

Abstract
Background & Aims
Activation of autoimmune pathways has been implicated as a contributing mechanism to the pathophysiology in some patients with chronic intestinal pseudoobstruction (CIP). In this study we tested the hypothesis that sera from a subpopulation of patients with CIP contain autoantibodies that activate autophagy via a Fas-dependent pathway in cultured human neuroblastoma SH-Sy5Y cells.
Methods
Twenty-five patients with established neurogenic CIP (20 F and 5 M; age range: 21–57 yrs) were investigated and circulating anti-neuronal antibodies to enteric neurons were found in 6 (24%) patients. The ability of anti-neuronal antibodies to induce autophagy was assessed using immunohistochemical, Western immunoblot and molecular techniques. The presence of autophagosomes was monitored using a specific immunohistochemical marker, anti-LC3 immunoreactivity and co-localization with mitochondrial- and Fas-activated death domain (FADD)-immunofluoresence using appropriate antibodies, in cells exposed to sera from matched healthy controls and patients with neurogenic CIP.
Results
Exposure of SH-Sy5Y cells to sera from patients with CIP containing anti-neuronal antibodies revealed increased binding of autoimmune immunoglobulin (IgG class) to the surface of SH-Sy5Y cells and increased formation of autophagosomes demonstrating co-localization with mitochondria and FADD compared to control sera. Pretreatment of sera with either protein L agarose beads or a soluble Fas receptor (extracellular domain) chimera prevented the stimulation of autophagy.
Conclusions
We provide novel evidence that anti-neuronal antibodies may contribute to neuronal dysfunction observed in a subset of patients with neurogenic CIP via autoantibody-mediated activation of autophagy involving the Fas receptor complex.
Introduction
Chronic intestinal pseudo-obstruction (CIP) is an uncommon, disabling disorder characterized by a severe impairment of gastrointestinal propulsion which results in a clinical picture resembling mechanical obstruction in the absence of any lesion occluding the gut.1–5 CIP typically passes unrecognized for long periods of time before the correct diagnosis is identified. The lack of appropriate medical recognition reflects, in part, poor understanding of the pathogenesis and the wide heterogeneity of CIP. When no underlying cause is identified that explains the gastrointestinal motor dysfunction, the etiology of CIP is defined as idiopathic (CIIP).1–5 Although familial cases have been described, the majority of CIIP in adults are sporadic.1–5
The origin of the underlying dysmotility in CIP is thought to be myogenic, neurogenic or mesenchymopathic depending on whether the abnormality involves the smooth muscle, the innervation (intrinsic or extrinsic) or the interstitial cells of Cajal in the gut.6 Neurogenic CIP can be classified into two major forms: (a) inflammatory neuropathies in which a significant inflammatory/immune response is identified within enteric ganglia and/or nerve processes; and (b) degenerative neuropathies characterized by evidence of neurodegenerative aspects in the absence of an identifiable inflammatory response. Inflammatory neuropathies are characterized by a dense infiltrate of lymphocytes and plasma cells involving either of the two major ganglionated plexuses of the enteric nervous system (ENS), although mainly the myenteric plexus, hence the term myenteric ganglionitis) and related axons appear to be involved by the inflammatory/immune response.4,5,7 In addition to cell-mediated immune injury, patients with lymphocytic myenteric ganglionitis develop a humoral response characterized by anti-neuronal antibodies (aNA), including anti-nuclear neuronal antibodies or anti-Hu (from the name of the molecular target recognized by these autoantibodies).8 In addition to these well defined aNA, emerging evidence indicates that other antibodies targeting either central or ENS neurons have been identified in a variety of autoimmune disorders,9,10 as well as dysmotility syndromes.7,11
In general, the presence of autoantibodies in the sera of patients with CIP has been viewed as a potentially useful biomarker for diagnosis of CIP. It is less clear whether autoimmune immunoglobulins, and specifically aNA, may contribute to the pathophysiology of CIP. In other disorders, for example diabetes mellitus, autoimmune immunoglobulins are present in the sera that bind and activate the cell-death receptor Fas(CD95) present on beta cells.12 An emerging body of evidence suggests that the Fas receptor complex is linked to a variety of cell functions including proliferation, in addition to activation of caspase-dependent cell death.13,14
Rapidly growing research data indicate that macroautophagy (also referred to as autophagy) plays a pivotal role in cellular response to inimical conditions.15,16 Autophagy is responsible for the sequestration of injured or senescent organelles, such as mitochondria, in autophagosomes. Also, it is considered an ubiquitous adaptive mechanism that allows cells to survive stressful conditions such as nutrient depletion and oxidative stress and appears to play an important role in the pathophysiology of neurodegenerative, cardiovascular, and muscular diseases, and some malignancies.17–19 Autophagy is a highly conserved pathway in eukaryotic organisms. In mammals, it involves the functional homologues of at least 16 gene products, studied comprehensively in yeast.20 Among these products is the specific marker of autophagosomes, LC3, a member of the microtubule-associated light chain family, a mammalian homologue of yeast Atg8.21 LC3 undergoes enzymatic cleavage and lipidation to generate the 16 Kd LC3-II form that is incorporated into autophagosomes,22 and whose immunohistochemical detection exhibits a punctate23 or ring-like pattern24 indicative of completed autophagosomes. Therefore, the detection of LC3-II by immunofluoresence or immunoblot, provides a specific marker for the assessment of macroautophagic activity, and allows the evaluation of the co-localization of LC3 with other events involved in cellular responses to stress.25
In this study, we examined the novel hypothesis that humoral autoimmune mechanisms may contribute to neuronal stress via a pathway involving activation of autophagy in patients with CIP. We found that the anti-LC3 immunohistochemical signal for autophagy was enhanced in human SH-Sy5Y neuroblastoma cells exposed to sera from a subset of patients with CIIP containing aNA compared to sera from CIP patients without aNA and sera from healthy individuals. Our results suggest that anti-neuronal autoimmune immunoglobulins play a potentially important role in the pathophysiology of neurogenic CIP via Fas-mediated activation of autophagy.
Material and Methods
Inclusion criteria and patients
Since no individual patient identification was involved and no study-driven clinical intervention was performed, a simplified Institutional Review Board approval by the St. Orsola-Malpighi University Hospital Ethics Committee was obtained and no patient consent was considered necessary.
Small bowel motility was evaluated in 34 patients with severe digestive symptoms referred to the Laboratory of Functional Gastrointestinal Disorders at the S.Orsola-Malpighi Hospital of the University of Bologna between 2003 and 2006. Of these, 25 patients (20 females and 5 males; age at entry in the study 37.3±10.9 years, mean ± SD; range 21–57 years) were affected by CIP, as defined previously.26 Seven were excluded because lacking acute episodes resembling mechanical occlusion and two because of secondary etiologies (1 paraneoplastic and 1 scleroderma).
Screening of sera
Sera from the 25 patients who met the diagnostic criteria for neurogenic CIP were screened for the presence of aNA as described previously.27 Briefly, aNA to the ENS were detected by indirect immunofluorescence on 5 µm cryostat sections of rat ileum and colon (Astra, Milan, Italy). Each patient’s serum was screened for its ability to induce autophagy using the vital lipophilic dye monodansyl cadaverine (MDC) which non-specifically labels intracellular vacuoles including autophagosomes as described previously.28 The subpopulation of CIP sera that stimulated autophagy via autoantibody-mediated activation of a Fas-dependent pathway was designated CIP with aNA. All the sera screened for their ability to induce autophagy using the MDC and anti-LC3 immunofluorescence assays were complement-inactivated (heated in a water bath at 56°C for 40 min).
Small bowel manometry
Small bowel manometry was performed in all included patients according to a previously described stationary, perfused, manometric technique and abnormal motility patterns identified and analyzed as described previously.6,26
Neuroblastoma cell culture
The human neuroblastoma cell line SH-Sy5Y (ATCC, Manassas, VA) was maintained in 150 cm2 Corning T-150 flasks in a 1:1 mixture of Dulbecco’s modified Eagle medium (DMEM) and Ham's F-12 containing 15% fetal bovine serum (FBS), penicillin (100 IU/mL), streptomycin (100 µg/ml), 2 mmol/L L-glutamine, and 15-mm Hepes buffer at 37°C with a 10% CO2 atmosphere. 28
Immunofluorescence assays
SY5Y cells were exposed to CIP sera with or without evidence of anti-neuronal antibodies or control sera (1:10 dilution in growth medium) for 16–24 h. Serial dilution studies were performed on a subset of sera that stimulated enhanced anti-LC3 immunoreactivity compared to control sera. After the initial screening of individual sera, some experiments were performed using pooled sera from three individuals using CIP with aNA that induced autophagy and colocalization with FADD compared to pooled sera from three healthy controls. Experiments using pooled sera were repeated two times and involved comparison of two separate pools of CIP with aNA and control sera. In some experiments, tumor necrosis factor-α (TNF-α) (100 ng/mL; Cell Sciences, Canton, MA) was used as a positive control to activate the Fas receptor complex.
Cells were plated on cover slips in Petri dishes (35×10mm) or 4 well cell culture chambers for 24 h at 37°C, and subsequently fixed in ice-cold 4% paraformaldehyde for 15 min, then washed with PBS and blocked with 10% serum containing 0.1% triton X-100 as described previously.28 The cells were subsequently exposed to the primary anti-LC3 antiserum (kindly provided by Dr. Tamotsu Yoshimori, Department of Cell Biology, National Institute for Basic Biology, Okazaki, Japan) at 1:400 dilution, anti-mitochondrial antibody (AMA) at 1:100 dilution (Chemicon International, Temecula, CA), anti-Fas activated death domain (FADD) antibody at 1:100 dilution (Cell Signaling, Tropix, Bedford MA) and secondary antibodies (Alexa Fluor 495, goat anti-rabbit IgG, 1:400, and Alexa Fluor 488, goat anti-mouse IgG, 1:400, Molecular Probes, Eugene, OR) depending on the donor species of the primary antibody, in blocking buffer for 2 h at room temperature. For the detection of IgG, the antibody utilized was goat anti-human IgG-Fc, HRP-conjugated (1:3000). The cells were then washed in PBS and mounted along with 10µL anti-fading gel (Molecular Probes, Eugene, OR).
Analysis of pixel density
Six to ten (x40) digital images of immunostained cells were acquired using a Zeiss LSM-510 confocol microscope. The number of pixels in each image corresponding to immunostained cells was counted by subtraction of the background from the total pixels using NIH image-J software. The cell area in each image was measured by outlining the cell edges. The pixel density was then normalized to pixels per 100 µm2 cell area.
Western immunoblotting
Cells were extracted using Tris-buffered saline (TBS), complete proteinase inhibitor cocktail (Roche, Mannheim, Germany), and 0.5% of Triton X-100. After SDS-PAGE (15% ready gel, Bio-Rad, Hercules, CA), proteins were transferred to membranes under 80 volts, for 2 h at 4 °C. The membranes were subsequently blocked with 5% non-fat milk for 1 h at room temperature. This was followed by an overnight incubation at 4°C with a 1:1000 dilution of anti-LC3 antiserum (kindly provided by Dr. Tamotsu Yoshimori, Research Institute for Microbial Diseases, Department of Cellular Regulation, 3-1 Yamadaaka; Suita Osaka, Japan) and processed as described previously.28 The signals were detected by chemiluminescence using the SuperSignal West Dura/Pico Kit (Pierce, Rockford, IL). Protein bands on X-ray film were quantified by densitometric scanning (ImageQuant 5.0) and analyzed by SigmaPlot 2001.
Immunoglobulin removal
Immunoglobulins were removed from human sera using the Protein L kit, (Protein L-agarose beads; Sigma, St. Louis, MO) according to the manufacturer’s instructions.
Immunoneutralization of sera with soluble Fas (sFas) chimera
Some experiments involved pre-incubation of human sera with a recombinant human Fas/Fc chimera/TNFRSF6 (R&D Systems, Inc., Minneapolis, MN. This chimera encodes the extracellular domain of human Fas antigen and has been shown to block the induction of apoptosis by Fas ligand.29 CIP with aNA or control sera were pre-incubated with Fas/Fc chimera (100 ng/µl) or vehicle for 3 h at 37°C in a rotating shaker. Following this preincubation, the sera were added to FBS-free incubation media to give a final concentration of sFas of 250 ng/mL. The sera were added to SH-Sy5Y cell cultures at a concentration of 10% vol/vol and the cells were incubated for an additional 16–24 h and processed for immunofluorescence or immunoblot assays as described above.
Statistical Analysis
Statistical analyses were performed using nonparametric Mann-Whitney test by GraphPad InStat 3 (GraphPad Software, Inc., CA). Differences with P<0.05 were considered significant.
Results
Study population
Demographic and clinical features of the study population are summarized in Table 1.
Table 1.
Demographic and clinical features of the patients with CIIP studied.
| N° | Patient’s initials | Sex | Age | ↓BMI | Feeding | Digestive symptoms | N° Surgery | Comments |
|---|---|---|---|---|---|---|---|---|
| 1. | GC | F | 28 | 1 | Modified oral | N, PPF, AD, AP, C | 1 | - |
| 2. | CM | F | 36 | 1 | Modified oral | N, PPF, AD, C | 5 | - |
| 3. | PF | F | 43 | 1 | Modified oral | AD, AP, C | 10 | Opioid depedence |
| 4. | GG | F | 43 | 1 | Modified oral | PPF, AD, AP, AB | 2 | Concomitant MGUS |
| 5. | DS | F | 38 | 1 | Free diet → PN | AD, AP, C | 3 | Seizure |
| 6. | EA | F | 48 | 0 | Free diet | N, AP, C | 3 | Onset at birth; seizure. |
| 7. | BMF | M | 43 | 1 | Modified oral → EN | N, AP, C | 4 | Opiod dependence |
| 8. | MA | F | 47 | 1 | PN | AD, AP,D | 2 | Dead |
| 9. | SG | F | 32 | 1 | Modified oral → PN | N, V, PPF, AD, AP, C | 1 | Urolithiasis |
| 10. | CAM | F | 38 | 1 | Modified oral → EN | AD, AP, C | 8 | Neurological symptoms |
| 11. | RV | M | 57 | 1 | Modified oral | PPF, AD, C | 0 | - |
| 12. | RE | F | 24 | 0 | Modified oral | N, V, PPF, AP | 4 | - |
| 13. | PS | F | 26 | 1 | Modified oral | N, PPF, AD, AP, D | 2 | Dead after SB transplatation |
| 14. | CI | F | 21 | 1 | EN | N, V, PPF, AD, AP, D | 3 | Transplantated, alive |
| 15. | SR | F | 31 | 0 | Modified oral | N, PPF, AD, AP, D | 5 | - |
| 16. | BM | F | 25 | 1 | Modified oral | N, PPF, AD, C | 4 | Onset after colectomy |
| 17. | SF | M | 55 | 1 | Modified oral → PN | SO | 1 | - |
| 18. | BF | F | 40 | 1 | EN | N, V, PPF, C | 2 | - |
| 19. | CM | M | 45 | 0 | Modified oral | AD, AP, D | 4 | Responsive to immunesuppressive therapy |
| 20. | BS | F | 25 | 1 | Modified oral → PN | N, F, AD, AP, C | 1 | Opiod dependence; transplantated, alive |
| 21. | CR | F | 33 | 1 | Modified oral | N, PPF, AD, AP, AB | 2 | - |
| 22. | SG | M | 56 | 0 | Free diet | SO | 0 | Megaduodenum |
| 23. | RM | F | 46 | 1 | Modified oral → EN | N, F, AD, AP, D | 4 | Opiod dependence; neurological symptoms; dead |
| 24 | ZM | F | 22 | 1 | Modified oral | N, V, AD, C | 0 | - |
| 25 | ML | F | 30 | 1 | Modified oral | N, PPF, AD, AP, ABB | 2 | - |
Note: CIIP patients with positive aNA (indicated in bold) included cases #11, #13 and #25 (+, titer 1:40); #4 and #19 (++, titer 1:80) and #24 (+++, titer 1:160).
↓ BMI: 1 = reduced BMI; 0 = normal BMI.
Feeding: PN = parenteral nutrition; EN = enteral nutrition;
Digestive symptoms: N = nausea; V = vomiting; PPF = postprandial fullness; AD = abdominal distension; AP = abdominal pain; C = constipation; D = diarrhea; AB = alternating bowel; SO = chronic sub-occlusive status; MGUS = monoclonal gammopathy of undetermined significance.
N surgery: number of times a laparotomy has been performed in a given patient.
Intestinal manometry
At least one obvious manometric abnormality (median: 2; range 1–4) was present in all patients. Relative frequencies of the different motor abnormalities recorded were: abnormal activity fronts in 60% of patients, bursts of uncoordinated contractions in 56%, clustered contraction in 52%, hypomotility in 20%, inability of an adequate meal to abolish interdigestive MMCs for at least 180 min in 8% of patients.
Screening of sera
Sera from 25 patients who met the criteria for CIP were selected to screen for their ability to induce autophagy using the MDC assay. Based on criteria described previously, we found that 6 out of 25 patients (24%; 4 females, 2 males; age range: 22–57 years) with CIP displayed aNA in their serum. The immunofluorescent pattern showed a bright staining in the nucleus and cytoplasm, including the plasma membrane, of all myenteric and submucosal neurons of the rat ileum and colon. The intensity of the aNA immunofluorescence signal was semi-quantitatively graded +, ++ and +++ which correlated with antibody titers 1:40, 1:80 and 1:160, respectively. Three patients demonstrated +, two patients ++ and one had +++ signal intensity. Sera from six individuals (4 females and 2 males; age range: 21–56 years) with CIP who did not demonstrate aNA were also screened for their ability to induce autophagy, in addition to healthy control sera from six individuals (4 females and 2 males; age range: 21–45 years).
Compared to sera from individuals with CIP without aNA and healthy controls, the six sera with aNA all demonstrated significant increases in the following markers: i) stimulation of cytoplasmic vacuole formation using the MDC assay; ii) binding of autoantibody(ies) (IgG class) to the surface membrane of SH-Sy5Y cells; and iii) induction of a punctate anti-LC3 immunofluorescence signal and co-localization with either anti-FADD or AMA immunofluorescence. In general, the intensity of the anti-IgG, anti-LC3 and co-localization with anti-FADD and AMA immunofluorescence signals correlated with the intensity and titer of the aNA signal, e.g. serum that demonstrated an aNA +++ (1:160 titer) response showed more intense binding of autoantibody and induction of autophagy compared to sera with aNA ++ (1:80) and aNA + (1:40) response. Some sera from CIP patients without aNA demonstrated a modest increase in the MDC assay and binding of autoantibody to surface membrane, but this did not reach statistical significance compared to control sera. Treatment of the CIP with aNA sera with either protein L agarose beads or the soluble Fas receptor (extracellular domain) chimera (sFas) prevented the induction of anti-LC3 and co-localization with anti-FADD and AMA immunofluorescence signals. Of interest, treatment of control sera with sFas also resulted in a decrease in the anti-LC3-II immunoblot signal.
Serial dilution studies using pooled CIP with aNA sera revealed the progressive loss of the effect to stimulate autophagy between 1:80 and 1:250. Induction of autophagy and colocalization with FADD were also observed after exposure of SH-Sy5Y cells to TNF-α. As negative control, co-treatment of cells with TNF-α and sFas did not prevent the induction of autophagy and co-localization with FADD (data not shown).
SH-Sy5Y cells exposed to sera from CIP patients with aNA demonstrated increased binding of autoimmune immunoglobulins and stimulation of autophagy
Exposure of SH-Sy5Y cells to CIP with aNA sera showed increased binding of autoimmune immunoglobulin (IgG class) to the surface membrane and stimulation of autophagy based on an increase in the anti-LC3 immunofluorescence signal compared to cells exposed to either control sera or growth medium not supplemented with sera (Figure 1A). Histograms summarizing the anti-IgG binding and anti-LC3 immunofluorescence data are shown in Figures 1B and 1C, respectively. Pixel density data were normalized to cell area (100 µm2; n=6, P< 0.05).
Figure 1. Exposure of SH-Sy5Y cells to sera from CIP patients with aNA demonstrated increased binding of autoimmune immunoglobulins and stimulation of autophagy.
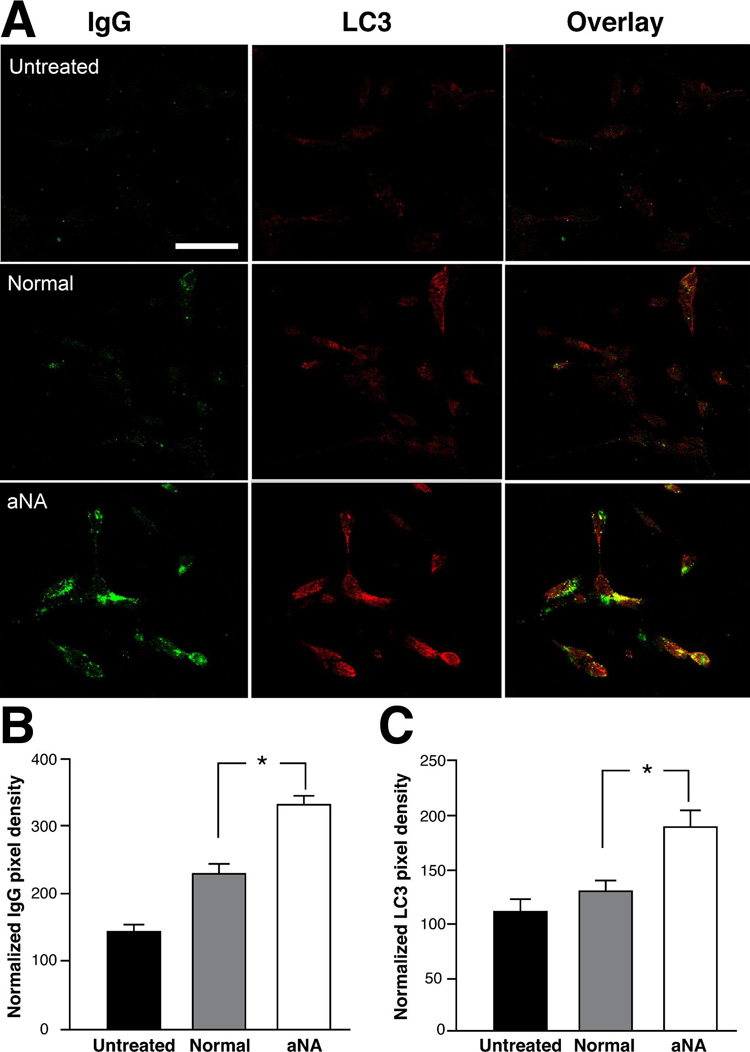
SH-Sy5Y cells exposed to CIP with aNA sera demonstrated increased binding of autoantibodies (IgG class) to the surface membrane and stimulation of autophagy based on an increase in the anti-LC3 immunofluorescence signal compared to cells exposed to either control sera or growth medium not supplemented with sera (Figure 1A). Histograms summarizing the anti-IgG immunofluorescence and anti-LC3 immunofluorescence are shown in Figure 1B and 1C, respectively. Pixel density data were normalized to cell area, 100 µm2 (n=6, P< 0.05).
Exposure of SH-Sy5Y cells to CIP with aNA sera stimulated autophagy and co-localization with mitochondria
Exposure of cells to CIP with aNA sera resulted in an increase in the anti-LC3 immunofluorescence signal and co-localization with AMA immunofluorescence in punctate cytoplasmic vacuoles consistent in appearance with autophagosomes, compared to cells exposed to control serum or growth medium not supplemented with serum. The results of a representative experiment is shown in Figure 2, see magnified view (n=6, P<0.05).
Figure 2. SH-Sy5Y cells exposed to CIP with aNA sera demonstrated increased autophagy and co-localization with mitochondria.
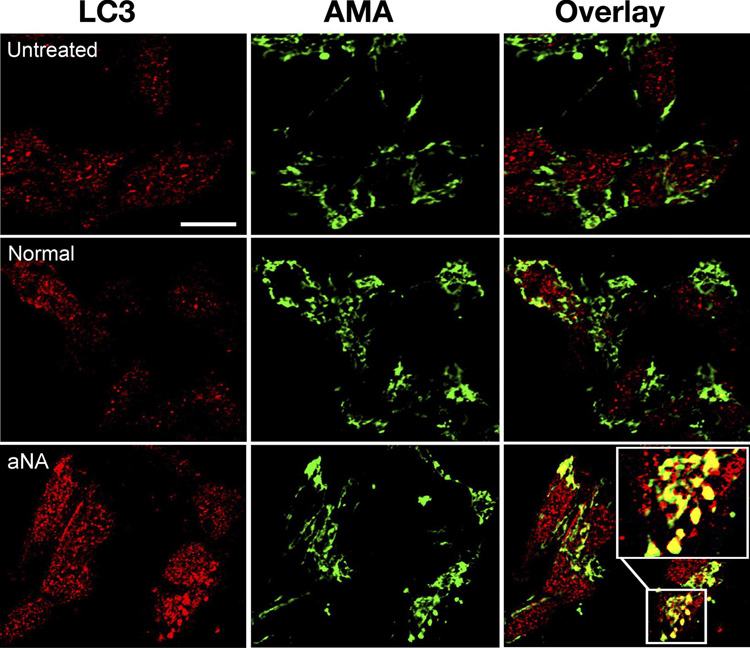
Treatment of SH-Sy5Y cells with sera from CIP patients containing aNA resulted in an increase in the anti-LC3 immunofluorescence signal and co-localization with AMA immunofluorescence in punctate cytoplasmic vacuoles consistent in appearance with autophagosomes, compared to cells exposed to control serum or growth medium not supplemented with serum (Figure 2, see magnified view) (n=6, P< 0.05).
Pretreatment of CIP with aNA sera with protein L agarose beads prevented the increased binding of autoimmune immunoglobulins
After exposure of CIP with aNA sera to protein L agarose beads to remove immunoglobulins, the increased binding of IgG autoantibodies to the surface membrane of SH-Sy5Y cells was not observed (Figure 3A). A summary histogram of the anti-IgG binding data is shown in Figure 3B. Of interest, binding of autoantibodies (IgG class) to SH-Sy5Y cells after exposure to sera from healthy controls was also increased compared to cells exposed to growth medium not supplemented with sera and decreased after exposure to protein L agarose beads. Pixel density data were normalized to cell area (100 µm2; n=6, P< 0.05).
Figure 3. Pretreatment of control and CIP with aNA sera with protein L agarose beads prevented the increased binding of autoimmune immunoglobulins.
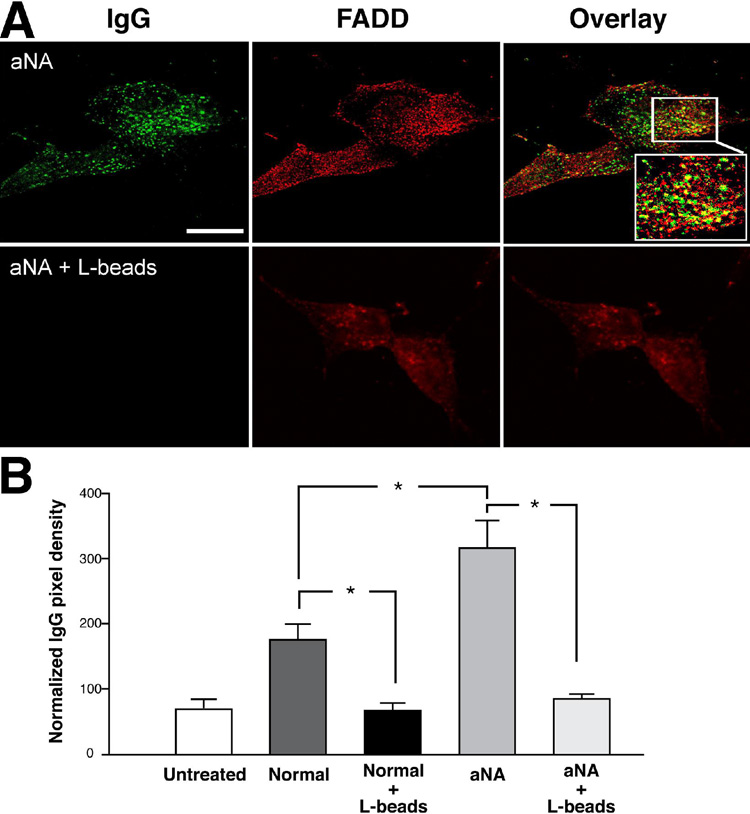
The increased binding of IgG autoantibodies to the surface membrane of SH-Sy5Y cells was not observed after exposure of sera from patients with CIP containing aNA to protein L agarose beads to remove immunoglobulins, Figure 3A. A summary histogram of the anti-IgG binding data is shown in Figure 3B. Binding of autoantibodies (IgG class) was also decreased after exposure of sera from healthy controls to protein L beads compared to cells exposed to growth medium not supplemented with serum. Pixel density data were normalized to cell area (100 µm2) (n=6, P< 0.05).
Pretreatment of CIP with aNA sera with sFas prevented activation of autophagy and co-localization with FADD
Figure 4 summarizes data demonstrating that pretreatment of CIP with aNA sera with sFas prevented the increase in anti-LC3 immunofluorescence (Figure 4C) and co-localization with anti-FADD (Figure 4B), compared to exposure of SH-Sy5Y cells to control sera or growth medium not supplemented with serum. Pixel density data were normalized to cell area (100 µm2; n=6, P< 0.05).
Figure 4. Pretreatment of sera from patients with CIP containing aNA with sFas prevented activation of autophagy and co-localization with FADD.
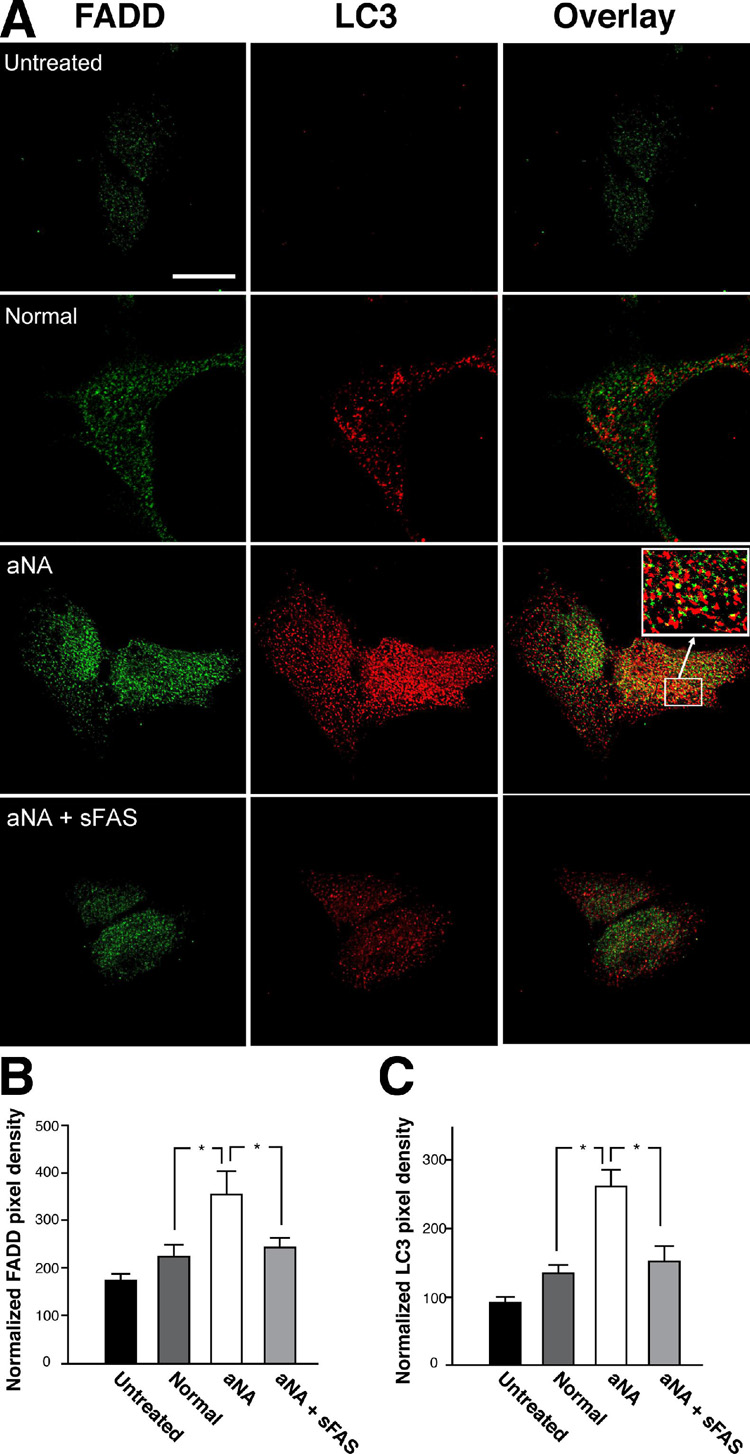
Exposure of SH-Sy5Y cells to CIP with aNA sera resulted in a significant increase in the anti-LC3 immunofluorescence signal and co-localization with anti-FADD immunofluorescence compared to cells exposed to control sera. Pretreatment of the sera with sFas prevented the increase in anti-LC3 immunofluorescence (Figure 4C) and co-localization with anti-FADD immunofluorescence (Figure 4B). Pixel density data were normalized to cell area (100 µm2; n=6, P< 0.05).
Pretreatment of control and CIP with aNA sera with sFas significantly decreased anti-LC3 II immunoblot density
To confirm that the sFas reagent prevents the induction of autophagy, we examined the effect of pretreatment of CIP with aNA and control sera on the induction of the anti-LC3-II using Western immunoblot. The pixel density of the anti-LC3-II immunoblot signal was significantly increased in SH-Sy5Y cells exposed to CIP with aNA sera. Pretreatment of sera with sFas resulted in a significant decrease in the anti-LC3-II band density in both control and CIP with aNA sera treated cells (Figure 5A). A summary histogram of the quantitative analysis of the normalized anti-LC3-II pixel density data is presented in Figure 5B. This experiment was performed using two pools of CIP with aNA sera, e.g. three individual’s sera in each pool and compared to two pools of sera (three individuals in each pool) from healthy controls (P< 0.05). Pixel density data were normalized to β-actin loading.
Figure 5. Pretreatment of sera from healthy controls and patients with CIP containing aNA with sFas significantly decreased anti-LC3 II immunoblot density.
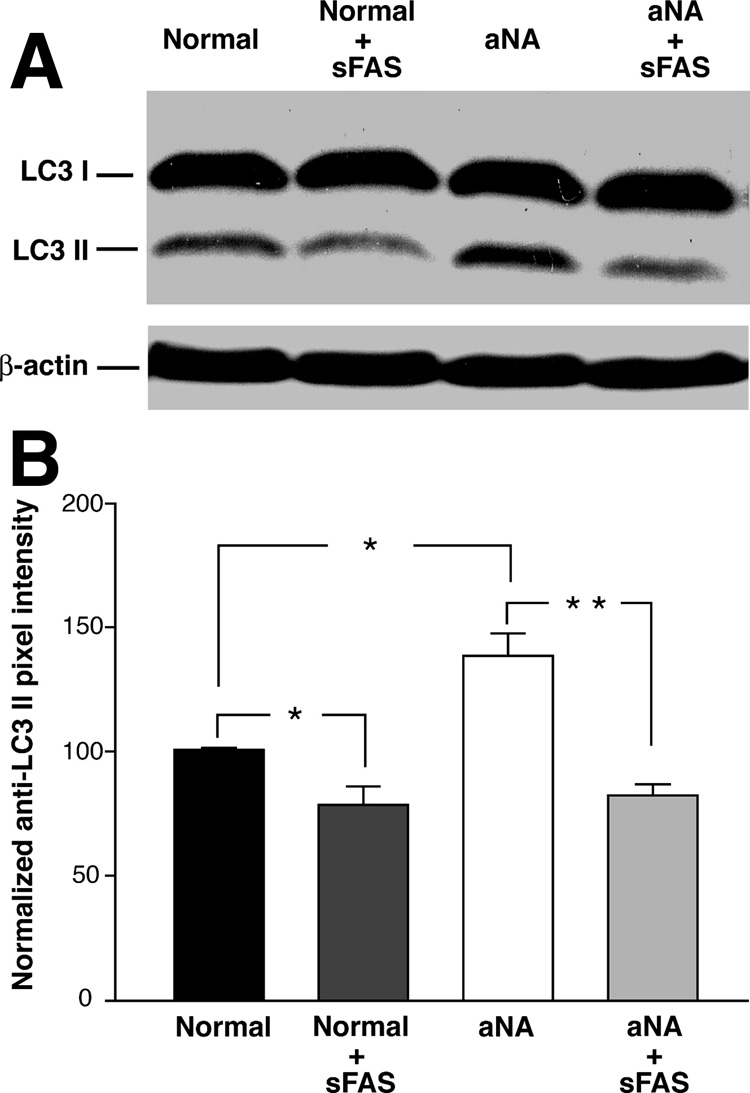
To confirm that CIP with aNA sera-mediated stimulation of autophagy in SH-Sy5Y cells involves binding to the Fas receptor complex, we examined the effect of pretreating control and CIP with aNA sera with the sFas reagent on induction of LC3 II immunoblot density using Western analysis. The pixel density of the anti-LC3 II immunoblot was significantly decreased after exposure of both CIP with aNA and control sera to sFas (Figure 5A). A quantitative analysis of the normalized anti-LC3 II pixel density data is presented in the summary histogram, Figure 5B. Loading conditions were evaluated using β-actin (P< 0.05).
Serial dilution of CIP with aNA sera resulted in graded attenuation in the ability to stimulate the anti-LC3 II immunoblot signal
Serial dilution studies using both control and CIP with aNA sera indicated that the ability of CIP with aNA sera to differentially stimulate anti-LC3-II immunoreactivity was reduced to control levels at 1:250 dilution (Figure 6). This experiment was performed with pooled sera from five patients with CIP with aNA, i.e. aNA + (1:40, n=3) or ++ (1:80, n=2). Pixel density data were normalized to β-actin loading.
Figure 6. Serial dilution of sera from patients with CIP containing aNA resulted in graded loss in the ability to differentially stimulate the anti-LC3 II immunoblot signal.
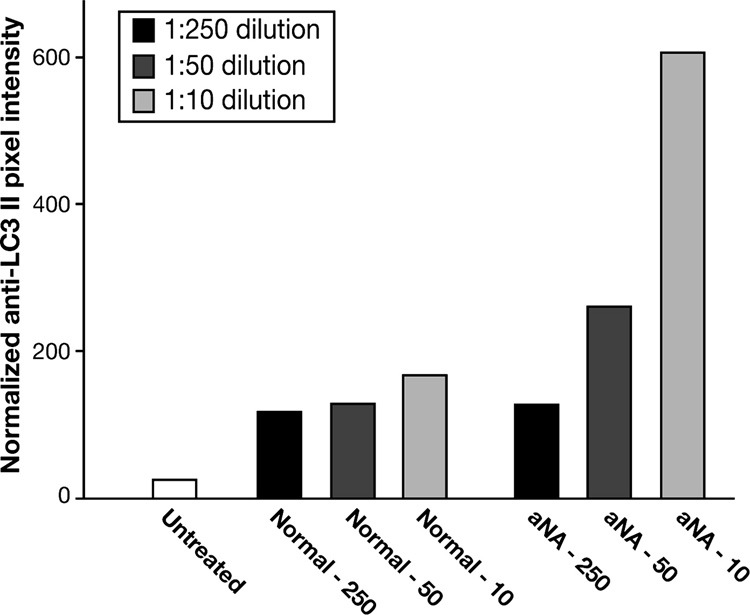
Serial dilution studies using both control and CIP with aNA sera revealed that the ability of CIIP with aNA sera to differentially stimulate anti-LC3 II immunoreactivity was reduced to control levels at 1:250 dilution. Pixel density data were normalized to β-actin loading.
DISCUSSION
We believe this is the first report supporting that a factor present in the sera of a subpopulation of patients with CIP exhibiting aNA stimulates autophagy and co-localization with AMA and anti-Fas immunoreactive signals. The increased signal for anti-LC3 immunoreactivity, co-localization with enhanced anti-IgG immunoreactivity and the significant reduction in these signals after exposure of the sera to either protein L agarose beads or a sFas receptor (extracellular domain) chimera suggest that autoimmune immunoglobulins (i.e., aNA) are involved in stimulating the increase in autophagy via a Fas-dependent pathway. Our data suggest that humoral autoimmunity, identified by circulating aNA, may contribute to the neurodegenerative abnormalities responsible for dysmotility in some CIP patients. Nonetheless, the potential significance of this pathway in the natural history of CIP and the mechanisms through which aNA may affect a highly preserved compartment such as the ENS and contribute to dysmotility remain to be fully elucidated. However, our previous report that SH-Sy5Y cells exposed to sera obtained from patients with diabetic neuropathy or active systemic lupus erythematosus also show increased autophagy, supports the notion that autoimmune disorders may, in general, be associated with activation of autophagy.28 It will be important to determine whether autophagy is differentially activated in the ENS of patients with CIP and aNA vs. CIP without aNA in future studies. In addition the current study does not exclude the possibilities that aNA sera may contain antibodies that bind to non-neuronal tissues or that there may be other mediators present in the sera of patients with CIP that contribute the pathophysiology of this syndrome.
The pathophysiological significance of enhanced autophagy and the role of sequestration of mitochondria in autophagosomes are an area of active investigation. The engulfment of mitochondria under conditions of cellular stress is known to occur.30–33 For example, mitochondria are known to generate reactive oxygen species (ROS) when cells are exposed to inimical conditions associated with increased oxidative stress.34 Increased mitochondrial engulfment in autophagosomes may be a cytoprotective response to oxidative stress.32 The potential significance of mitochondrial dysfunction, enhanced oxidative stress and sequestration of mitochondria in autophagosomes in CIP requires further exploration. We hypothesize that autophagy is an “early” cytoprotective response to the binding of autoimmune immunoglobulins that activate cellular pathways (such as Fas) associated with mitochondrial dysfunction and increased formation of ROS. If confirmed, a logical extension of this hypothesis is that the increased presence of autoimmune immunoglobulins and, perhaps, other activators of autophagy over time could be associated ultimately with cell injury and death when the balance of cytoprotective and cytotoxic mechanisms favors the latter. Our previous reports suggest that sera from patients with diabetes and neuropathy can induce a low level of apoptosis in neural tissue.35,36 A key issue for future studies is to examine the potential linkage between activation of autophagy and apoptotic vs. non-apoptotic cell death.
Our results suggest that aNA-mediated stimulation of autophagy can involve a Fas-dependent pathway. Relevant to this report, other studies suggest that the interaction of FADD with the autophagic molecule, Atg5, is necessary for autophagosomal development.37 It is well established that Atg5 is necessary for early autophagosomal assembly but it is not known to be a component of the mature autophagosome. Notably, our data, showing LC3 co-localization with FADD, suggests that there may be a later, more permanent association of FADD with the autophagosome, because LC3-II is part of the mature autophagic vacuole.38 This observation implies that FADD may be engulfed in the autophagosome as part of a well conserved cellular pathway that modulates death receptor signal transduction. Although SH-Sy5Y cells demonstrate low basal expression in procaspase 8, a “sequence of hits” by cytokines or antibodies can increase Fas or caspase 8 expression and lead to executioner caspase activation.39,40 Therefore, FADD engulfment may be an “early” cytoprotective mechanism since cytokines that induce Fas are known to be elevated in chronic inflammatory conditions.
The participation of Fas in aNA-mediated stimulation of autophagy is supported by the removal of this effect after treating sera with a soluble Fas receptor (extracellular domain) reagent. We believe it is noteworthy that there was a significant increase in the anti-LC3 immunofluorescence signal in cells exposed to control serum compared to growth medium not supplemented with serum and that the anti-LC3-II immunoblot signal in cells exposed to control sera was significantly decreased by pretreatment with the sFas (extracellular domain) chimera. This observation supports the presence of naturally occurring agonist anti-Fas autoantibodies in the sera of healthy individuals.41 We hypothesize that under circumstances of chronic inflammation or active autoimmune disorders the titer of agonist vs. antagonist anti-Fas autoantibodies favors production of the former.
Other autoantibodies have been identified in the sera of some patients with CIP, including anti-Hu. High titers of aNA targeting Hu molecules (i.e. a group of RNA-binding proteins involved in neuronal maintenance and survival) have been mainly identified in the sera of patients with paraneoplastic syndrome.8 In that study, we provided evidence that exposure of SH-Sy5Y neuroblastoma cells and isolated myenteric neurons to anti-HuD antibodies evoked a significant degenerative response characterized by caspase-3 activation and apoptosis, as identified by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) technique. Based on the important neurobiological functions exerted by the Hu RNA binding proteins, we proposed that HuD protein abrogation, induced by autologous autoantibodies, may contribute to enteric neuron degeneration in patients with paraneoplastic gut dysmotility. Although apoptosis and autophagy may be independently activated, it is quite possible that both processes are activated by oxidative stress evoked by humoral autoimmunity and contribute to neurodegeneration in the ENS. It is noteworthy that, compared to anti-Hu antibodies, aNA present in the subgroup of CIP patients have a lower titer and do not recognize specific molecular antigen(s) (Volta and De Giorgio unpublished data). The fact that autophagy can be evoked by sera with undetermined aNA further strengthens the concept that humoral autoimmune-mediated pathways can contribute to neuronal damage in CIP.
In conclusion, our results indicate that autoimmune immunoglobulins are present in a subpopulation of patients with CIP that activate autophagy via a Fas-dependent pathway supporting a potential novel link between the presence of autoantibodies and induction of a cellular pathway(s) implicated in both cytoprotection and neuronal injury. Elucidating the role of autophagy will likely help to take the term “idiopathic” out of CIIP and generate novel therapeutic targets for this severe, highly disabling syndrome.
Acknowledgments
Grant support: This work was supported by the following Grants from the National Institutes of Health (to J.W.W): R01-056997, R01-052387, M01-RR-00042, UL1RR024986 and Grants from the Italian Ministry of University and Research (COFIN Projects n° 2003064378_003, 2004062155_003) to G.B. and R. De G. and R.F.O. (to R. De G., V.S., G.B., R.C.) funds from University of Bologna. R. De G. is a recipient of a grant from “Fondazione Del Monte di Bologna e Ravenna”, Bologna, Italy.
Abbreviations used in this paper
- aNA
anti-neuronal antibodies
- AMA
anti-mitochondrial antibody
- CIIP
Chronic idiopathic intestinal pseudoobstruction
- ENS
enteric nervous system
- DMEM
Dulbecco’s modified Eagle medium
- FADD
Fas-activated death domain
- FBS
fetal bovine serum
- LC-3
microtubule-associated light chain
- MDC
monodansyl cadaverine
- MMCs
migrating motor complexes
- sFas
soluble Fas receptor chimera (extracellular domain)
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor-α
- trypsin-EDTA
trypsin-ethylene diamine tetraacetic acid
- TBS
Tris-buffered saline
- TUNEL
terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There is no conflict of interest to disclose
References
- 1.Stanghellini V, Corinaldesi R, Barbara L. Pseudo-obstruction syndromes. Baillieres Clin Gastroenterol. 1988;2:225–254. doi: 10.1016/0950-3528(88)90029-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Lorenzo C. Pseudo-obstruction: Current approaches. Gastroenterology. 1999;116:980–987. doi: 10.1016/s0016-5085(99)70082-x. [DOI] [PubMed] [Google Scholar]
- 3.Coulie B, Camilleri M. Intestinal pseudo-obstruction. Annu Rev Med. 1999;50:37–55. doi: 10.1146/annurev.med.50.1.37. [DOI] [PubMed] [Google Scholar]
- 4.De Giorgio R, Guerrini S, Barbara G, Cremon C, Stanghellini V, Corinaldesi R. New insights into human enteric neuropathies. Neurogastroenterol Motil. 2004;16 suppl 1:143–147. doi: 10.1111/j.1743-3150.2004.00491.x. [DOI] [PubMed] [Google Scholar]
- 5.De Giorgio R, Camilleri M. Human enteric neuropathies: Morphology and molecular pathology. Neurogastroenterol Motil. 2004;16:515–531. doi: 10.1111/j.1365-2982.2004.00538.x. [DOI] [PubMed] [Google Scholar]
- 6.Stanghellini V, Camilleri M, Malagelada J-R. Chronic idiopathic intestinal pseudoobstruction: clinical and intestinal manometric findings. Gut. 1987;28:5–12. doi: 10.1136/gut.28.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Giorgio R, Guerrini S, Barbara G, Stanghellini V, De Ponti F, Corinaldesi R, Moses PL, Sharkey KA, Mawe G. Inflammatory neuropathies of the enteric nervous system. Gastroenterology. 2004;126:1872–1883. doi: 10.1053/j.gastro.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 8.King PH, Redden D, Palmgren JS, Nabors LB, Lennon VA. Hu antigen specificities of ANNA-I autoantibodies in paraneoplastic neurological disease. J Autoimmun. 1999;13:435–443. doi: 10.1006/jaut.1999.0337. [DOI] [PubMed] [Google Scholar]
- 9.Volta U, De Giorgio R, Petrolini N, Stanghellini V, Barbara G, Granito A, De Ponti F, Corinaldesi R, Bianchi FB. Clinical findings and anti-neuronal antibodies in coeliac disease with neurological disorders. Scand J Gastroenterol. 2002;37:1276–1281. doi: 10.1080/003655202761020542. [DOI] [PubMed] [Google Scholar]
- 10.Cervio E, Volta U, Verri M, Boschi F, Pastoris O, Granito A, Barbara G, Parisi C, Felicani C, Tonini M, De Giorgio R. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology. 2007;133:195–206. doi: 10.1053/j.gastro.2007.04.070. [DOI] [PubMed] [Google Scholar]
- 11.Latiano A, De Giorgio R, Volta U, Palmieri O, Zagaria C, Stanghellini V, Barbara G, Mangia A, Andriulli A, Corinaldesi R, Annese V. HLA and enteric antineuronal antibodies in patients with achalasia. Neurogastroenterol Motil. 2006;18:520–525. doi: 10.1111/j.1365-2982.2006.00772.x. [DOI] [PubMed] [Google Scholar]
- 12.Pearl-Yafe M, Kaminitz A, Yolcu ES, Yaniv I, Stein J, Askenasy N. Pancreatic islets under attack: cellular and molecular effectors. Curr. Pharm. Des. 2007;13:749–760. doi: 10.2174/138161207780249155. [DOI] [PubMed] [Google Scholar]
- 13.Park SM, Schickel R, Peter ME. Nonapoptotic functions of FADD-binding death receptors and their signaling molecules. Curr Opin Cell Biol. 2005;17:610–616. doi: 10.1016/j.ceb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Grivennikov SI, Suprash DV, Liu ZG, Nedospasov SA. Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: From simple paradigms to complex mechanisms. Int Rev Cytol. 2006;252:129–161. doi: 10.1016/S0074-7696(06)52002-9. [DOI] [PubMed] [Google Scholar]
- 15.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–424. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 19.Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 20.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 21.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori TJ. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. Erratum in: EMBO J 2003; 22:4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1011. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanghellini V, Cogliandro RF, De Giorgio R, Barbara G, Morselli-Labate AM, Cogliandro L, Corinaldesi R. Natural history of chronic idiopathic intestinal pseudo-obstruction in adults: a single center study. Clin Gastroenterol Hepatol. 2005;3:449–458. doi: 10.1016/s1542-3565(04)00675-5. [DOI] [PubMed] [Google Scholar]
- 27.Volta U, De Giorgio R, Petrolini N, Stanghellini V, Barbara G, Granito A, De Ponti F, Corinaldesi R, Bianchi FB. Clinical findings and anti-neuronal antibodies in coeliac disease with neurological disorders. Scand J Gastroenterol. 2002;37:1276–1281. doi: 10.1080/003655202761020542. [DOI] [PubMed] [Google Scholar]
- 28.Towns R, Kabey Y, Yoshimori T, Guo C, Hong S, Kaplan M, Klionsky D, Wiley JW. Sera from patients with type 2 diabetes and neuropathy induce autophagy and colocalization with mitochondria in SH-SY5Y cells. Autophagy. 2005;1:163–170. doi: 10.4161/auto.1.3.2068. [DOI] [PubMed] [Google Scholar]
- 29.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 30.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–240. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 31.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Enriquez S, He L, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–2472. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 34.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Srinivasan S, Stevens MJ, Sheng H, Hall KE, Wiley JW. Serum from patients with type 2 diabetes with neuropathy induces complement-independent, calcium-dependent apoptosis in cultured neuronal cells. J Clin Invest. 1998;102:1454–1462. doi: 10.1172/JCI2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–1938. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- 37.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. Essential role of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 38.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnsen JI, Pettersen I, Ponthan F, Sveinbjornsson B, Flaegstad T, Kogner P. Synergistic induction of apoptosis in neuroblastoma cells using combination of cytostatic drugs with interferon-gamma and TRAIL. Int J Oncol. 2004;25:1849–1857. [PubMed] [Google Scholar]
- 40.Tong HX, Lu CW, Zhang JH, Ma L, Zhang JH. Combination of gamma-interferon with TRAIL and cisplatin or etoposide induces apoptosis in human neruoblastroma cell line SH-SY5Y. Clin Med Sci J. 2007;22:38–43. [PubMed] [Google Scholar]
- 41.Takata-Tomokuni A, Ueki A, Shiwa M, Isozaki Y, Hatayama T, Katsuyama H, Hyodoh F, Fujimoto W, Ueki H, Kusaka M, Arikuni H, Otsuki T. Detection, epitope-mapping and function of anti-Fas autoantibody in patients with silicosis. Immunology. 2005;116:21–29. doi: 10.1111/j.1365-2567.2005.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]