

Abstract
We report the first demonstration of rapid electrophoretic monitoring of homocysteine thiolactone-induced protein oligomerization (HTPO), a unique type of post-translational protein modification that may have clinical significance as an indicator of cardiovascular and neurovascular diseases. HTPO of the model protein bovine cytochrome c was initiated in vitro. The relative monomer and aggregate levels of the resultant protein mixtures were determined following separation using capillaries coated with the cationic polymer, poly(diallyldimethylammonium chloride). UV detection provided adequate sensitivity for the monitoring of higher order species, which exist at relatively low concentrations in the protein reaction mixture as compared to the monomeric species. Separations performed under standard injection conditions were optimized on the basis of applied voltage and sample denaturation conditions. Separations performed using short-end injection allowed for more rapid analyses, typically in less than 70 s. Relative errors for run-to-run migration times were less than 0.5%. This novel oligomeric system provides a rapid and straightforward in vitro method to screen therapeutic agents for their ability to inhibit HTPO. Changes in peak area for monomer and aggregate species were used to assess HTPO inhibition as a function of pyridoxal 5-phosphate (PLP) concentration. PLP was shown to effectively inhibit HTPO in vitro. Rapid analysis times of ~1.5 min were achieved for inhibition screening.
Hyperhomocysteinemia is a disorder characterized by elevated plasma levels of the naturally occurring amino acid homocysteine (Hcy), which has been identified as an independent risk factor for cardiovascular disease.1,2 Although the exact function of Hcy in the pathogenesis of cardiovascular disease remains unknown, there is a growing body of evidence that suggests Hcy thiolactone (HTL), the thioester metabolite of Hcy, is a contributing agent.3–6 HTL is produced in vivo via enzymatic aminoacyl-tRNA synthase pathways at a rate proportional to extracellular Hcy concentration.7 Thus, it is likely that HTL production is enhanced to some degree in individuals with hyperhomocysteinemia.
Post-translational protein modification by HTL involves the site-specific acylation of lysine residues at ε-amino moieties to yield free thiols in the form of protein homocystamide (Scheme 1).8,9 If left untreated, protein homocystamide can undergo spontaneous in vitro intermolecular disulfide bonding to yield oligomeric species.10 Protein oligomerization can give rise to a range of adverse physiological affects. For example, oligomeric proteins often exhibit decreased biological activity, decreased solubility,8 and increased immunogenicity.11 As one would expect, the extent of physiological dysfunction caused by oligomerization varies based on the size and degree of modification. Experimental evidence of HTL-induced protein oligomerization (HTPO) under physiological conditions has been reported for various proteins including low-density-lipoprotein,12 cytochrome c, and human serum protein.8
Scheme 1. Homocysteine Thiolactone-Induced Protein Modificationa.
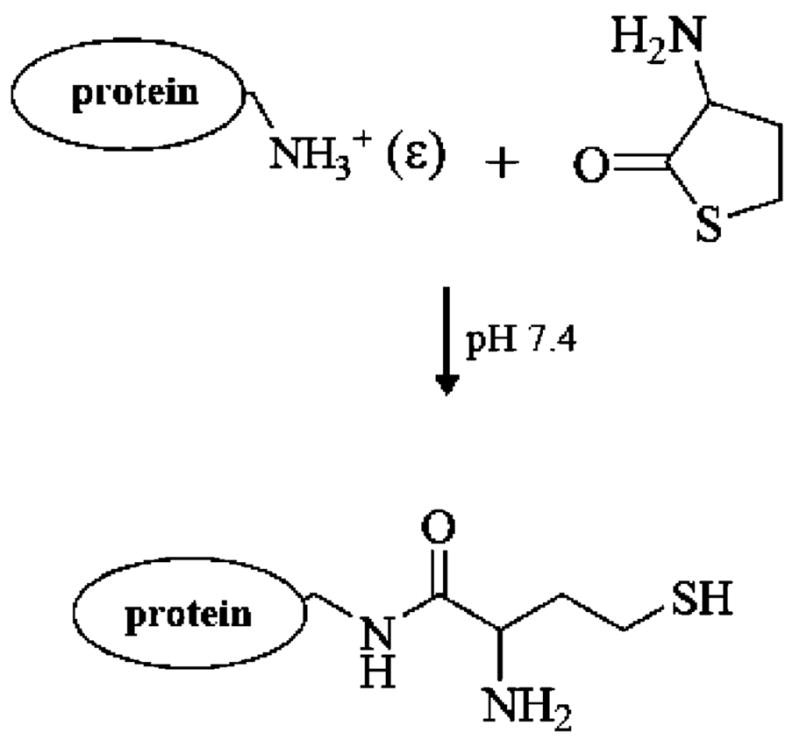
a Under physiological pH conditions, homocysteine thiolactone can preferentially acylate protein lysine residues at ε-amino groups to yield protein homocystamide.
Increased in vivo HTL production may also contribute to vascular damage and the formation of atherosclerotic lesions at the onset of cardiovascular disease.3,13 Moreover, a recent clinical study suggested that the level of HTL-modified protein in plasma could be a more appropriate risk indicator for cardiovascular disease than plasma Hcy levels.5 Given the emerging clinical significance of HTPO as a biomarker for cardiovascular disease, the development of new analytical methods for studying modification-induced protein aggregation is of increasing interest.
Conventional SDS-PAGE is perhaps the most widely used technique for routine protein separations. However, lengthy analysis times are a hindrance to its use in rapid, high-throughput screening applications. Another potential problem with conventional SDS-PAGE is the tedious and time-consuming staining procedures required to visualize the gel separation. In contrast, capillary electrophoresis (CE) can often provide rapid, highly efficient separations without the necessity for time-consuming dye staining or radioisotope labeling. Recent advances in instrumentation and column technology have made CE an attractive alternative to conventional electrophoretic techniques such as SDS-PAGE for clinical and pharmaceutical protein separations.14–16 In fact, reports in the literature of CE-based protein separation techniques have steadily increased over the past decade.17–20 This trend is due in part to the ongoing development of polyelectrolyte coatings, which provide a practical and effective remedy for analyte–wall adsorption.21 Our laboratory has demonstrated the utility of neutral22 and polyelectrolyte coatings23–27 for improved capillary electrophoretic separations. Our most recent studies have employed various amino acid-functionalized molecular micellar stationary phases for open tubular–capillary electrochromatography. These novel coatings allow for facile column fabrication and exceptional separation performance for both chiral and achiral analytes.23–27
Herein, we report the development of a rapid CE method for studying in vitro post-translational modification-induced protein oligomerization. Capillaries coated with poly(diallyldimethylammonium chloride) (PDADMAC) were used to separate homooligomeric cytochrome c species. We also demonstrate the potential use of CE for rapidly monitoring in vitro HTPO inhibition by pyridoxal 5-phosphate, a potentially therapeutic agent for offsetting the effects of protein homocysteinylation.
EXPERIMENTAL SECTION
Reagents
Bovine cytochrome c (cyt c), L-HTL hydrogen chloride, pyridoxal 5-phosphate hydrogen chloride (cofactor B6), SDS-PAGE protein molecular weight markers, PDADMAC 20% w/v water solution (Mw 200 000–350 000), tris(hydroxymethyl)-aminomethane (TRIS), and all other reagents used for preparing reaction and background electrolytes were obtained from Sigma-Aldrich (St. Louis, MO) at the highest purity available and used without further purification. Ultrapure water (18.2 MΩ) was obtained by use of an Elga model Purelab Ultra water filtration system.
In Vitro Protein Modification
Protein reaction mixtures were prepared by dissolving HTL (2.5 mM), and cyt c (10 mg/mL) in pH 7.4, 100 mM sodium phosphate, 0.2 mM ethylenedi-aminetetraacetic acid (EDTA) buffer, according to procedures previously reported by Jakubowski.8 It was determined that 12 h is sufficient time for detectable oligomerization of cyt c to occur in the protein reaction mixture at 25 °C. However, protein mixtures reacted beyond 24 h tended to form precipitates. Samples used for CE studies were reacted at room temperature while mixing for 12 h. The modification reaction was quenched by removing the protein from the reaction mixture using a Millipore (Bedford, MA) Amicon Ultra, 10 000 kDa molecular weight cutoff (MWCO) filter device centrifuged at 3600 rpm for 20 min or until completion. The protein retentate was rinsed with 4 mL of reaction buffer to remove nonspecifically bound material. Protein and filtrates were stored at −20 °C for no more than one week. Unless otherwise noted, the protein retentates were reconstituted in either the background electrolyte (50 mM pH 8.5 TRIS buffer) or a mixture of background electrolyte and organic solvent. All protein and buffer solutions were passed through a 0.45-μm nylon syringe filter prior to analysis. Protein reaction mixtures for HTPO inhibition studies were prepared and treated in the same manner as described above, except the desired quantity of pyridoxal 5-phosphate stock solution was simultaneously added to the HTL and protein solution to obtain the desired concentrations.
Gel Electrophoresis
SDS-PAGE experiments were conducted using a Mini-Protean 3 gel electrophoresis unit with 1000-V power supply. Polyacrylamide gels (4–20%) were purchased from Bio-Rad (Hercules, CA). The sample buffer was prepared by combining 0.2 mL of 0.5% (w/v) bromophenol blue, 2.0 mL of 10% (w/v) SDS, 2.5 mL of glycerol, 1.25 mL of pH 6.8, 0.5 M TRIS hydrochloride, and 3.55 mL of bioscience grade water to obtain a total volume of 9.5 mL. Protein samples were diluted to 0.1 μg/mL with sample buffer and thermally denatured under nonreducing conditions at 90 °C for 5 min prior to SDS-PAGE analyses. The stock running buffer was prepared by dissolving 15.15 g of TRIS-base, 72 g of glycine, and 10.0 g of SDS in bioscience grade water to obtain a final volume of 1 L and working pH of 6.8. The stock run buffer was diluted 10-fold to obtain the final running buffer. SDS-PAGE separations were run at 200 V for 30 min at room temperature, followed by staining for 1 h with SimplyBlue SafeStain Coomassie G-250, obtained from Invitrogen (Carlsbad, CA). The gels were destained with water for at least 12 h to obtain optimum band contrast. A Kodak Gel Logic 100 digital imaging system purchased from Eastman Kodak Co. (Rochester, NY) was used for gel imaging.
Capillary Electrophoresis
A Beckman Coulter (Fullerton, CA) P/ACE MDQ capillary electrophoresis system equipped with a photodiode array detector was used for CE separations. Fused-silica capillaries (50-μm i.d.) were purchased from Polymicro Technologies (Phoenix, AZ) and cut to 32-cm total length, yielding an effective length of 22 cm. Capillaries were modified with 20 mg/mL PDADMAC dissolved in pH 8.5, 50 mM TRIS buffer. A capillary coating was deposited for a 12-h period using alternating rinse cycles of PDADMAC solution at high pressure (20 psi) for 5 min and low pressure (0.5 psi) for 120 min as described by Wang and Dubin.28 Upon completion of the coating process, capillaries were rinsed with the background electrolyte solution (50 mM TRIS, pH 8.5 buffer) at 5 psi for 40 min to remove excess PDADMAC. Under normal polarity conditions (+ to − with reference to the inlet), electroosmotic flow is reversed due to the positively charged PDADMAC-coated capillary surface. Therefore, electrokinetic injections and separations were performed under reversed polarity conditions. All separations were performed at 25 ± 1 °C. The capillary was rinsed with the background electrolyte for 2 min at 5.0 psi prior to each run. Separations were simultaneously monitored from 200 to 320 nm using a photodiode array detector. Data acquired at 200 nm were exported to a spreadsheet and fit to Gaussian peaks. Determinations of migration time, peak width, and peak area for CE separations were made based on the best-fit data.
Resolution (Rs), full width at half-height (fwhh), and number of theoretical plates (N) were calculated using the following equations:
| (1) |
| (2) |
| (3) |
where tr,1 and tr,2 are the migration times for the earlier and later eluting peaks, respectively, w is peak width at baseline for each peak, w(1/2) is fwhh, and σ is the standard deviation of the Gaussian-fitted peaks.
Short-End Injection
Short-end injection is an approach used in CE to obtain shorter analysis times by circumventing instrumental limitations on minimum capillary length.29–32 The shortest capillary readily accommodated by the Beckman MDQ CE instrument used in this work is 32 cm (22-cm effective length). Short-end injections and separations were performed by applying voltage in reversed polarity with reference to the outlet in order to obtain EOF toward the detection window, resulting in an effective capillary length of 10 cm with no change in total capillary length (32 cm). Short-end electrokinetic injections were performed from the outlet end of the capillary using 5 kV for 7 s.
RESULTS AND DISCUSSION
Confirmation of Oligomerization in the Protein Reaction Mixtures
Recall that homocysteine thiolactone-induced protein oligomerization (HTPO) involves site-specific acylation of protein lysine residues by HTL. Consequently, the degree of reactivity of HTL toward a given protein is largely dependent on lysine content. Cyt c (12 327 kDa) is a small, highly conserved metalloprotein found associated with the mitochondria of most cells. Cyt c was found to be an ideal model protein for demonstration of the modification-induced oligomerization. This is because the relatively high lysine content (~20% w/w)33 makes it susceptible to post-translational modification by HTL. HTL-modified cyt c has been shown to form a range of soluble oligomeric species in vitro.8
SDS-PAGE analysis was used in this study to confirm aggregation in the protein reaction mixtures prior to proceeding to CE analyses. As expected, monomer and aggregate bands were visible in gel images after dye staining with coomasie blue (Figure 1a). The three relatively broad bands were identified as monomeric (I), dimeric (II), and trimeric (III) oligomeric cyt c species as determined by comparison with SDS-PAGE protein molecular weight markers. The gel separation profiles were consistent with SDS-PAGE separations of oligomeric cyt c previously reported in the literature.8 Treatment of the protein reaction mixture with 5 mM dithiothreitol, a disulfide-reducing agent, resulted in elimination of the oligomer bands (data not shown). Additional characterization of the protein reaction mixture using MALDI TOF-MS (Figure S1 in Supporting Information) allowed detection of monomeric and oligomeric species in the protein reaction mixture, having signal intensities consistent with those observed with SDS-PAGE. Collectively, these data are indicative of modification-induced oligomerization due to intermolecular disulfide formation.
Figure 1.

Representative electrophoretic separations of the model oligomeric protein mixture. (a) Black and white photograph of coomasie blue-stained SDS-PAGE minislab gel after separation shows three bands corresponding to monomeric, dimeric, and trimeric cyt c species, (b) electropherogram of nondenatured protein reaction mixture separation monitored at 200 nm showing five distinct components corresponding to monomeric and oligomeric species (I–V), and (c) electropherogram obtained for the separation of a thermally denatured sample showing improved peak shapes. Plot of electrophoretic migration time versus species molecular weight typically obtained for protein mixture separations (inset). The x-axis was generated based on integer multiples of the molecular weight for monomeric bovine cyt c as reported by the manufacturer. Roman numerals (I–V) indicate monomeric, dimeric, trimeric, tetrameric, and pentameric species, respectively.
CE Separation of the Oligomeric Protein Mixtures
Figure 1b is a typical electropherogram resulting from the separation of cyt c (pI ~10) protein reaction mixtures using cationic PDADMAC-coated capillaries. The increased sensitivity provided by UV detection allowed for detection of two additional peaks attributed to tetrameric (IV) and pentameric (V) cyt c species that were not visible in the SDS-PAGE image shown in Figure 1a. Analysis time for the separation shown in Figure 1b was less than 8 min, including the rather significant tailing of the monomer peak. Thermal denaturation of the protein reaction mixture is shown to significantly improve peak symmetry (vide infra).
The migration order was determined to be pentamer (V), tetramer (IV), trimer (III), dimer (II), and monomer (I), respectively. Under reversed polarity conditions, the species with the largest electrophoretic mobility has the smallest apparent mobility and elutes last. Thus, the largest electrophoretic mobility is observed for the monomer. It is interesting to note that migration times for oligomeric protein species were inversely related to aggregate number (molecular weight) with a strong linear correlation for species I–V.
This unique electrophoretic behavior is not easily explained in terms of classical electrophoresis theory. In the simplest case (the absence of analyte–capillary interactions, spherical particle, continuous medium, etc.), the electrophoretic behavior of a charged species can be explained in terms of Debey–Huckel–Henry theory, i.e.
| (4) |
where μ is electrophoretic mobility, q is the charge on the particle, η is the viscosity of the background electrolyte, and r is the Stokes radius.34 However, this simple relation is not generally sufficient to predict the CE behavior of proteins due in part to difficulties in accurately predicting Stokes radii and net charges associated with proteins.
Even if reasonable values for q and r are available, the prediction of protein mobilities is further complicated by the sometimes large effect of protein–wall interactions. For example, it is well-established that the adsorption of proteins to the capillary wall is a major problem in the CE separation of proteins. A positively charged wall coating was used to minimize protein adsorption in this study. However, it has also been shown that PDADMAC-coated capillaries may provide for selective protein interactions leading to improved resolution.28 In particular, coating procedures that use a combination of high ionic strength and high molecular weight PDADMAC have been found to provide enhanced surface coverage and charge inversion and may facilitate the formation of “loops and tails” structures on the capillary surface, with which protein analytes may interact. Studies performed using such coated capillaries have demonstrated an enhancement in selectivity for bovine serum albumin and β-lactoglobulin28 as compared to unmodified capillaries. A similar coating, utilizing PDADMAC with a molecular weight range of 200 000–350 000 was employed in our investigation of the oligomeric protein mixtures.
In addition, modification-induced charge neutralization may also influence the electrophoretic behavior of oligomeric protein species investigated in this work by changing their effective charge. Recall that HTL-induced protein modification results in a net loss of positive charge for lysine ε-amino groups upon the formation of protein homocystamide (Scheme 1),8 thereby causing a net reduction in effective charge. Regardless of the underlying mechanism, the unique behavior of this oligomeric system provides a rapid and straightforward method for screening therapeutic agents for HTPO inhibition activity (vide infra).
Effect of Thermal Denaturation on the Oligomer Separation
The rather significant band broadening observed in Figure 1b is consistent with the expected electrophoretic behavior of post-translationally modified proteins. It is known that post-translational protein modification often results in the introduction of additional structural heterogeneity to protein systems.34 Specifically, HTL-induced protein modification can lead to a generic loss of positive charge at lysine residues upon the formation of protein homocystamide,8 resulting in less structural homogeneity, altered electrophoretic mobility, and band broadening (Figure 1b).
Thermal denaturation may provide a means for minimizing structural heterogeneity in protein mixtures. Thermal denaturation involves exposing protein molecules to temperatures well above the protein melting point (usually ~90 °C) for several minutes in order to disrupt the native conformation. In doing so, near-complete and irreversible denaturation of the protein molecules is usually achieved, effectively providing more uniformity in protein structure. Therefore, the effect of thermal denaturation on CE separation performance was investigated. In this study, thermal denaturation was performed by heating the protein reaction mixtures at 90 °C for 5 min under nonreducing conditions prior to separation by CE. Figure 1c shows the significant improvement in peak symmetry that resulted from thermal denaturation. Note that there is a significant improvement in the peak shape and band broadening previously observed for nonthermally denatured samples. In addition, thermal denaturation allowed for better detection of a minor pentameric species. The analysis time for the thermally denatured sample was ~7 min. These results suggest that thermal denaturation is an effective means of improving band broadening and overall CE separation performance.
The inset in Figure 1c shows a plot of protein molecular weight versus migration time generated from the average of five consecutive runs of thermally denatured samples containing species I–V. Note that the x-axis was generated based on integer multiples of the molecular weight for monomeric bovine cyt c as reported by the manufacturer. Migration times determined from best-fit data exhibited great precision, having standard deviations less than 0.5% for all detected species. Furthermore, the homooligomeric system exhibited remarkable linearity and yielded a surprisingly distinct “stepladder” electrophoretic migration profile.
The effect of protein denaturation by organic solvents including methanol, ethanol, acetonitrile, and acetone was also examined. In general, the use of volatile organic solvents adversely affected sample preparation and CE analyses. Relatively high organic solvent content, ~1:1 buffer/organic solvent, was usually required to achieve results comparable to thermally denatured samples. However, the addition of organic solvents to the samples caused protein precipitation, often leading to diminished analytical signal and poor reproducibility for separations (data not shown). In addition, protein precipitation also caused column fouling and reduced column lifetime. For these reasons, organic solvents were excluded from use in sample preparation procedures in this study.
Voltage Optimization Study
The effect of applied voltage on migration time and peak efficiency was investigated for the separation of oligomeric mixture using 21-cm effective length columns. Figure 2 illustrates the effect of a range of applied voltages on the separation of bovine cyt c aggregates using a freshly prepared capillary. As expected, the shortest analysis time (2.33 min) was obtained at −15 kV. Applied voltages higher than −15 kV often resulted in unstable current and poor reproducibility. In addition, separation efficiency improved with increasing voltage as indicated by a roughly 2-fold improvement in peak width observed from −8 to −15 kV. Resolution also improved significantly with increasing voltage. Resolution (Rs) between the monomer and dimer peaks at −8 and −15 kV were 1.04 and 1.20, respectively. These data suggest that peak efficiency was limited primarily by longitudinal band broadening under the conditions used for these studies.
Figure 2.

Effect of applied voltage on CE separations of oligomeric protein mixture components: electropherograms acquired at −15, −12, −10, and −8 kV. Separations were performed using a freshly prepared column; all other experimental parameters are provided in the text.
Short-End Injection CE Reproducibility Studies
Optimization studies for this work were conducted using standard injection conditions on capillaries columns having a total length of 32 cm (21-cm effective capillary length). In order to demonstrate the high-throughput capability of the method, the effective length of the capillary was decreased to 10 cm by use of short-end injection mode. Figure 3 shows a representative short-end electropherogram where separation of the protein reaction mixture containing species I–V was achieved in less than 70 s.
Figure 3.

Electropherogram of thermally denatured oligomeric protein mixture containing cyt c species I–V separated in less than 70 s using short-end injection. Separation performed with an applied voltage of 8 kV (22-μA current) and a 50-μm-i.d., 32-cm total length, 10-cm effective length capillary column.
In order to assess the stability of the short-end injection method, reproducibility and electrophoretic efficiency were evaluated for five successive runs without regenerating the PDADMAC coating. We note that a 2-min buffer rinse cycle was performed between runs in order to minimize sample carryover. Various column performance values are provided in Table 1. Run-to-run reproducibility for migration times was generally excellent, having RSD values of <0.5% for all monomer, dimer, and trimer peaks. Good precision was observed for peak area values, indicating reproducible injection volumes and near-ideal eletrophoretic behavior with respect to minimized analyte–wall adsorption. The relatively high number of theoretical plates (N), in some instances approaching 1 million plates, indicates exceptional column efficiency for separating monomer and aggregate species. A table of representative data resulting from replicate analyses is provided in the Supporting Information (Table S1). In addition, satisfactory column-to-column reproducibility was also achieved as indicated by RSD values of <1% (n = 10) for migration times obtained from five consecutive runs performed with two different PDADMAC-coated capillaries (data not shown). Overall, short-end injection provided acceptable separation performance as well as a ~6-fold increase in sample throughput over standard injection conditions.
Table 1.
Separation Performance and Reproducibility Statistics for Short-End CE Separations of Protein Reaction Mixturea
| protein mixture component | migration time, min | peak area | number of theoretical plates (N) × 105 |
|---|---|---|---|
| monomer (I) | 1.92 (0.31) | 220.54 (3.90) | 8.70 (3.24) |
| dimer (II) | 1.75 (0.42) | 63.76 (4.38) | 5.75 (5.93) |
| trimer (III) | 1.63 (0.45) | 14.20 (13.98) | 8.53 (12.77) |
Average migration time, peak area, and number of theoretical plates (N) values calculated for successive runs (n = 5) without regeneration of the PDADMAC coating. Percent relative standard deviation (% RSD) values are given in parentheses. Separations performed using 8 kV applied voltage.
Monitoring Inhibition of HTPO by Pyridoxal 5-Phosphate (PLP)
PLP is the aldehyde-functionalized cofactor analogue of vitamin B6. The antiatherogenic properties of PLP for lowering plasma Hcy levels and risk of cardiovascular disease are documented in the medical literature.35,36 However, to our knowledge, the effect of PLP on in vitro HTPO has not been formally investigated. Recently, mechanistic studies have been reported that suggest the possibility of pyridoxal tetrahydrothiazine formation via condensation of HTL with PLP as a means of detoxifying HTL in vivo.37 Similarly, we propose that in situ pyridoxal tetrahydrothiazine formation could possibly disrupt HTL-induced protein modification in the protein reaction mixture, thereby inhibiting HTPO. To further investigate this hypothesis, the previously optimized short-end CE method was applied to monitoring oligomer levels in protein reaction mixtures pretreated with PLP.
Representative results obtained from the analysis of protein reaction mixtures treated with PLP are summarized in Figure 4. Panels a and b in Figure 5 show representative electropherograms acquired for protein reaction mixtures treated with 0.25 and 2.5 mM PLP after 24 h. It is quite evident that the relative amount of oligomerization detected in the protein reaction mixtures prepared for the inhibition study is greater than was previously detected in samples prepared for the optimization studies. The primary reason for this discrepancy is the innate and characteristic variability of the post-translational modification reaction. Even the slightest variations in sample preparation, incubation temperature, or reaction time can greatly influence the yield of the oligomerization reaction. Consequently, one would not expect to obtain a consistent degree of oligomerization in the protein reaction mixtures from batch to batch. Fortunately, the inclusion of an internal control allows one to account for the natural inconsistency of yields in the protein reaction mixtures.
Figure 4.

Results for oligomerization inhibition study represented as (a) percent of total protein peak area determined by integration of best fit data, and (b) percent change for respective mixture component peak areas normalized to an experimental control versus pyridoxal 5-phosphate concentration. Oligomeric mixture components are denoted as monomeric, I (●); dimeric, II (◆); trimeric, III (▲); and tetrameric, IV (□); respectively. The total of aggregates (■) is obtained by taking the sum of the respective contributions of mixture components II–IV.
Figure 5.

Representative short-end CE electropherogram resulting from the separation of the protein reaction mixture pretreated with (a) 0.25 mM pyridoxal 5-phosphate and (b) 2.5 mM pyridoxal 5-phosphate. Separation performed using an applied voltage of 8 kV (22-μA current) and a 50-μm-i.d., 32-cm total length, 10-cm effective length capillary column.
The experimental control sample was found to contain monomeric and oligomeric species I–IV, in which oligomers (II–IV) represented 77.12 ± 1.72% of total bovine cyt c (Figure 4a). All aggregate species decreased with increasing concentrations of PLP to a minimum value of 55.58 ± 1.42% observed at 2.5 mM PLP (Figure 4b). At the same time, there was a complementary increase in the monomeric species with increasing concentrations of PLP. The trimer species exhibited the greatest change of total protein (−11.79 ± 0.94%) followed by the dimer (−8.32 ± 1.60%). The changes observed for the minor tetrameric component with respect to the experimental control were within experimental error. Note the overall reduction in aggregate species for the sample treated with 2.5 mM PLP. Comparable inhibition trends were also visible in SDS-PAGE separation of PLP-treated protein reaction mixtures (Figure S2 in Supporting Information). Collectively, these results suggest that HTPO is effectively inhibited by PLP in vitro.
Analysis times for PLP-treated samples under optimized conditions were typically ~1.5 min, ~20 s longer than those obtained for untreated samples. It was found that PLP, which has a phosphate group, binds nonspecifically with the protein resulting in decreased cationic character and increased electrostatic interactions between the protein and the PDADMAC coating. The increased band broadening observed for the separation of PLP-treated samples is consistent with increased analyte–wall interactions. Despite efforts to thoroughly rinse the protein prior to analysis, some PLP remained rather tightly associated with the protein as indicated by the distinctive reddish-orange color of the reaction solutions. Nevertheless, adequate resolution was obtained to achieve a quantitative analysis.
In situ pyridoxal tetrahydrothiazine formation was initially proposed as the mechanism for HTPO inhibition by PLP. Therefore, efforts were made to determine whether pyridoxal tetrahydrothiazine could be detected in the protein reaction mixtures. Protein reaction mixtures (500 μL) containing 10 mg/mL bovine cyt c, 2.5 mM HTL, and the desired quantity of PLP were prepared as previously described and allowed to react for 12 h. For comparison, a set of control reaction mixtures containing identical amounts of PLP and HTL (without protein) was also prepared. All reaction mixtures were passed through a 10-kDa MWCO centrifugation filter to remove the protein. Reaction mixture filtrates and control samples were diluted 10-fold and analyzed using UV–vis spectroscopy. Individual reaction components, PLP and pyridoxal tetrahydrothiazine, were monitored at 390 and 330 nm,37 respectively.
Tetrahydrothiazine formation in the control reactions increased linearly with PLP concentration (Figure S3a in the Supporting Information). In situ tetrahydrothiazine formation in protein filtrates also increased in response to increasing PLP concentration until a maximum was reached at 1.88 and 2.5 mM (Figure S3b in the Supporting Information). The maximum absorption for the tetrahydrothiazine product for protein samples was ~2-fold less than the maximum observed for the control reaction. This reduction in product absorption for filtrate samples is primarily attributed to nonspecific binding of PLP or pyridoxal tetrahydrothiazine to the protein, which was indicated by a subtle color change in the retentate from red to reddish-orange. It is also possible that some small amount of pyridoxal tetrahydrothiazine is lost due to nonspecific adsorption to the molecular weight cutoff filter. Nonetheless, these results suggest that in situ pyridoxal tetrahydrothiazine formation is involved in the inhibition of HTL-induced protein modification and oligomerization in vitro.
CONCLUSIONS
We have shown that CE with UV detection provides a practical alternative to SDS-PAGE for monitoring homocysteine thiolactone-induced protein aggregation. Capillaries coated with PDADMAC provided good column stability and reproducibility. Under optimized conditions, short-end injection typically allowed the separation of thermally denatured protein reaction mixtures in less than 70 s.
In this study we have also demonstrated rapid screening of HTPO inhibition in vitro with analysis times of ~1.5 min, which is a considerable improvement over the day time frame typically required to perform a comparable SDS-PAGE analysis. We have also shown that PLP effectively inhibits HTPO in vitro, presumably due to in situ pyridoxal tetrahydrothiazine formation. The rapid and reproducible CE separations obtained for the model oligomeric system investigated in this study suggest that this method may be amenable to high-throughput preclinical therapeutic drug screening.
Supplementary Material
Additional experimental data including mass spectrometry, SDS-PAGE, and UV–vis spectroscopy characterizations of the protein mixture components. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Tara Lavigne (Belaire High School, Baton Rouge, LA), a 2006 ACS Project SEED summer research scholar, for SDS-PAGE electrophoresis analyses conducted in support of these studies. A.T.G is the recipient of a fellowship from the National Science Foundation (NSF) GK-12 and a Dow Chemical Company fellowship through the National Organization of Black Chemist and Chemical Engineers. I.M.W. acknowledges the NSF, National Institutes of Health, and the Philip W. West Endowment for their support of this study.
References
- 1.Clarke R, Daly L, Robinson K, Naughten E, Cahalane S, Fowler B, Graham I. N Engl J Med. 1991;324:1149–1155. doi: 10.1056/NEJM199104253241701. [DOI] [PubMed] [Google Scholar]
- 2.Taylor LM, Defrang RD, Harris EJ, Porter JM. J Vasc Surg. 1991;13:128–136. doi: 10.1067/mva.1991.24913. [DOI] [PubMed] [Google Scholar]
- 3.Mercie P, Garnier O, Lascoste L. Apoptosis. 2000;5:403–411. doi: 10.1023/a:1009652011466. [DOI] [PubMed] [Google Scholar]
- 4.Jakubowski H, Ambrosius WT, Pratt JH. FEBS Lett. 2001;491:35–39. doi: 10.1016/s0014-5793(01)02143-3. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, Gao Y, Zhou J. Clin Chim Acta. 2006;364:230–234. doi: 10.1016/j.cccn.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Jakubowski H. Biomed Pharmacother. 2001;55:443–447. doi: 10.1016/s0753-3322(01)00085-3. [DOI] [PubMed] [Google Scholar]
- 7.Jakubowski H. Biochemistry. 1997;36:11077–11085. doi: 10.1021/bi970589n. [DOI] [PubMed] [Google Scholar]
- 8.Jakubowski H. FASEB J. 1999;13:2277–2283. [PubMed] [Google Scholar]
- 9.Jakubowski H. J Nutr. 2000;130:377S–381S. doi: 10.1093/jn/130.2.377S. [DOI] [PubMed] [Google Scholar]
- 10.Hop ECA, Bakhtiar R. Rapid Commun Mass Spectrom. 2002;16:1049–1053. doi: 10.1002/rcm.681. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg A. AAPSJ. 2006;8:E501–E507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naruszewicz M, Mirkiewicz E, Olszewski AJ, McCully KS. Nutr Metab Cardiovasc. 1994;4:70–77. [Google Scholar]
- 13.Austin RC, Lentz SR, Werstuck GH. Cell Death Differ. 2004;11:S56–S64. doi: 10.1038/sj.cdd.4401451. [DOI] [PubMed] [Google Scholar]
- 14.Jabeen R, Payne D, Wilktorowicz J, Mohammad A, Petersen J. Electrophoresis. 2006;27:2413–2438. doi: 10.1002/elps.200500948. [DOI] [PubMed] [Google Scholar]
- 15.Shihabi ZK. J Liq Chromatogr Relat Technol. 2000;23:79–95. [Google Scholar]
- 16.Jenkins MA, Guerin MD. J Chromatogr, B. 1996;682:23–34. doi: 10.1016/0378-4347(95)00552-8. [DOI] [PubMed] [Google Scholar]
- 17.Dolnik V. Electrophoresis. 2006;27:126–141. doi: 10.1002/elps.200500567. [DOI] [PubMed] [Google Scholar]
- 18.Hutterer K, Dolnik V. Electrophoresis. 2003;24:3998–4012. doi: 10.1002/elps.200305635. [DOI] [PubMed] [Google Scholar]
- 19.Dolnik V, Hutterer KM. Electrophoresis. 2001;22:4163–4178. doi: 10.1002/1522-2683(200111)22:19<4163::AID-ELPS4163>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Dolnik V. Electrophoresis. 1997;18:2353–2361. doi: 10.1002/elps.1150181226. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez N, Elvira C, San Roman J, Cifuentes A. J Chromatogr, A. 2003;1012:95–101. doi: 10.1016/s0021-9673(03)01175-0. [DOI] [PubMed] [Google Scholar]
- 22.Harrell CW, Dey J, Shamsi SA, Foley JP, Warner IM. Electrophoresis. 1998;19:712–718. doi: 10.1002/elps.1150190519. [DOI] [PubMed] [Google Scholar]
- 23.Zhu XF, Kamande MW, Thiam S, Kapnissi CP, Mwongela SM, Warner IM. Electrophoresis. 2004;25:562–568. doi: 10.1002/elps.200305713. [DOI] [PubMed] [Google Scholar]
- 24.Kapnissi CP, Valle BC, Warner IM. Anal Chem. 2003;75:6097–6104. doi: 10.1021/ac034452g. [DOI] [PubMed] [Google Scholar]
- 25.Kamande MW, Zhu XF, Kapnissi-Christodoulou C, Warner IM. Anal Chem. 2004;76:6681–6692. doi: 10.1021/ac049313t. [DOI] [PubMed] [Google Scholar]
- 26.Kamande MW, Kapnissi CP, Zhu XF, Akbay C, Warner IM. Electrophoresis. 2003;24:945–951. doi: 10.1002/elps.200390137. [DOI] [PubMed] [Google Scholar]
- 27.Kamande MW, Fletcher KA, Lowry M, Warner IM. J Sep Sci. 2005;28:710–718. doi: 10.1002/jssc.200500006. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Dubin PL. Anal Chem. 1999;71:3463–3468. [Google Scholar]
- 29.Cahours X, Viron C, Morin P, Renimel I, Andre P, Lafosse M. Anal Chim Acta. 2001;441:15–21. [Google Scholar]
- 30.Souverain S, Geiser L, Rudaz S, Veuthey JL. J Pharm Biomed Anal. 2006;40:235–241. doi: 10.1016/j.jpba.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 31.Fan LY, Cheng YQ, Li YQ, Chen HL, Chen XG, Hu ZD. Electrophoresis. 2005;26:4345–4354. doi: 10.1002/elps.200500013. [DOI] [PubMed] [Google Scholar]
- 32.Altria KD, Kelly MA, Clark BJ. Chromatographia. 1996;43:153–158. [Google Scholar]
- 33.Nakashima T, Higa H, Matsubara H, Benson A, Yasunobu K. J Biol Chem. 1966;241:1166–1177. [PubMed] [Google Scholar]
- 34.Landers JP, editor. Handbook of Capillary Electrophoresis. 2. CRC Press; Boca Raton, FL: 1997. [Google Scholar]
- 35.Endo N, Nishiyama K, Otsuka A, Kanouchi H, Taga M, Oka T. Br J Nutr. 2006;95:1088–1093. doi: 10.1079/bjn20061764. [DOI] [PubMed] [Google Scholar]
- 36.Verhoef P, Kok FJ, Kruyssen DA, Schouten EG, Wittenman JC, Grobbee DE, Ueland PM, Refsum H. Arterioscler Thromb Vasc Biol. 1997;5:989–995. doi: 10.1161/01.atv.17.5.989. [DOI] [PubMed] [Google Scholar]
- 37.Jakubowski H. Chem Eur J. 2006;12:8039–8043. doi: 10.1002/chem.200600785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional experimental data including mass spectrometry, SDS-PAGE, and UV–vis spectroscopy characterizations of the protein mixture components. This material is available free of charge via the Internet at http://pubs.acs.org.