wells were filled with PBS and left in an incubator overnight. After washing with PBS, the wells were filled with growth media for 30 minutes prior to cell seeding.
Human sarcoma osteoblast-like cells (SaOS-2, ATCC, Manassas, VA) were grown in media comprised of Modified McCoy's Medium (ATCC) with 10% fetal bovine serum (FBS, Hyclone, Logan, UT) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA). SaOS-2 cells were seeded on the polymer films at 50,000 cells per well. The cells were cultured for 24 or 48 hours (media changed daily) and fixed in 2.5% glutaraldehyde (Polysciences, Warrington, PA) for 15 minutes. Phase contrast photomicrographs were taken using an inverted microscope (Axiovert, Zeiss, Oberkochen, Germany) and a digital camera (Axiovision, Zeiss). The total cell number was determined by counting adhered cells in at least 5 random fields on 3 individual films for each composition at each time point. Cell area was measured using NIH ImageJ software (3 samples per polymer/ratio, 5 pictures per well, 20 cells per picture, at least 300 cells per composition).
Statistical analysis
Statistical analysis was performed using ANOVA with Tukey's post-hoc among the groups with significance defined as a confidence level of 0.05. All values are reported as the mean and the standard deviation of the mean.
RESULTS AND DISCUSSION
Macromer synthesis and characterization
Three macromers (E1, C12, and J6) were selected from our previously reported library of acrylate terminated PBAE macromers28 and synthesized as shown in Figure 1. These specific macromers were chosen because they illustrated potential as scaffolding for tissue engineering due to their gradual mass loss and overall degradation times (~4 months). For certain tissues, such as bone, this would allow for sufficient cell and tissue infiltration prior to complete degradation, yet would not inhibit tissue growth.30 The initial macromer library incorporated a large range of chemical reagents (amines and diacrylates) and the influence of chemical alterations on polymer mechanics and degradation was investigated, however only one diacrylate to amine molar ratio was examined (i.e., 1.2). This ratio controls the extent of polymerization that occurs before the amines are consumed and all macromer ends are terminated with acrylate groups (with a ratio > 1). It is anticipated that simple alterations in the ratio can lead to changes in the MMW. In this study, the diacrylate to amine ratio was varied at 1:1, 1.05:1, 1.1:1, 1.2:1, and 1.4:1 in order to change the MMW of the macromer and investigate the influence of MMW on polymer properties and cell interactions. It should be noted that the objective of this study is not to illustrate that one of these macromers is ideal for tissue engineering, but to investigate the influence of chemical and structural alterations of the macromer on the final properties. This information can then aid in development of biodegradable PBAEs for a specific application.
The resulting macromers were characterized with 1H-NMR and representative spectra are shown in Figure 2. These spectra demonstrate the chemical diversity of the different macromers, illustrate the complete consumption of amine protons during synthesis, and show the maintenance of peaks for acrylate reactive moieties. Protons from the amine group are represented by a broad peak found around ~1.1 ppm and are found in the spectra of the initial amine reagents but are not present in any of the synthesized macromer spectra. The area of acrylate peaks (labeled in Figure 2 as a and b at ~6.4, 6.0, and 5.7 ppm for one ratio) changed with variations in the diacrylate to amine ratios (i.e., smaller peaks for lower ratios) indicating variations in the MMWs (results not shown).
Figure 2.
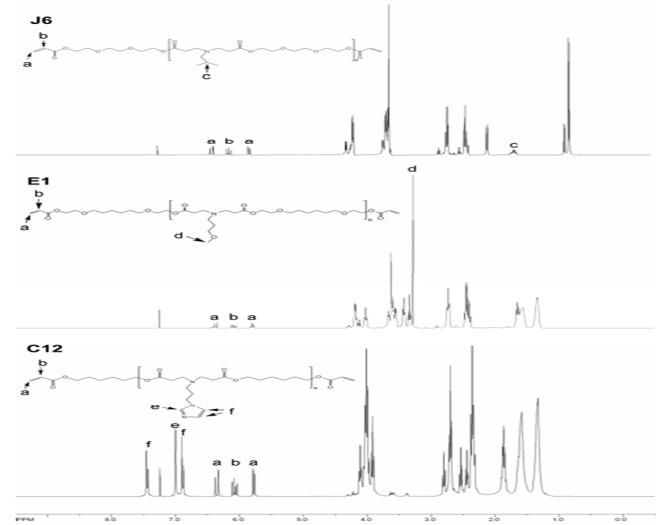
NMR spectra of macromers J6, E1, and C12 synthesized at a diacrylate to amine ratio of 1.2:1. Of interest is the maintenance of acrylate proton peaks (~6.4, 6.0, 5.7 ppm) and the disappearance of the amine proton peaks (~1.1 ppm).
The MMW was calculated with 1H-NMR, via end group analysis (comparison of acrylate protons to backbone protons), and the results are shown in Table 1. The area of the acrylate peaks (representing 6 hydrogen atoms, 3 per macromer end group) was divided by the area of a peak corresponding to distinct protons in the backbone (labeled c, d, e, and f). This gives the average number of repeat units, and this value can be used to calculate the number average molecular weight (Table 1). For all cases, the MMW increases as the diacrylate to amine ratio decreases, as expected since the reaction can proceed further prior to termination when a more equivalent molar ratio is used. The calculated number average molecular weight is anticipated to be an overestimation of the MMW, since this analysis assumes that all end groups are diacrylates. However, impurities in the original diacrylate reagents (which were 85-90% pure) can lead to end-groups that are not acrylates, but hydroxyl groups.
Table 1.
Number of repeat units (n) and number average molecular weight (MMW) determined with end group analysis using 1H-NMR, as well as the sol fraction of the photopolymerized networks polymerized from the various macromers and MMWs.
| Macromer | Ratio | n | MMW | Sol Fraction (%) |
|---|---|---|---|---|
| J6 | 1:1 | 17.5 | 6070 | 47.5 ± 1.1 |
| 1.05:1 | 12.2 | 4317 | 36.9 ± 1.0 | |
| 1.1:1 | 11.4 | 4049 | 24.5 ± 0.3 | |
| 1.2:1 | 5.8 | 2182 | 12.8 ± 1.9 | |
| 1.4:1 | 3.5 | 1406 | 2.2 ± 0.3 | |
| C12 | 1:1 | 13.8 | 5080 | 47.2 ± 0.4 |
| 1.05:1 | 10.6 | 3945 | 44.9 ± 3.8 | |
| 1.1:1 | 8.3 | 3159 | 33.8 ± 0.9 | |
| 1.2:1 | 5.7 | 2230 | 17.4 ± 0.6 | |
| 1.4:1 | 3.6 | 1486 | 10.9 ± 0.4 | |
| E1 | 1:1 | 9.1 | 3982 | 48.9 ± 0.6 |
| 1.05:1 | 8.0 | 3531 | 33.9 ± 0.4 | |
| 1.1:1 | 6.9 | 3111 | 23.2 ± 0.2 | |
| 1.2:1 | 5.7 | 2596 | 12.0 ± 0.7 | |
| 1.4:1 | 3.6 | 1768 | 4.3 ± 0.9 | |
Additionally, the ATR-FTIR spectra of the various macromers at the different ratios were obtained to assess variations in the acrylate peak (~1630 cm-1) and are shown in Figure 3 for macromer E1. The acrylate region (Figure 3A) is suppressed as the ratio decreases, indicating once again that the MMW is increasing. Figure 3B shows the area of the acrylate peak normalized to the area of the carbonyl peak (~1730 cm-1). Again, these values illustrate an increase in acrylate concentration with an increase in the diacrylate:amine ratio during synthesis. Due to the highly cationic nature of the macromers and interaction between the macromers and gel permeation chromatography (GPC) columns, GPC measurements of polydispersities and weight average molecular weights were not possible (even though several solvent systems were investigated to this end).
Figure 3.
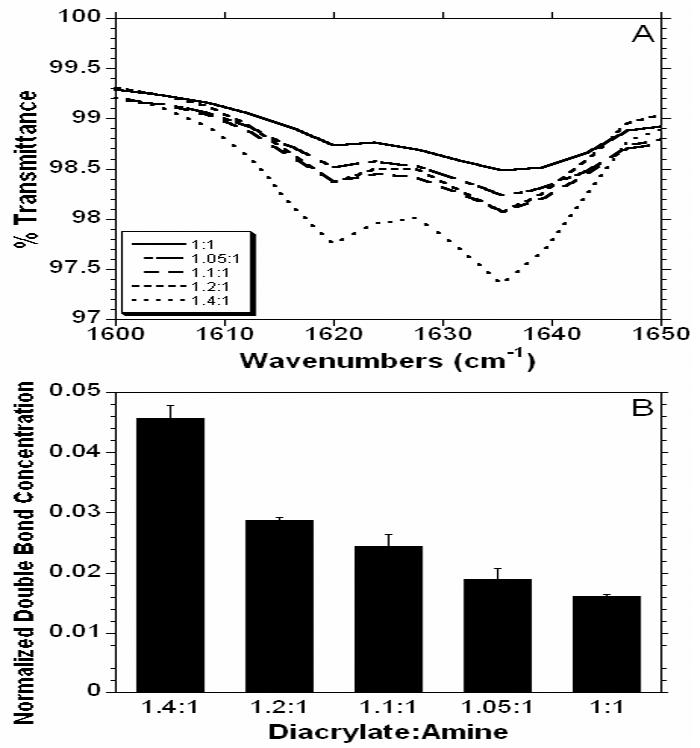
Initial ATR-FTIR spectra for the acrylate double bond (~1630 cm-1) with varying ratios of diacrylate to amine (A) and the normalized double bond concentration calculated as the ratio of the acrylate to carbonyl (~1730 cm-1) peak areas.
Photopolymerization and network characterization
Free radical polymerizations of the synthesized macromers were initiated by ultraviolet light exposure of the macromer with 0.5% DMPA as the initiator. Although a solvent was used to add the initiator to the macromer, the solvent was evaporated prior to polymerization. This reaction was monitored in real time using ATR-FTIR to determine the real-time conversion of the acrylate double bonds that form network kinetic chains and ultimately lead to a crosslinked polymer. The conversion profiles (Figure 4A) of macromer E1 for all MMWs indicate that lower MMWs react much faster and to higher double bond conversions (Figure 4B) after 10 minutes than the higher MMWs. This is likely due to the increase in the concentration of reactive groups for the lower MMWs and an increase in initial diffusion limitations for the higher MMWs. The overall reaction behavior and ultimate double bond conversion is important because it is correlated to network mechanical properties and unreacted macromer that may leach from the network and be toxic. The sol fraction, listed in Table 1, is also an indicator of the lack of the complete acrylate conversion with the high MMW. The sol fraction for the 1:1 ratio is over 47% for all three macromer chemistries. The high concentration of soluble factors is likely due to these unreacted double bonds found during monitoring of the reaction with ATR-FTIR. These molecules are never reacted into the 3-dimensional network and are released when the network is swollen in methylene chloride. Also, impurities in the original diacrylate reagents produce macromers with one or both ends without acrylate moieties, which increases the sol content.
Figure 4.
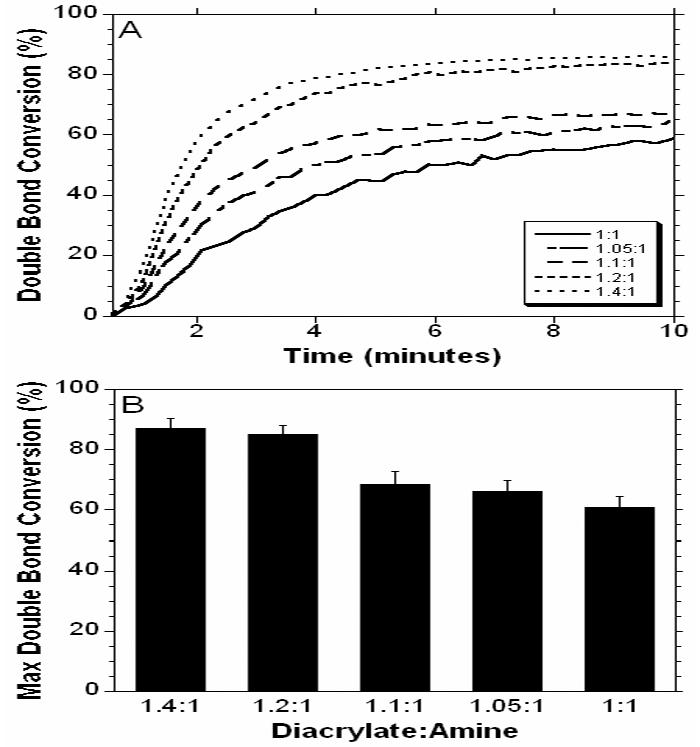
Real-time double bond conversion of macromer E1 synthesized at various MMWs and polymerized on a zinc selenium crystal with exposure to ultraviolet light measured with ATRFTIR (A) and the double bond conversion after 10 minutes (B) of ultraviolet light exposure.
The mechanical properties of polymers J6, C12, and E1 formed from all MMWs were measured in both tension and compression. The tensile moduli for J6, C12, and E1 for the different MMWs are shown in Figure 5A. For all of the polymers, a general trend is seen between the tensile modulus and the MMW (i.e. the moduli increases with increasing diacrylate:amine ratio). For instance, J6 at the 1:1 ratio had a modulus of 0.22 ± 0.012 MPa and a modulus of 4.50 ± 1.08 MPa for the 1.4:1 ratio, which represents a 20 fold increase in modulus. The mean moduli were statistically different between ratios of the same polymer for all cases except 1:1 and 1.05:1 for C12 and J6, 1:1 and 1.1:1 for J6, and 1.05:1 and 1.1:1 for all polymers. Lower MMWs (greater diacrylate:amine ratio) led to networks with a higher crosslinking density, which corresponds to a greater modulus. Variations in the chemistry also contributed to the tensile properties, as the moduli for the C12 polymers were significant greater than the corresponding ratios of all E1 polymers and the 1:1, 1.05:1, and 1.4:1 ratios of J6. This is attributed to the polymer chemistry and not the actual MMW since the MW for the C12 polymer fell between the J6 and E1 polymer at all ratios (see Table 1). The J6 polymers are significantly larger that the E1 for the 1.2:1, 1.1:1, and 1:1 ratios.
Figure 5.
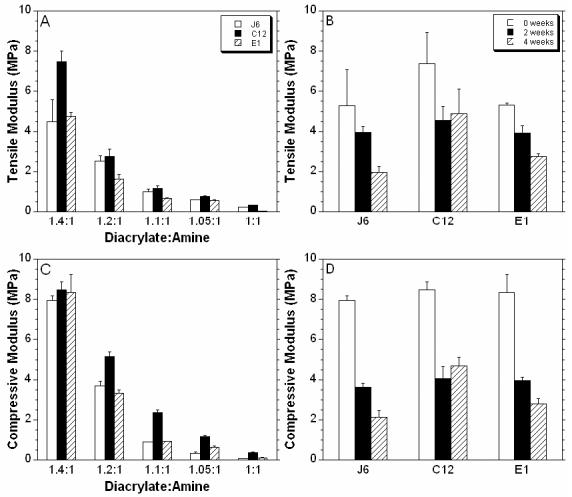
Moduli calculated in tension (A) and compression (C) for polymers formed from the J6, C12, and E1 macromers at various MMWs. The tensile (B) and compressive (D) moduli measured after 2 and 4 weeks of degradation in PBS at 37°C for the 1.4:1 ratio of all three polymers.
The compressive moduli (Figure 5C), shows a similar trend as the tensile moduli with respect to MMW. For example, the compressive moduli for the J6 1.4:1 and 1:1 were found to be 7.93 ± 0.23 and 0.075 ± 0.003 MPa, respectively. At the highest ratio, there was no significant difference between the three chemistries, but for the other ratios, the C12 polymers were significantly stiffer. For the 1.05:1 ratio, the modulus for J6 was significantly larger than E1. The lower MMWs correspond to higher reactive group concentrations and subsequently, higher crosslinking densities. The reaction behavior indicates that this also leads to greater double bond conversions. These properties correspond to alterations and control over the bulk mechanics of the polymer. The macromer chemistry can also play a role and the C12 macromers have a large imidazole ring in each repeat unit, which intuitively may alter polymer mechanics.
The ability to change mechanical properties by altering the MMW could be useful for fine tuning these properties in order to properly adapt to a desired application. This is useful in tissue engineering applications as different cell types prefer different extracellular environments. Cells have been shown to exert forces on their environment in order to sense the stiffness of their environment17 and subsequently respond to it. Thus, material properties can play a role in cellular interaction behavior. Recently Engler and coworkers31 reported that matrix elasticity directs mesenchymal stem cells (MSC) differentiation. When MSCs were grown on polyacrylamide gels of different stiffnesses, but in the same media, the cells grown on the soft gels expressed neurogenic markers, stiffer gels were myogenic, and the stiffest gels displayed osteogenic markers. Therefore, the ability to control matrix stiffness can be used to alter MSC differentiation without soluble factors.
Polymer degradation
The degradation of the various polymers was monitored over 17 weeks and the mass loss profiles are shown in Figure 6. Typically, degradation began with a period of rapid mass loss, likely due to unreacted macromer release as indicated by the sol fraction from Table 1. The rate of degradation increased with a decrease in the diacrylate:amine ratios. For example, for the C12 polymer 88.2% of the mass is lost after 17 weeks for the 1.05:1 ratio, whereas only 58.5% is lost for the 1.4:1 ratio. As the MMW increases, there are more hydrolytically susceptible bonds between the crosslinks. Therefore, there is a greater probability that multiple bonds are hydrolyzed in the crosslink and a polymer fragment (i.e., degradation product) can be released into solution. With this control, the degradation rate can be tuned to a particular application.
Figure 6.
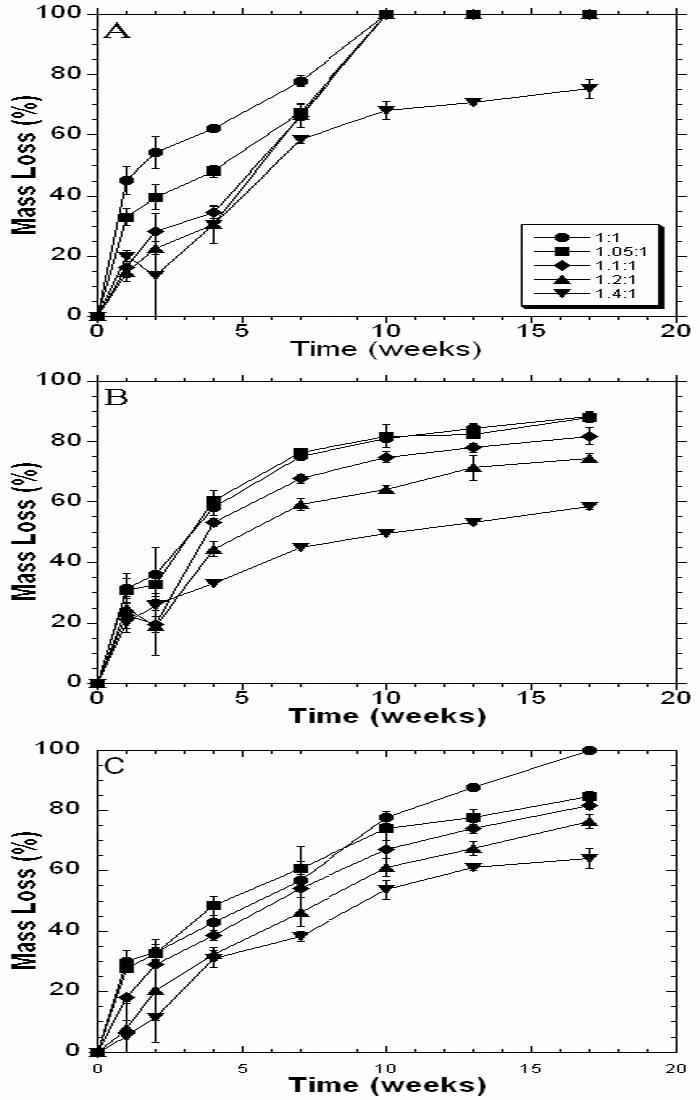
Network mass loss with degradation in PBS at 37°C for the various MMWs of J6 (A), C12 (B), and E1 (C).
The influence of polymer chemistry on degradation is apparent in the mass loss profiles. In Figure 6, the degradation of the J6 polymers occurs much faster than the C12 or E1 polymers. After 9 weeks, all of the ratios, except the 1:1.4 ratio, have degraded completely for the J6 polymer. The C12 polymer (Figure 6B) has distinct separation between the ratios as degradation proceeds, but the mass loss appears to plateau, which may indicate that there are non-degradable crosslinks incorporated in the network. E1 polymer degradation for all MMWs is fairly linear. The influence of macromer chemistry on degradation was reported previously during the initial development of the PBAE macromer library, but now more control over the degradation behavior is afforded by alterations in MMW.
In general, mass loss correlates with a decrease in both the compressive and tensile moduli as shown in Figure 5 (B,D). J6 and E1 polymers were consistently weaker with degradation, for the ratios (1.4:1 shown in Figure 5) studied. The C12 polymer showed an initial decrease in mechanics after 2 weeks, but no change was observed between 2 and 4 weeks. This may be due to a portion of non-degradable crosslinks in the network as mentioned previously. The decrease in polymer moduli with time is typical of a bulk-degrading polymer, which may be useful for tissue engineering applications since the surface is not shed with degradation and could potentially support cellular adhesion and growth.
The ability to control degradation can be utilized in a number of medical applications. Since molecule release is typically controlled by both diffusion and degradation, degradation can be used to alter and optimize the release of therapeutic molecules from degradable polymers. The local and controlled release of drugs and growth factors can be used to treat diseased or damaged tissues without the side effects associated with larger, systemic treatments.32,33 For example, this has been demonstrated in slow release of chemotherapeutics in neuronal implants, drug eluding stents, and microsphere treatments for prostate cancer.33 In tissue engineering, control over degradation can be used to optimize extracellular matrix production and distribution.34 The desired degradation rate is likely specific to tissue type, as some tissues exhibit regenerative capabilities like skin and bone, while others have limited regenerative potential, such as cartilage.35 Thus, the ideal degradation can be adjusted by varying the MMW of these polymers or others from our initial PBAE library.
Cell interaction studies
SaOS-2 cells, a human osteoblast-like cell, were seeded onto polymer films to see how well anchor dependent cells attached and spread on the polymers with changes in the polymer chemistry and structure (i.e., MMW). This experiment gives some indication of the toxicity of the polymers and how substrate properties influence cellular interactions. As a control, cells were also cultured under typical conditions on tissue culture polystyrene (TCPS).
Figure 7 shows photomicrographs of cells grown on the surface of the J6 polymer at each ratio (Figure 7B-F) as well as a control on standard tissue culture polystyrene (TCPS) (Figure 7A) after 24 hours in culture. While cells adhered to all surfaces, cells were in a more rounded morphology on the lower ratios (i.e., higher MMWs) and showed no spreading on the lowest ratio, unlike the controls. As the ratio increased, the cells appeared to spread out more. A similar trend was seen on the E1 polymers. However, cell viability was low on the C12 polymers, potentially due to changes in the pH as indicated by color changes in the phenol-red containing media. This limits the use of this polymer for tissue engineering applications, but fortunately there are other potentially viable candidates from the polymer library. Quantification of cell adhesion is shown in Figure 8A. There was no statistical difference in the numbers of cells adhered to the films with variation in the MMW.
Figure 7.
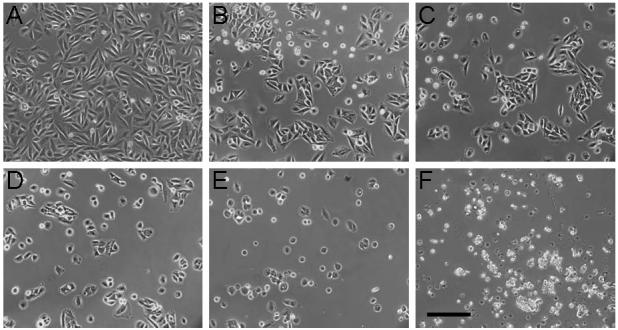
Photomicrographs of SaOS-2 cells 24 hours after seeding on TCPS (A), and films of polymers from the J6 macromers synthesized at diacrylate:amine ratios of 1.4:1 (B), 1.2:1 (C), 1.1:1 (D) , 1.05:1 (E), and 1:1 (F). The scale bar represents 200 μm.
Figure 8.
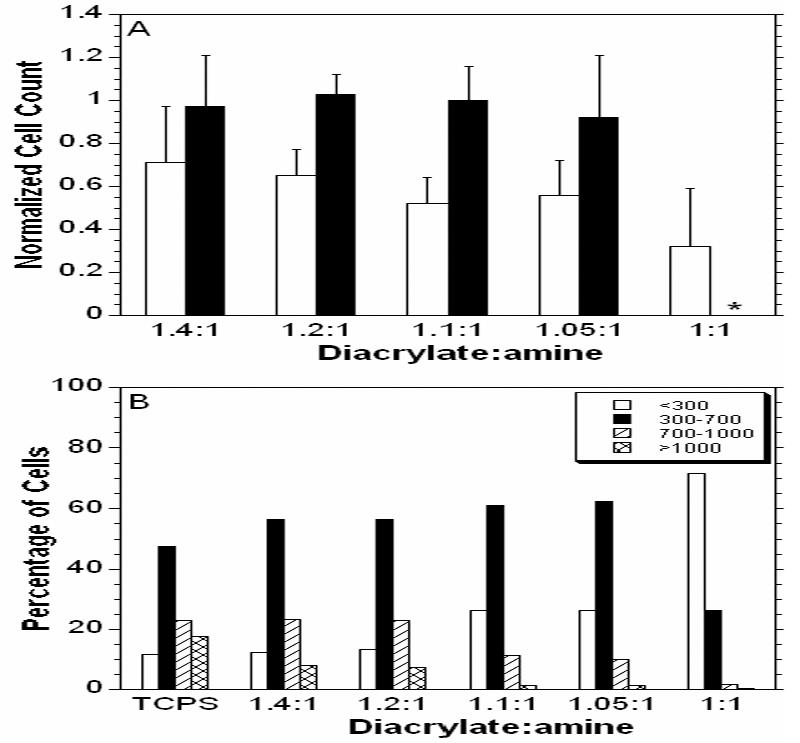
Number of cells adhered to J6 (white) and E1 (black) normalized to the number of cells adhered to the TCPS control (A). The C12 films created an acidic environment and there were no cells adhered to the polymer after 24 hours. Histogram of the percentage of cells in ranges of spreading area for polymer J6 (B). * denotes that the polymers was not transparent at that composition and could not be analyzed.
The areas of cell spreading on films of polymer J6 are plotted as a histogram in Figure 8B. For higher MMWs the cells, while similar in number in Figure 8A, are dominated by cells with spread areas less than 300 μm2. As the ratio increases the cells are more distributed with larger areas. The higher ratios have more of the most spread cells (>1000 μm2), but still not as many as the TCPS cells. While these are just short term measurements (24 hr), they indicate that chemistry can play a role in cell adhesion, as the cells appear to adhere to the E1 polymers more readily than the J6 polymers. The MMW (i.e., diacrylate to amine ratio) influenced the spreading of cells on the polymer films to morphologies similar to that of the TCPS controls. This could be due to a few differences caused by changing the MMW. First, many anchorage dependent cells have more organized cytoskeletons and more stable adhesions to stiffer environments17 (hence why attachment to the stiff TCPS is so high) and therefore spread better on the stiffer matrices of the more highly crosslinked networks. Also, polymers from higher MMWs had a higher sol fraction, which may release more potentially toxic components into the media.
CONCLUSIONS
We have shown that by varying the ratio of diacrylates to amines in forming three different PBAE macromers, we can adjust the MMW. By increasing the diacrylate to amine ratio (i.e., decreasing the MMW), the photopolymerization is faster and more complete. This leads to increased mechanical stiffness and decreased rates of mass loss. For given ratios, the chemical structure of the networks can also affect the mechanical properties and degradation. The mechanical properties decreased with degradation, which is indicative of bulk degradation. Two of the polymers were found to support attachment and spreading of human SaOS-2 cells, and spreading was heavily dependent on MMW, with greater cells spreading on polymers formed from lower MMWs. Although no claims can be made regarding the suitability of these specific compositions for tissue regeneration applications, particularly due to high sol fractions, this work is beneficial in better understanding fundamental structure-function relationships and can be used for better designing photocrosslinkable biomaterials.
Acknowledgments
The authors acknowledge funding through the National Institutes of Health (K22 DE-015761). Also, the authors are grateful to Dr. Daniel Anderson for helpful discussions.
References
- 1.Anseth KS, Goodner MD, Reill MA, Kannurpatti AR, Newman SM, Bowman CN. The influence of comonomer composition on dimethacrylate resin properties for dental composites. Journal of Dental Research. 1996;75(8):1607–1612. doi: 10.1177/00220345960750081301. [DOI] [PubMed] [Google Scholar]
- 2.Hillwest JL, Chowdhury SM, Sawhney AS, Pathak CP, Dunn RC, Hubbell JA. Prevention of Postoperative Adhesions in the Rat by in-Situ Photopolymerization of Bioresorbable Hydrogel Barriers. Obstetrics and Gynecology. 1994;83(1):59–64. [PubMed] [Google Scholar]
- 3.Anseth KS, Burdick JA. New directions in photopolymerizable biomaterials. Mrs Bulletin. 2002;27(2):130–136. [Google Scholar]
- 4.Chung C, Mesa J, Miller GJ, Randolph MA, Gill TJ, Burdick JA. Effects of auricular chondrocyte expansion on neocartilage formation in photocrosslinked hyaluronic acid networks. Tissue Engineering. 2006;12(9):2665–2673. doi: 10.1089/ten.2006.12.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung C, Mesa J, Randolph MA, Yaremchuk M, Burdick JA. Influence of gel properties on neocartilage formation by auricular chondrocytes photoencapsulated in hyaluronic acid networks. Journal of Biomedical Materials Research Part A. 2006;77A(3):518–525. doi: 10.1002/jbm.a.30660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elisseeff J, McIntosh W, Anseth K, Riley S, Ragan P, Langer R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. Journal of Biomedical Materials Research. 2000;51(2):164–171. doi: 10.1002/(sici)1097-4636(200008)51:2<164::aid-jbm4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 7.Fisher JP, Dean D, Engel PS, Mikos AG. Photoinitiated polymerization of biomaterials. Annual Review of Materials Research. 2001;31:171–181. [Google Scholar]
- 8.Leach JB, Bivens KA, Patrick CW, Schmidt CE. Photocrosslinked hyaluronic acid hydrogels: Natural, biodegradable tissue engineering scaffolds. Biotechnology and Bioengineering. 2003;82(5):578–589. doi: 10.1002/bit.10605. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2002;23(22):4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 10.Smeds KA, Grinstaff MW. Photocrosslinkable polysaccharides for in situ hydrogel formation. Journal of Biomedical Materials Research. 2001;54(1):115–121. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 11.Anseth KS, Metters AT, Bryant SJ, Martens PJ, Elisseeff JH, Bowman CN. In situ forming degradable networks and their application in tissue engineering and drug delivery. Journal of Controlled Release. 2002;78(13):199–209. doi: 10.1016/s0168-3659(01)00500-4. [DOI] [PubMed] [Google Scholar]
- 12.Burdick JA, Peterson AJ, Anseth KS. Conversion and temperature profiles during the photoinitiated polymerization of thick orthopaedic biomaterials. Biomaterials. 2001;22(13):1779–1786. doi: 10.1016/s0142-9612(00)00347-1. [DOI] [PubMed] [Google Scholar]
- 13.Beebe DJ, Moore JS, Yu Q, Liu RH, Kraft ML, Jo BH, Devadoss C. Microfluidic tectonics: A comprehensive construction platform for microfluidic systems. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(25):13488–13493. doi: 10.1073/pnas.250273097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albrecht DR, Underhill GH, Wassermann TB, Sah RL, Bhatia SN. Probing the role of multicellular organization in three-dimensional microenvironments. Nature Methods. 2006;3(5):369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- 15.Khademhosseini A, Eng G, Yeh J, Fukuda J, Blumling J, III, Langer R, Burdick JA. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. Journal of Biomedical Materials Research Part A. 2006;79A(3):522–532. doi: 10.1002/jbm.a.30821. [DOI] [PubMed] [Google Scholar]
- 16.Burg KJL, Porter S, Kellam JF. Biomaterial developments for bone tissue engineering. Biomaterials. 2000;21(23):2347–2359. doi: 10.1016/s0142-9612(00)00102-2. [DOI] [PubMed] [Google Scholar]
- 17.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310(5751):1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 18.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23(22):4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 19.Hern DL, Hubbell JA. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. Journal of Biomedical Materials Research. 1998;39(2):266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 20.Burdick JA, Chung C, Jia XQ, Randolph MA, Langer R. Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules. 2005;6(1):386–391. doi: 10.1021/bm049508a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher JP, Holland TA, Dean D, Engel PS, Mikos AG. Synthesis and properties of photocross-linked poly(propylene fumarate) scaffolds. Journal of Biomaterials Science-Polymer Edition. 2001;12(6):673–687. doi: 10.1163/156856201316883476. [DOI] [PubMed] [Google Scholar]
- 22.Anseth KS, Shastri VR, Langer R. Photopolymerizable degradable polyanhydrides with osteocompatibility. Nature Biotechnology. 1999;17(2):156–159. doi: 10.1038/6152. [DOI] [PubMed] [Google Scholar]
- 23.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: synthetic ECM analogs for tissue engineering. Biomaterials. 2001;22(22):3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 24.Burdick JA, Mason MN, Hinman AD, Thorne K, Anseth KS. Delivery of osteoinductive growth factors from degradable PEG hydrogels influences osteoblast differentiation and mineralization. Journal of Controlled Release. 2002;83(1):53–63. doi: 10.1016/s0168-3659(02)00181-5. [DOI] [PubMed] [Google Scholar]
- 25.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible Hydrogels Based on Photopolymerized Poly(Ethylene Glycol)-Co-Poly(Alpha-Hydroxy Acid) Diacrylate Macromers. Macromolecules. 1993;26(4):581–587. [Google Scholar]
- 26.Lynn DM, Langer R. Degradable poly(beta-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. Journal of the American Chemical Society. 2000;122(44):10761–10768. [Google Scholar]
- 27.Akinc A, Lynn DM, Anderson DG, Langer R. Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery. Journal of the American Chemical Society. 2003;125(18):5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 28.Anderson DG, Tweedie CA, Hossain N, Navarro SM, Brey DM, Van Vliet KJ, Langer R, Burdick JA. A combinatorial library of photocrosslinkable and degradable materials. Advanced Materials. 2006;18(19):2614–+. [Google Scholar]
- 29.Soltz MA, Ateshian GA. Experimental verification and theoretical prediction of cartilage interstitial fluid pressurization at an impermeable contact interface in confined compression. Journal of Biomechanics. 1998;31(10):927–934. doi: 10.1016/s0021-9290(98)00105-5. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal CM, Ray RB. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. Journal of Biomedical Materials Research. 2001;55(2):141–150. doi: 10.1002/1097-4636(200105)55:2<141::aid-jbm1000>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 31.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 32.Langer R. Drug delivery and targeting. Nature. 1998;392(6679):5–10. [PubMed] [Google Scholar]
- 33.Langer R, Peppas NA. Advances in biomaterials, drug delivery, and bionanotechnology. Aiche Journal. 2003;49(12):2990–3006. [Google Scholar]
- 34.Bryant SJ, Anseth KS. Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly(ethylene glycol) hydrogels. Journal of Biomedical Materials Research. 2002;59(1):63–72. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- 35.Miller MD. Review of Orthopaedics. Saunders; Philadelphia: 2004. p. 7004. [Google Scholar]