

Abstract
Focal Adhesion Kinase (FAK) is a non-receptor kinase that is overexpressed in many types of tumors. We developed a novel cancer-therapy approach, targeting the main autophosphorylation site of FAK, Y397 by computer modeling and screening of the National Cancer Institute (NCI) small molecule compounds database. More than 140,000 small molecule compounds were docked into the N-terminal domain of the FAK crystal structure in 100 different orientations that identified 35 compounds. One compound 14 (1,2,4,5-Benzenetetraamine tetrahydrochloride) significantly decreased viability in most of the cells to the levels equal or higher than control FAK inhibitor, 1a (2-[5-Chloro-2-[2-methoxy-4-(4-morpholinyl)phenylamino]pyrimidin-4-ylamino]-N-methylbenzamide; TAE226) from Novartis, Inc. The compound 14 specifically and directly blocked phosphorylation of Y397-FAK in a dose- and time-dependent manner. It increased cell detachment and inhibited cell adhesion in a dose-dependent manner. Furthermore, 14 effectively caused breast tumor regression in vivo. Thus, targeting the Y397 site of FAK with 14 inhibitor can be effectively used in cancer therapy.
Keywords: Focal Adhesion Kinase, Y397 site, autophosphorylation, tumor, inhibitor
Introduction
FAK is a 125 kDa protein that localizes to focal adhesions 1 and is activated and tyrosine phosphorylated in response to integrin clustering 2. Tyrosine 397 is an autophosphorylation site of FAK and is a critical component in downstream signaling 3, providing a high-affinity binding site for the SH2 domain of Src family kinases 4, 5. The interaction between Y397-activated FAK and Src leads to a cascade of tyrosine phosphorylation of multiple sites in FAK (-576, -577, -925), as well as other signaling molecules such as p130CAS and paxillin, resulting in cytoskeletal changes and activation of other downstream signaling pathways 6. Y397 is also a site of binding PI3 kinase, growth factor receptor binding Grb-7, Shc, and other proteins. Thus, the Y397 site is one of the main phosphorylation sites that activate FAK signaling in the cells.
Focal adhesion kinase is involved in multiple cellular functions such as cell proliferation, survival, motility, invasion, metastasis, and angiogenesis 7. Different approaches to inhibit FAK with FAK anti-sense oligonucleotides 8, dominant-negative C-terminal domain of FAK, FAK-CD or FRNK 9, 10 or FAK siRNA 11, 12 caused decreased cellular viability, growth inhibition or apoptosis. Recently, FAK has been proposed to be a new potential therapeutic target in cancer 13, 14. Three novel kinase inhibitors of FAK, blocking FAK catalytic activity, were developed and reported recently, one by Novartis: 1a (2-[5-Chloro-2-[2-methoxy-4-(4-morpholinyl)phenylamino]pyrimidin-4-ylamino]-N-methylbenzamide, NVP-TAE226 (TAE226)) 15 16, 17 and another two by Pfizer: 2a (6-(4-(3-(methylsulfonyl)benzylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-3,4-dihydroquinolin-2(1H)-one C22H20F3N5O3S (PF-573,228 or PF-228)) and 3a (N-Methyl-N-(3-{[2-(2-oxo-2,3-dihydro-1H-indol-5-ylamino)-5-trifluoromethyl-pyrimidin-4-ylamino]-methyl}-pyridin-2-yl)-methanesulfonamide (PF-562,271))19. The first inhibitor, 1a 16, inhibited glioma and ovarian tumor growth in vivo 16, 17, although it also inhibited IGFR kinase 17. The efficacy of the 2a 18 inhibitor on tumor growth in vivo has not been reported, it inhibited only motility and did not inhibit cell growth and survival in vitro 18. 3a 19 inhibitor effectively blocked kinase activity of FAK and decreased tumor growth19. All inhibitors effectively blocked Y397-FAK autophosphorylation.
Since the Y397 site is important for FAK survival function, we performed a computer modeling approach, described in 20, to specifically target the Y397-site of FAK and to find potential small-molecule compounds that inhibit FAK function and decrease cell viability and tumor growth.
To identify a novel FAK inhibitor, we employed computer modeling and functional approaches. We used DOCK5.1 program and tested 140,000 small molecule compounds to target Y397 site of FAK. We found that compound 14 targets the Y397 site, directly and specifically decreases Y397-phosphorylation of FAK in vitro, inhibits cancer cell viability in vitro, causes detachment, decreases cell adhesion and blocks tumor growth in vivo. Thus, targeting the Y397 site can be an effective therapy approach for developing future novel FAK inhibitors.
Materials and Methods
Cell lines and culture
BT474 breast carcinoma cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS), 5 μg/ml insulin, and 1 μg/ml penicillin/streptomycin. The MCF-7 cell line was obtained from ATCC and maintained according to the manufacturer’s protocol. Other cancer and normal cell lines were maintained according to ATCC protocol.
Structure-Based In Silico Molecular Docking of FAK Small-Molecule Inhibitors
We used a structure-based approach combining molecular docking with functional testing. 140,000 small-molecule inhibitors following the Lipinski rules were docked into the N-terminal domain of FAK domain of the human FAK crystal structure in 100 different orientations using DOCK5.1 program, as described in 20. The crystal structure of FAK, N-terminal FERM domain 21 (PDB ID:2AL6) was used for docking of FAK inhibitors. All water molecules were removed from the crystal structure, and SYBYL (Tripos, St, Louis, MO) was used to protonate and add charges to the residues. The spheres describing the target pocket of FAK were created using DOCK 5.1 suite program SPHGEN. 24 spheres were ultimately selected to constrain small molecule orientations to within 5 angstroms of Y397 site. Docking calculations were performed on the University of Florida High Performance Computing supercomputing cluster using16 processors (http://hpc.ufl.edu).
Computational Docking
All docking calculations were performed with the University of California, San Francisco DOCK 5.1. program, using a clique-matching algoritm to orient small molecule structures with sets of spheres that describe the target Y397 site. Orientations were optimized using a simplex minimization algorithm, 100 orientations were created for each small molecule in the target site that were independently scores using DOCK5.1 grid-based scoring function. Briefly the three dimensional coordinates of the 140,000 compounds of the National Cancer Institute, Developmental Therapeutics Program (NCI/DTP) database were obtained from NCI. The files for hydrogen atoms and partial charges were created using SYBDB program.
Small-Molecule Compounds
The top thirty-five compounds that were detected by the DOCK5.1 program to best fit into the Y397 site of FAK were ordered from the NCI/DTP database free of charge. Each compound was solubilized in water at concentration of 25 mM and stored at -20°C and -80°C. The compound 14 (Table 1, shown in bold) was ordered from Sigma for biochemical analyses in vitro and injection into mice and in vivo studies.
Table 1.
Top scoring Y397 targeting compounds
| Comp. No | Comp. Label | NSC No | Formula | Mol. Wt |
|---|---|---|---|---|
| 1. | Y2 | 47228 | C4H12N4 | 116 |
| 2. | Y3 | 53040 | C9H12N6 | 204 |
| 3. | Y4 | 55937 | C4H12N6 | 144 |
| 4. | Y5 | 66330 | C2H6N4S | 118 |
| 5. | Y6 | 81462 | C9H18N4 | 182 |
| 6. | Y7 | 87061 | C15H24N4 | 260 |
| 7. | Y8 | 100649 | C6H18N4.3ClH | 256 |
| 8. | Y9 | 168639 | C9H16BrN4.C9H16BrN4.2Br | 680 |
| 9. | Y10 | 77977 | C16H13NO5 | 299 |
| 10. | Y11 | 206142 | C8H17N4O.Br | 265 |
| 11. | Y12 | 243780 | C4H13N5.2ClH | 204 |
| 12. | Y13 | 337807 | C4H12N4.4BrH | 440 |
| 13. | Y14 | 408480 | C16H13NO5 | 141 |
| 14. | Y15 | 667249 | C6H10N4.4ClH | 284 |
| 15. | Y17 | 10333 | C7H6FNO2 | 155 |
| 16. | Y18 | 21220 | C9H21N3O | 187 |
| 17. | Y19 | 23434 | C8H12N2O.ClH | 189 |
| 18. | Y20 | 26493 | C4H7N5 | 125 |
| 19. | Y21 | 80640 | C13H18BrN4.Br | 390 |
| 20. | Y23 | 84628 | C6H16N2O4.2BrH | 342 |
| 21. | Y24 | 88220 | C8H20N2O2 | 176 |
| 22. | Y25 | 112847 | C6H14N2O2.2ClH | 219 |
| 23. | Y26 | 114704 | C6H4F4N2 | 180 |
| 24. | Y27 | 116289 | C14H25N5O2 | 295 |
| 25. | Y28 | 125186 | C4H8N8 | 168 |
| 26. | Y29 | 145046 | C4H8N4.ClH | 149 |
| 27. | Y30 | 172826 | C6H12N4O2S | 204 |
| 28 | Y31 | 175751 | C5H10N6 | 154 |
| 29. | Y32 | 193871 | C9H14N4O2 | 210 |
| 30. | Y33 | 240745 | C8H12N2O.H3O4P | 250 |
| 31. | Y35 | 281680 | C4H12N4.2ClH | 189 |
| 32. | Y36 | 304712 | C7H15N3OP.I | 315 |
| 33. | Y37 | 374666 | C5H12N4O2.ClH | 197 |
| 34. | Y38 | 403347 | C6H12N4.Cu | 204 |
| 35. | Y39 | 664332 | C9H16BrN4.Br | 340 |
FAK inhibitors
FAK kinase inhibitor, 1a was obtained from Novartis Inc16. FAK kinase inhibitor 2a was obtained from Pfizer Inc18. Both 1a and 2a inhibitors were dissolved at DMSO at 25 mM. The structure and the therapeutic effect of compound are described in 15-17. The 1a and 2a inhibitors used as a control of FAK inhibition in the experiments.
Antibodies
Monoclonal anti-FAK (4.47) antibody to N-terminal FAK and monoclonal anti-paxillin antibody were obtained from Upstate Biotechnology, Inc. Polyclonal anti-phospho-Tyr397-FAK, anti-phospho-Tyr118-Paxillin, anti-phospho-Tyr402-Pyk-2 were from Biosource Inc. Total Pyk-2, paxillin and PARP antibodies were from Cell Signaling Inc. Monoclonal anti-β-actin antibodies were obtained from Sigma.
Cell Viability Assay
The cells were treated with compounds at different concentrations for 24 hours. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium compound from Promega Viability kit (Madison, IL) was added, and the cells were incubated at 37C for 1-2 hours. The optical density on 96-plate was analyzed with a microplate reader at 490 nm to determine cell viability.
Propidium iodide staining
Treated with the compound cells were collected, and propidium iodide 10 μg/ml in 1xPBS was added for 10 minutes. Hoechst 33342 (10 μg/ml) was added to the cells for detecting nuclei. Then cells were analyzed under fluorescent microscope to detect dead propidium iodide-stained red cells.
Cell adhesion assay
The poly-L-Lysine or Collagen (5 μg/ml) coated 96-well plates were blocked with the blocking buffer (medium with 0.5% BSA) for 1 hour at 37C. The cells were pre-treated with the inhibitor for 3 hours, collected and plated for adhesion assay at 4×10 5 cells on a 96-well plate. Cells were incubated at 37°C for 1 hour, fixed in 3.7% formaldehyde, washed in 0.1% BSA in PBS and stained with crystal violet (5 mg/ml in 2% ethanol) for 10 minutes. Then 2% SDS was added to the dried plated and OD at 590 nm was measured for detecting cell adhesion.
Western Blotting
Cells or homogenized tumor samples were washed twice with cold 1xPBS and lysed on ice for 30 minutes in a buffer containing: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton-X, 0.5% NaDOC, 0.1% SDS, 5mM EDTA, 50 mM NaF, 1mM NaVO3, 10% glycerol and protease inhibitors: 10 μg/ml leupeptin, 10 μg/ml PMSF and 1 μg/ml aprotinin. The lysates were cleared by centrifugation at 10,000 rpm for 30 minutes at 4°C. Protein concentrations were determined using a Bio-Rad Kit. The boiled samples were loaded on Ready SDS-10% PAGE gels (Bio Rad, Inc) and used for Western blot analysis with the protein-specific antibody. Immunoblots were developed with chemiluminescence Renaissance reagent (NEN Life Science Products, Inc). For quantification, densitometry of protein bands was performed with NIH Scion Image software.
Immunoprecipitation
Immunoprecipitation was performed according to the standard protocol. In brief, the pre-cleared lysates with equal amount of protein were incubated with 1 μg of primary antibody and 30 μl A/G agarose beads overnight at 4°C. The precipitates were washed with lysis buffer three times and re-suspended in 2xLaemmli buffer. The boiled samples were used for Western blotting, as described above.
Detachment Assay
Cells were plated with and without inhibitors for 24 hours, and detached and attached cells were counted in a hemocytometer. We calculated the percent of detachment by dividing the number of detached cells by the total number of cells. The percent of detached cells was calculated in three independent experiments.
Apoptosis Assay
Detached cells were collected and fixed in 3.7% formaldehyde in 1xPBS solution for the apoptosis assay. Detection of apoptosis was done with Hoechst 33342 staining. The percent of apoptotic cells was calculated as a ratio of apoptotic detached cells divided by the total number of cells in three independent experiments in several fields with the fluorescent microscope. For each experiment 300 cells per treatment were counted.
In vitro Kinase Assay
For in vitro kinase assay and detecting FAK autophosphorylation activity, 0.1 μg of purified full length FAK protein was used in a kinase buffer (20 mM HEPES, pH 7.4, 5 mM MgCl2, 5 mM MnCl2) with 10 μCi of [γ-32P]-ATP. For detecting FAK kinase activity, GST-paxillin N1C3 protein, the N-terminal paxillin domain containing the two binding sites for FAK, called GST-paxillin (a kind gift of Dr. M. Schaller, UNC, Chapel Hill, NC) was used for in vitro kinase assay. GST-paxillin (2.7 μg) was added as a substrate in the above kinase reaction. The kinase reaction with either FAK alone or with FAK and paxillin was performed for 10 minutes at room temperature and stopped by addition of 2 x Laemmli buffer. Isolated Pyk-2 protein was used in a kinase buffer (50 mM Tris, pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 10 μM ATP) with 10 μCi of [γ-32P]-ATP. The kinase reaction was performed as described above. Proteins were separated on a Ready SDS-10% PAGE gel. The gels were fixed in 7% acetic acid and 20% methanol and dried. The phosphorylated protein bands were visualized by autoradiography.
KinaseProfiler screening
Kinase specificity screening was performed with KinaseProfiler™ Service (Millipore) available on http://www.millipore.com/ drugdiscovery/dd3/KinaseProfiler. The screening was performed with 1 μM compound 14, 10 μM ATP and kinase substrates on 9 recombinant kinases according to Millipore protocol (http://www.millipore.com/drugdiscovery/dd3/KinaseProfiler).
Tumor Growth in Nude Mice in vivo
Female nude mice, 6 weeks old, were purchased from Harlan Laboratory. The mice were maintained in the animal facility and all experiments were performed in compliance with NIH animal-use guidelines and IACUC protocol approved by the UF Animal Care Committee. BT474 cells were injected, 2×106 cells/injection subcutaneously. In preliminary experiment different doses of the compound were introduced into the mice, and 30 mg/kg was chosen as optimal, non-toxic dose. The day after injection, the compound was introduced by IP injection at 30 mg/kg dose daily 5 days/week for 3 weeks. Tumor diameters were measured with calipers and tumor volume in mm3 was calculated using this formula = (width)2 × Length/2. At the end of experiment, tumor weight and volume was determined.
Immunohistochemistry and immunostaining
FAK staining was performed with Y397-antibody on slides with paraffin-embedded tumor samples or on coverslips with fixed cells, as described previously 22.
Statistical Analyses
Student’s t test was performed to determine significance. The difference between data with P<0.05 was considered significant.
Results
Targeting Y397 site of FAK by structure-based molecular docking approach and NCI database screening reveals Y397 compound that significantly decreased cell viability
The crystal structure of the N-terminal (FERM) domain of FAK has been recently identified 21 and was available in Protein Data Bank. Instead of high-throughput screening, we used a rapid structure-based approach combining molecular docking and functional testing. More than 140,000 compounds with known three-dimensional structure were docked into the structural pocket of FAK containing Y397 site. This approach combined the NCI/DTP (atomic coordinates and small molecules) database with improved molecular docking and scoring algorithms of DOCK 5.1 program 20. Each of 140,000 small-molecule compounds was docked in 100 different orientations using DOCK 5.1.0. As an example, one of such docked compounds in the Y397 site of FAK is shown on Figure 1A, B. The 35 compounds of >1400,000 compounds that had the highest scores of binding energy of interaction with Y397 site of FAK (Table 1) were ordered from NCI and tested for the effect on cancer cell viability by MTT assay.
Figure 1.
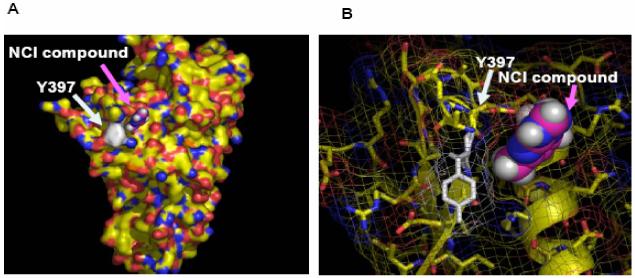

A, B. Targeting of Y397 site of FAK by structure-based molecular docking approach. A, The crystal structure of FAK (FERM) domain, reported in 21 with one of the NCI compound targeting Y397 site of FAK (shown by arrows). B, Zoomed image of Y397 site and this example compound (B).
C, D. The effect of compounds targeting Y397 site on viability of breast cancer and melanoma cell lines. C, The 35 compounds, 1-35 (Table 1) were added to the cells for 24 hours at 100 μM dose and MTT assay was performed, as described in Materials and Methods. The known FAK inhibitor, 1a 16 (Novartis), was used as a control. T47D breast cancer cell line (C) and C8161 (D) melanoma cell line. Bars show means ± standard deviations. Compound 14 decreased maximally cancer cell viability (shown by arrow).
We tested 6 different cancer cell lines: BT474, T47D, MCF-7 breast cancer, HT29 colon cancer, C8161 melanoma, and A549 lung cancer cell lines with the 35 compounds, targeting the Y397 site of FAK. One of these compounds, compound 14 (labeled Y15 in bold, Table 1) maximally decreased cell viability in all cancer cell lines (Figure 1 C, D). Compound 14 decreased viability in T47D breast cancer (Figure 1C) and C8161 melanoma cancer cells (Figure 1D) and is compared with the known FAK catalytic inhibitor 1a16 from Novartis Inc. Some compounds such as compounds 19 and 27 (Table 1) decreased viability of some cancer cell lines, (melanoma C8161 cell line, Figure 1D), but the effect was less in other cancer cell lines (Figure 1C) due to cancer and cell type specific-differences. Importantly, compound 14 was effective on all tested cancer cell lines. The structure of compound 14, docking to Y397 site, and the chemical name of this compound are shown on Figure 2 A. The predicted model of compound 14 interaction with Y397 site of FAK is shown on Figure 2B with four hydrogen bonds (red dashed lines and one hydrophobic interaction (blue dashed line). The compound 14 with molecular weight of 284 and chemical formula shown in (Table 1) was ordered from Sigma for the functional studies (below).
Figure 2.


A. The structure of 14 compound. Upper panel: Compound 14 targets Y397 site of FAK. Lower panel: Chemical structure and name of the 14 compound.
B. The closeup view of compound 14 interaction in the Y397 site of FAK. The secondary structure of alpha helices and beta sheets of the FAK-N-terminal domain are shown. 14 compound is shown in purple. The red dashed lines show hydrogen bonds and blue dashed line shows hydrophobic interaction. Y397 residue is shown in orange.
The compound 14 inhibits cancer cell viability in a dose-dependent manner
To determine whether 14 inhibits cell viability in a dose-dependent manner MTT assay was performed with different doses 0, 0.1; 1; 10, 30; 50, and 100 μM of compound 14 (Figure 3A). The viability of BT474 cells started to decrease at 10 μM dose and was significantly blocked at 50-100 μM doses of 14 inhibitor. The inhibitor 14 was less effective in several normal cells: breast epithelial MCF10A cells and normal fibroblast cell lines compared with BT474 cell line (not shown). Thus, 14 blocks cancer cell viability in a dose-dependent manner.
Figure 3.




A, B, C. The compound 14 inhibits cell viability and decreases Y397 FAK phosphorylation in a dose-dependent manner. A, BT474 breast cancer cells were treated with different doses of 14 inhibitor for 24 hours and MTT assay was performed to test the effect on cell viability. Bars show mean of triplicate determinations ± standard deviations. 14 inhibits cell viability in a dose-dependent manner. *P<0.05 viability of 14-treated cells versus control untreated cells.
B, Left upper panel: 14 specifically inhibits Y397 phosphorylation of FAK. BT474 cells were treated with 14 at 100 μM for 24 hours and Western blotting with Y397 and Y118 paxillin antibodies was performed to detect the level of phosphorylated FAK and paxillin, respectively. Western blotting with total FAK, paxillin and β-Actin was performed to detect expression of proteins in the cells. 14 effectively inhibited phosphorylation of Y397 and FAK substrate, Y118-paxillin. B, Right upper panel: Immunostaining with Y397-FAK antibody was performed on BT474 cells either untreated or treated with 14 at 100 μM for 24 hours. Immunostaining was performed with primary Y397-FAK antibody and Rhodamine-conjugated secondary antibody. FITC-BodipyFL-phallacidin was used for actin staining. 14 significantly decreased Y397-FAK in 14-treated cells.
B, left lower panel: 14 blocked total phosphorylation of FAK. Immunoprecipitation was performed with FAK antibody and Western blotting with phosphotyrosine antibody was performed. The blot was stripped and probed with FAK antibody. 14 blocked phosphorylation of FAK.
C, 14 decreases Y397 phosphorylation in a dose-dependent manner. Cells were treated with different doses of 14 inhibitor and Western blotting was performed with Y-397 and then with FAK antibody. Western blotting with Y402-Pyk-2 antibody was performed to detect Y402-Pyk-2 phosphorylation. Western blotting with β-Actin antibody was performed to control equal protein loading.
D. The compound 14 inhibits FAK autophosphorylation in a time-dependent manner. Cells were treated with 100 μM of 14 inhibitor for 1, 1, 4, 8 and 24 hours. Treatment with 1a inhibitor at 100 μM for 24 hours was used as a control. Western blotting with Y397 was performed to detect Y397-FAK level. Then the blot was stripped and Western blotting with FAK and β-Actin was performed. The compound 14 inhibits Y397-FAK phosphorylation in a dose-dependent manner.
E. Quantification of inhibition of Y397-phosphorylation, caused by 14 compound. Densitometry analysis was performed of 14-treated and untreated cells at 100 μM dose at three independent experiments using Scion Image, NIH software. Total FAK and Y397-FAK were expressed relatively to β-Actin, and then ratio of total FAK and Y397-FAK levels was calculated in -treated cells and expressed relative to untreated cells. Bars show mean ratio of 14-treated/untreated cells ± standard errors. * p<0.05 Y397-FAK compared to total FAK.
The compound 14 specifically blocks Y397-FAK phosphorylation
To test the effect of 14 on Y397 phosphorylation, we treated BT474 breast cancer cells with 14 at 100 μM dose and performed Western blotting with Y397 FAK antibody (Figure 3 B, left upper panels). The compound 14 specifically inhibited Y397 phosphorylation of FAK and also phosphorylation of FAK down-stream substrate paxillin, Y118-paxillin (Figure 3B, upper panel). 14 did not inhibit phosphorylation of other proteins, such as VEGFR-3 and c-Src (not shown). In addition, we performed immunostaining of FAK with Y397-FAK antibody on BT474 cells, treated with 14 (Figure 3B, right upper panel). 14 significantly decreased Y397-FAK phosphorylation on attached cells. Thus, the effect of 14 was specific to FAK. To test the effect of 14 on total FAK autophosphorylation activity, we immunoprecipitated FAK and performed Western blotting with P-tyrosine antibody (Figure 3B, left lower panels). 14 inhibited total phosphorylation of FAK. Thus, 14 that targets Y397 of FAK and decreases cell viability specifically inhibits Y397 and total FAK phosphorylation.
The compound 14 blocks FAK autophosphorylation in a dose-dependent manner
Next we analyzed the effect of 14 on inhibiting FAK Y397 phosphorylation in a dose-dependent manner. We treated BT474 breast cancer cells with 0, 0.1, 1, 10, 50 and 100 μM of 14 for 24 hours and performed Western blotting with Y397 antibody (Figure 3C). The decreased Y397-FAK phosphorylation was observed at 10 μM dose, and 14 decreased Y397 phosphorylation in a dose-dependent manner with high and maximal inhibition detected at 50 μM and 100 μM doses respectively that is consistent with the effect on cell viability. At 100 μM dose 14 had the same FAK inhibition as 1a16 (Novartis) (Figure 3C). To detect the effect of 14 on autophosphorylation activity of FAK homologue, Pyk-2, we performed Western blotting with Y402-Pyk2 antibody. Importantly, 14 did not inhibit Y402-Pyk2 levels compared to Y397-FAK level (Figure 3C). High doses of 14 (100 μM) decreased also the level of total Pyk-2 and FAK similar to 1a inhibitor. Thus, 14 inhibits FAK autophosphorylation in a dose-dependent manner.
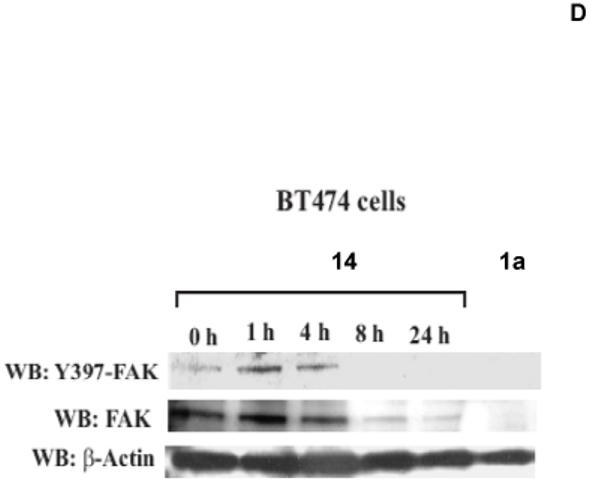
The compound 14 blocks FAK autophosphorylation in a time-dependent manner
Next we analyzed whether 14 inhibits FAK Y397 phosphorylation in time-dependent manner. We treated BT474 cells with 100 μM of 14 for 0, 1, 4, 8 and 24 hours, then Western blotting was performed with Y397 antibody (Figure 3D). The result shows that treatment with 14 at 4 hours did not significantly decreased Y397 phosphorylation, but 8 hours was enough to completely block Y397-phosphorylation and to down-regulate FAK. The control inhibitors 1a 16 at 100 μM also completely blocked Y397 phosphorylation. Similarly to 14 inhibitor, 1a16 also down-regulated total FAK (Figure 3D). We performed densitometry analysis of 14-treated cells, and show that 14 decreased more Y397-FAK level than total FAK (Figure 3E). Thus, the data demonstrate that 14 inhibits FAK Y397 phosphorylation and down-regulates FAK in a time-dependent manner.
The compound 14 is a direct FAK autophosphorylation inhibitor
To test, whether 14 is a direct inhibitor of FAK, we performed in vitro kinase assay with recombinant isolated with Baculovirus system purified FAK protein, described in 23. We performed in vitro kinase assay with 1-100 μM doses of 14. We used 1a16 inhibitor as a positive control. 14 directly blocked autophosphorylation activity of FAK starting from 1 μM dose, as well as control 1a16 (Figure 4A, upper panel). In addition, we performed in vitro kinase assay with 1-100 μM doses of 14 and a recombinant baculoviral purified Pyk-2 protein, homologous to FAK, described in 23. 14 did not significantly block autophosphorylation activity of Pyk-2 compared to FAK activity (Figure 4A, lower panel). 14 did not significantly inhibit Pyk-2 at high 100 μM dose in contrast to 1a16 inhibitor. The amount of 14 required to inhibit >50% of the FAK autophosphorylation (IC50) in this assay is equal to 1 μM range.
Figure 4.


A, B. The compound 14 directly blocks in vitro catalytic autophosphorylation and kinase activity of FAK. A, upper panel. 14 blocks FAK catalytic autophosphorylation activity. In vitro kinase assay was performed with γ-ATP32, 0.1 μg of purified recombinant FAK protein and different doses of 14 inhibitor for 10 minutes at room temperature, as described in Materials and Methods. 14 directly blocks FAK autophosphorylation activity in a dose-dependent manner. A, lower panel. 14 has no inhibiting effect on Pyk-2 catalytic autophosphorylation activity. In vitro kinase assay was performed with γ-ATP32, 0.1 μg of purified recombinant Pyk-2 protein and different doses of 14 inhibitor for 10 minutes at room temperature, as described in Materials and Methods. 14 did not affect significantly Pyk-2 autophosphorylation. The data are from three independent experiments. B. The compound 14 blocks FAK kinase activity in vitro. GST-paxillin was added as a substrate in the kinase assay reaction. The reaction was performed with γ-ATP32, 0.1 μg of purified recombinant FAK protein and different doses of 14 inhibitor for 10 minutes at room temperature. 14 inhibits phosphorylation of paxillin in a dose-dependent manner.
C. Kinase profile of compound 14. The compound 14 was screened against nine commercially available recombinant enzymes and catalytically active FAK kinase domain (411-686 a.a.) without the N-terminal domain, containing Y397 site by Kinase Profiller Service (Millipore/Upstate Biotechnology Inc.). The assay was performed with 1 μM dose of 14 that inhibited activity of FAK (Figure 4 A and B), 10 μM ATP and kinase substrates as described in Materials and Methods (http://www.millipore.com/drugdiscovery/dd3/KinaseProfiler). 14 effectively blocks full length FAK kinase activity (Figure 4A, B) and it doesn’t affect activities of homologous Pyk-2, FAK kinase domain and other kinases. The bars show percentage of inhibition of the enzyme activity (14/Untreated). The mean and standard errors of two independent experiments are shown.
To test whether inhibition of FAK autophosphorylation activity will affect FAK kinase activity, we used paxillin as a substrate, and performed in vitro kinase assay, as described in Materials and Methods) (Figure 4B). FAK effectively phosphorylates paxillin in vitro (Figure 4B, lane 3). 14 inhibited FAK autophosphorylation and paxillin phosphorylation starting with 1 μM dose (Figure 4B). Thus, 14 blocked FAK autophosphorylation and kinase activity of FAK. In addition, 14 was screened by in vitro kinase assay with 9 other recombinant commercially available kinases (c-RAF, c-Src, EGFR, VEGFR-3, IGF-1, Met, PDGFR-α, Pyk2 (homologue of FAK), PI3K (p110δ/p85α) (Upstate Biotechnology, Inc), as described in Materials and Methods (Figure 4C). In this assay, 14 significantly decreased catalytic activity of the full length FAK, while it did not significantly affected kinase activities of the other kinases (Figure 4C). 14 did not decrease kinase activity of the FAK kinase domain (411-686 a.a), with the deleted N-terminal domain, containing Y397 site, and did not affect kinase activity of the FAK homologue, Pyk2 protein (Figure 4C). Thus, 14 is a direct and specific inhibitor of FAK Y397-phosphorylation.
The compound 14 causes dose-dependent cell detachment in cancer cells
To test the effect of 14 inhibitor on breast cancer cells, we treated BT474 cells with 14 at 1 and 100 μM for 24 hours. We performed analysis of detachment and apoptosis in 14-treated BT474 cells and compared with TAE-226-treated cells (Figure 5A). 14 caused dose-dependent detachment in BT474 cells (Figure 5A). At 10 μM dose, 14 caused only 8% detachment in BT474 cells, while at 50 μM dose, detachment was equal to 38%. At 100-200 μ M doses, detachment reached 64%-66% respectively. 14 caused less detachment than 1a16 inhibitor, which induced 30% detachment at 10 μM and >80% detachment at 50 μM dose. Thus, 14 effectively caused dose-dependent cellular detachment. In addition, we performed analysis of cell detachment in normal mammary epithelial MCF-10A cells (Figure 5 B). 14 caused less detachment in MCF10A cells than in BT474 cells at high 100 μM dose (15% in MCF10A cells versus 64% in BT474 cells) (Figure 5B, upper panel). The morphology of MCF10A cells treated with 14 at 100 μM dose is shown on Figure 5B, lower panel. Thus, normal MCF10A breast cells are less detached than breast cancer BT474 cells (Figure 5B).
Figure 5.






A. The compound 14 causes dose-dependent cell detachment in BT474 cells. BT474 cells were treated with different doses of 14 inhibitor. The detachment was determined on a hemacytometer, as described in Materials and Methods. Bars show means ± standard errors in three independent experiments. 14 significantly decreased cell detachment. Bars show means ± standard errors in three independent experiments. *P<0.05 versus untreated cells.
B. Detachment caused by 14 in normal breast MCF10A cells. Upper panel: Normal breast MCF10A cells were treated with different doses of 14 inhibitor. The detachment was determined, as described in Figure 5A. Bars show means ± standard errors in three independent experiments. Lower panel: A phase-contrast microscopy of MCF10A cells treated with 14 at 100 μM dose. Detachment is shown in BT474 and MCF10A cells treated with 100 μM of 14. No significant detachment was caused by 14 in MCF10A cells compared with BT474 cells.
C. The compound 14 doesn’t cause significant apoptosis in BT474 cells. Hoechst staining was performed on BT474 cells at 24 hours treatment with different doses of 14 and 1a 16 inhibitors, as described in Materials and Methods. No significant apoptosis was detected with 14 compared to 1a inhibitor. Bars represent means ± standard deviations in four independent experiments. *P<0.05 versus untreated cells.
D. Upper panel: Hoechst staining of 14-treated BT474 cells. BT474 cells treated as above were analyzed by Hoechst staining. Apoptotic nuclei stained with Hoechst are shown. No apoptotic nuclei were observed with 14 inhibitor at 100 μM dose compared to 1a 16 inhibitor at 20 μM dose. Lower panel: PARP cleavage in 14-treated and 1a-treated cells. Western blotting on BT474 cells, treated with 14 or 1a inhibitors at different doses was performed with PARP antibody. 1a caused significantly higher levels of cleaved PARP (89 kDa) compared with 14-treated cells. No cleaved 89 kDa PARP was detected at 100 μM dose in 14-treated cells compared with 1a-treated cells.
E. Propidium iodide staining of 14-treated cells. BT474 cells treated with 14 at 10 and 100 μM as above, were collected and stained for 10 minutes with 10 μg/ml of propidium iodide, detecting dead necrotic cells and with Hoechst (10 μg/ml), detecting nuclei. Cells were analyzed under the Zeiss fluorescent microscope. Propidium iodide staining was increased in a dose-dependent manner and reached maximum at 100 μM dose.
F. The compound 14 blocks cell adhesion in a dose-dependent manner. BT474 cells were treated with 14 at different concentration and cell adhesion was measured as described in Materials and Methods. 1a 16 and 2a 18 inhibitors at 100 μM were used as a control. 14 significantly blocked cell adhesion in a dose dependent manner. Bars show means ± standard errors in four independent experiments*, P<0.05, cells adhesion in 14 treated cells less than in control untreated cells.
The compound 14 causes less apoptosis than 1a inhibitor
To test the effect of 14 on apoptosis, we performed Hoechst staining on untreated and 14-treated cells. At high 50-100 μM dose, 14 did not cause significant apoptosis in BT474 cells, with apoptotic levels less than 5% (Figure 5C). In contrast, 1a inhibitor (Novartis)16 caused higher levels of apoptosis than 14, that were equal to 35% at 10 μM and reached 69% at 20 μM dose (Figure 5C). Hoechst stained nuclei of 14 and 1a 16-treated cells are shown in Figure 5D. No apoptotic nuclei were detected in treated cells with 14 at 100 μM dose in contrast to 1a-treated cells at 20 μM doses (Figure 5D, upper panel). To confirm apoptosis data, we performed analysis of PARP, poly(ADP-ribose) polymerase cleavage in 14-treated and 1a-treated cells (Figure 5D, lower panel). 1a -treated cells had cleaved PARP 89 kDa fragment starting at 1 μM dose, while cells treated with 14 did not have the cleaved PARP 89 kDa protein. At 10-20 μM doses, 14-treated cells had significantly less cleaved 89 kDa PARP than 1a-treated cells (Figure 5D, lower panel). At high 100 μM dose, both 14 and 1a-treated cells expressed less uncleaved 116 kDa PARP (Figure 5D, lower panel). However, 14-treated cells did not express cleaved 89 kDa PARP protein in contrast to 1a-treated apoptotic cells (Figure 5D, lower panel). 14-treated cells expressed 55 kDa cleavage PARP fragments (not shown) that have been reported to be present in necrotic cells. To detect necrosis and confirm decreased viability data in 14-treated BT474 cells at 100 μM dose, we stained the cells with propidium iodide that detects necrotic dead cells. We found dose-dependent increase of propidium iodide stained cells, reaching maximum at 100 μM dose (Figure 5E). Thus, 14 induces non-apoptotic cell death by necrosis at 100 μM dose that decreased viability in the cells. Thus, 14 inhibitor causes less apoptosis than 1a inhibitor at 10-20 μM dose, and induces necrosis at high 100 μM dose.
The compound 14 inhibits cell adhesion in a dose-dependent manner
To test the effect of 14 on cell adhesion, we treated BT474 with different doses of 14 and with 100 μM 1a16 on collagen-coated plates and measured adhesion. 14 inhibited cell adhesion in a dose-dependent manner (Figure 5F). Starting with 50 μM cell adhesion was significantly decreased that is consistent with Y397-decreased FAK phosphorylation at these doses. At 100 μM dose 14 significantly inhibited cell adhesion, as well as 1a inhibitor (Figure 5F). Thus, 14 effectively blocks cell adhesion.
The compound 14 inhibits breast tumor growth in vivo and decreases Y397-FAK phosphorylation
To detect in vivo effect of 14, we introduced BT474 cells subcutaneously into nude mice. Initially we determined that dose 30 mg/kg is the optimal non-toxic dose. We treated mice with 30 mg/kg dose of 14 for 5 days/week and compared tumor growth with untreated mice. No animal weight loss or death was observed in any tumor growth inhibition experiment for 23 days. 14 significantly blocked tumor growth in treated mice compared with untreated mice in vivo (Figure 6A). 14 significantly reduced tumor weight compared to untreated mice (Figure 6B, upper panel) and tumor volume was significantly less than in untreated samples (Figure 6B, lower panel). We isolated tumors from untreated mice and mice treated with 14 and probed for Y397 levels by Western blotting. Tumors from untreated mice had significantly higher levels of Y397 phosphorylation than tumors treated with 14, while total FAK levels were the same (Figure 6C). Similar result was obtained by immunohistochemical staining of tumors with Y397 antibody (Figure 6D). Tumors from 14-treated mice had less Y397-FAK phosphorylation than tumors from untreated mice. Thus, 14 significantly suppressed breast tumorigenesis that is consistent with in vitro viability and biochemical data.
Figure 6.




A, B. The compound 14 significantly blocks tumor growth in vivo. A, BT474 breast cancer cells were subcutaneously injected in 5 mice. The day after injection, 14 compound at 30 mg/kg was added daily 5 days per week. Five untreated mice were used as a control group. Tumor volume was measured with calipers. 14 significantly blocked tumor growth in vivo. Bars represent means ± standard deviations (n=5) P<0.05, Student’s t-test.
B, The compound 14 significantly blocks tumor weight and volume. At day 23 after breast cancer cell injection, tumors were extracted, and weight and volume was determined in grams (upper panel) and in mm3 (lower panel), respectively. 14 significantly blocked tumor weight (upper panel) and volume (lower panel). Bars represent means ± standard deviations. * p<0.05, Student’s t-test.
C, D. The compound 14 decreases Y397-FAK phosphorylation in tumors. C, We isolated tumors from untreated mice and from mice, treated with 14 compound. Cell lysates were prepared, and Western blotting was performed with Y397 antibody. FAK and β-actin antibodies were used for detecting total FAK and β-Actin levels. D, Immunohistochemical staining analysis was performed on untreated and 14-treated tumors with Y397-antibody. Two representative tumors from untreated and 14 treated mice groups are shown (marked as T1 and T2 tumors in each group) (C, D). 14 decreased Y397FAK phosphorylation in tumors treated with 14 (30 mg/kg) compared with untreated mice.
Discussion
FAK has been shown to be important for survival signaling, angiogenesis, motility, metastasis and has been shown to be overexpressed in a number of tumors 7. FAK is proposed recently to be a novel target for developing anti-cancer therapy drugs13. We developed a novel approach to target Focal Adhesion Kinase by targeting its main autophosphorylation site by computer modeling and functional approaches. Thus, targeting Y397 of FAK with structure-based molecular docking and National Cancer Institute database screening approaches revealed 35 compounds out of 140,000 different compounds that best fit in this pocket. Among these compounds, compound 14 was the most effective in decreased cell viability in several cancer cell lines. Importantly, this compound decreased Y397 phosphorylation, and total FAK phosphorylation. It directly decreased FAK autophosphorylation and inhibited FAK kinase activity on paxillin substrate in vitro. In BT474 cancer cells 50 μM of 14 almost completely decreased FAK autophosphorylation. 14 decreased FAK phosphorylation in a dose- and time dependent manner. 14 increased cell detachment and decreased cell adhesion. In vitro kinase assay demonstrated that 1 μM. dose of 14 was enough to significantly and specifically block FAK authophosphorylation and kinase activity. In cell culture higher dose required to inhibit Y397 phosphorylation that can be explained by differences in sensitivity of Western blotting and in vitro radioactive kinase assay. Treatment with 14 induced cell detachment in BT474 cells. Importantly, that decreased phosphorylation of Y397-FAK occurred prior to significant detachment and cell death, as we detected decreased Y397-phosphorylation in 14-treated attached cells. In addition in vivo, 14 compound significantly inhibited tumor growth at 30 mg/kg dose in BT474 breast cancer cells, subcutaneously injected in mice. Tumors from mice treated with 14 had decreased Y397-phosphorylation of FAK. Thus, 14 inhibitor, targeting Y397 site of FAK, can be affective inhibitor in anti-cancer therapy.
Thus, this report shows that the DOCK program shows proof-of principle for using silico-based strategies for identifying novel inhibitors of FAK. This method was successfully used before for Jak2 kinase 20, but for the FAK kinase and for targeting the main phosphorylation site of FAK it was never reported. We showed that using DOCK5.1 program in conjunction with functional testing, we can successfully identify novel small molecule inhibitors of FAK by database screening. It is possible to use this approach for testing other sites and pockets of FAK in future. Thus, screening compounds that target other phosphorylation can also provide novel inhibitors that can be potentially used in therapy.
The molecular structure of 14 is known, and it contains a single aromatic ring. Thus, this single aromatic ring compound can serve as a potential lead compound for future chemically synthesized derivatives and novel FAK inhibitors.
The most important finding is that 14 blocked tumorigenesis in mice in vivo, showing potential in therapy for these inhibitors. Two other novel FAK inhibitors were reported, 2a 18 by Pfizer and 1a 16 by Novartis. Both inhibitors have their own limitations; the first inhibitor 2a has no effect on cell viability and no report on tumorigenesis is known. The second one, 1a has an inhibiting effect on IGFR-1, MAPK and AKT 17. 1a caused induced apoptosis in vitro15. In contrast, 14 did not cause significant apoptosis similar to Pfizer inhibitor 2a18, but caused detachment and loss of adhesion. The authors suggest that scaffolding function of FAK can be important in regulating apoptosis17. We detected increased cell death by trypan blue (not shown) and propidium iodide staining in 14-treated cells, thus decreased viability caused by 14 can be explained by non-apoptotic mechanism such as necrosis that will be studied in future reports. Since FAK and TNF receptor pathways are linked in cells7, the TNF-induced non-apoptotic cell death mechanism may be important in 14-treated cells. Thus, 14 can have therapeutic effect in apoptosis-resistant tumor cells. The difference between 14 and 1a in causing apoptosis at 10-20 μM doses can be explained by higher specificity of 14 to FAK. For example, 14 did not inhibit kinase activity of IGFR, while 1a has been shown to effectively inhibit IGFR-1 activity16. 14 also did not affect phosphorylation of PYK-2 homologue that can be explained by only 43% amino acid identity between N-terminal domains of FAK and Pyk-2 proteins21. In addition, amino-acids, involved in interaction with 14 are not conserved between FAK and Pyk-2. We performed computer modeling and docking of 14 into the predicted PYK2 domain and found that 14 was not docked into Pyk-2 structure, as well as into FAK. 14 is more specific than 1a16 inhibitor, as 1a at 100 μM dose decreased Y402- Pyk-2 autophosphorylation, while 14 did not affect its activity. Increased selectivity of 14 to FAK, but not to homologous Pyk-2 can be useful in the therapy of FAK-overexpressing tumors. 14 inhibitor blocked cell adhesion in a dose-dependent manner. The fact that 14 inhibited less cell adhesion and caused less detachment than 1a inhibitor, can be explained by less-specificity of 1a to FAK, as it cross-reacts with other kinases. Normal breast MCF10A cells were more resistant to 14 than cancer cells similar to 1a-caused detachment in MCF10A15. This can explain that tumors that overexpressed FAK were sensitive to 14 inhibitor in vivo. 14 specifically blocked Y397 phosphorylation of FAK, inhibited FAK kinase activity and did not inhibit kinase activity of other enzymes. 14 that targets Y397 site blocked FAK autophosphorylation and phosphorylation of its substrate, paxillin that is consistent with data on critical role of Y397 site in phosphorylation of paxillin in vitro24. 14 significantly blocked Y397 inside cancer cells at <50 μM, and 1 μM dose is enough to significantly block FAK autophosphorylation in a tube by in vitro kinase assay. Importantly, both assays demonstrated FAK as a specific target of 14. The 14 and 1a16 inhibitors caused cell death by different mechanisms, 1a caused more apoptosis than 14 due to differences in specificities. 14 blocked tumorigenesis at doses lower than 1a16. In glioma tumors, 1a was used at 50-75 mg/kg 17. In the ovarian tumor model, 1a alone at 30 mg/kg was not so effective and required additional docetaxel treatment to reduce tumor growth 16. In the breast cancer model, 14 alone effectively inhibited >74% of tumor growth at 30 mg/kg dose. The third inhibitor of FAK 3a from Pfizer19 was effective in human xenograft models at doses 25-50 mg/kg that was accompanied with decreased FAK phosphorylation 19. Thus, developing novel and more specific inhibitors of FAK that will block tumorigenesis is important to the field.
Thus, this report led to the identification of compound 14, a small-molecule FAK inhibitor that directly targeted the Y397 autophosphorylation site of FAK and decreased its phosphorylation, inhibited cell viability and adhesion in vitro and blocked tumorigenesis in vivo. Thus, this compound and its derivatives may be important for future therapies.
Acknowledgments
We would like to thank Dr. Ann Dontao Fu and members from Molecular Pathology Core, Department of Pathology, UF for technical help in immunohistochemical analysis of tumors. We would like to acknowledge Dr. Shinji Hatakeyama (Novartis Inc.) for providing chemical name of 1a inhibitor. We would like to acknowledge the University of Florida High-Performance Computing center for providing computational resourses and support. The work was supported by Susan G. Komen for the Cure grant BCTR0707148 (V.M. Golubovskaya) and NIH grant CA65910 (W.G. Cance).
Abbreviations
- FAK
Focal Adhesion Kinase
- PARP
poly(ADP-ribose) polymerase
- FAK-CD (FRNK)
C-terminal domain of FAK
References
- 1.Schaller MD. The focal adhesion kinase. J Endocrinol. 1996;150(1):1–7. doi: 10.1677/joe.0.1500001. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. Journal of Cell Biology. 1993;123(4):993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xing Z, Chen HC, Nowlen JK, Taylor SJ, Shalloway D, Guan JL. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5(4):413–21. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK. Molecular & Cellular Biology. 1994;14(1):147–55. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular & Cellular Biology. 1994;14(3):1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19(2):137–45. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 7.Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol. 2007;263:103–53. doi: 10.1016/S0074-7696(07)63003-4. [DOI] [PubMed] [Google Scholar]
- 8.Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang X, Cance WG. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005;15(5):357–62. doi: 10.1097/00008390-200510000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Xu L.-h., Yang X.-h., Bradham CA, Brenner DA, Baldwin AS, Craven RJ, Cance WG. The focal adhesion kinase suppresses transformation-associated, anchorage-Independent apoptosis in human breast cancer cells. J. Biol. Chem. 2000;275:30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 10.Golubovskaya V, Beviglia L, Xu LH, Earp HS, 3rd, Craven R, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;277(41):38978–87. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- 11.Halder J, Landen CN, Jr., Lutgendorf SK, Li Y, Jennings NB, Fan D, Nelkin GM, Schmandt R, Schaller MD, Sood AK. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin Cancer Res. 2005;11(24 Pt 1):8829–36. doi: 10.1158/1078-0432.CCR-05-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, Vella J, Cance WG, Golubovskaya VM. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282(17):12503–16. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- 13.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5(7):505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 14.van Nimwegen MJ, van de Water B. Focal adhesion kinase: A potential target in cancer therapy. Biochem Pharmacol. 2006 doi: 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Golubovskaya VM, Virnig C, Cance WG. TAE226-Induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol Carcinog. 2007 doi: 10.1002/mc.20380. [DOI] [PubMed] [Google Scholar]
- 16.Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G, Sood AK. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67(22):10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 17.Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, Jackson D, de Groot J, Yung WK. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6(4):1357–67. doi: 10.1158/1535-7163.MCT-06-0476. [DOI] [PubMed] [Google Scholar]
- 18.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282(20):14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 19.Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, Lorenzen M, Berliner M, Luzzio M, Patel N, Schmitt E, LaGreca S, Jani J, Wessel M, Marr E, Griffor M, Vajdos F. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68(6):1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg EM, Ma X, He K, Frank SJ, Ostrov DA, Sayeski PP. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of jak2 tyrosine kinase autophosphorylation [correction of autophophorylation] J Med Chem. 2005;48(7):2526–33. doi: 10.1021/jm049470k. [DOI] [PubMed] [Google Scholar]
- 21.Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281(1):252–9. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- 22.Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, Cance WG. p53 regulates FAK expression in human tumor cells. Mol Carcinog. 2007 doi: 10.1002/mc.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280(26):25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]