

Abstract
We previously demonstrated that synthetic peroxisome proliferator activated receptor gamma (PPARγ) ligands inhibit non-small cell lung carcinoma (NSCLC) cell growth through multiple signaling pathways. Here, we show that dietary compounds, such as fish oil (which contains certain kinds of fatty acids like ω3 and ω6 polyunsaturated fatty acids), also inhibits NSCLC cell growth by affecting PPARγ and by inhibiting the expression of integrin-linked kinase (ILK). Exogenous expression of ILK overcame, while silencing ILK enhanced the inhibitory effect of fish oil on cell growth. The inhibitor of p38 MAPK, SB239023, abrogated the inhibitory effect of fish oil on ILK expression, whereas the inhibitor of ERK, PD98059, had no effect. Transient transfection experiments showed that fish oil reduced ILK promoter activity and this effect was abolished by AP-2α siRNA and by SB239023, and by deletion of a specific portion of the ILK gene promoter. Western blot analysis and Gel mobility shift assay demonstrated that fish oil significantly induced AP-2α protein expression and AP-2 DNA binding activity in the ILK gene promoter, and that this was dependent on PPARγ activation. Blockade of AP-2α abrogated the effect of fish oil on ILK expression and on cell growth, while exogenous expression of AP-2α enhanced cell growth in the setting of fish oil exposure. Taken together, these findings demonstrate that fish oil inhibits ILK expression through activation of PPARγ- and p38 MAPK-mediated induction of AP-2α. In turn, this leads to inhibition of NSCLC cell proliferation. This study unveils a novel mechanism by which fish oil inhibits human lung cancer cell growth.
Keywords: PPARγ, ILK, p38 MAPK, AP-2, human lung carcinoma cells
INTRODUCTION
Lung cancer is the leading cause of cancer death in the world with an estimated 213,000 new cases and 160,000 deaths in 2007 in the United States (1). Despite marked improvements in understanding the molecular biology of cancer and in advances in diagnostics, surgery, and chemotherapy the 5-year poor survival rate (<15%) has not changed substantially (1, 2). New approaches to the treatment and prevention of lung cancer are clearly indicated, but depend greatly on a better understanding of the molecular mechanisms that control oncogenesis and tumor growth in lung. Fish oils are enriched with polyunsaturated fatty acids (PUFAs) of the ω3 family, and are believed to have protective effects against cancer (3). Several investigators have demonstrated that diets enriched with fish oil can reduce the growth rates of implanted tumors in vivo (4–6). However, the mechanisms responsible for the anti-cancer effects of fish oil remain incompletely elucidated.
Both n-6 PUFAs and n-3 PUFAs modulate peroxisome proliferator-activated receptor gamma (PPARγ) and decrease cell growth in human lung cancer cells (7). PPARγ is a member of the ligand-inducible nuclear transcription factors that heterodimerize with retinoid X receptors and bind to peroxisome proliferator response elements (PPRE) located in the promoter region of PPAR target genes (8). These lipid-sensitive receptors can be activated in a variable isotype-specific manner by natural/dietary ligands including long chain polyunsaturated fatty acids which are found in fish oil (e.g. n-3-PUFA, n-6-PUFA), various eicosanoids (e.g. 15d-PGJ2), lipid hydroperoxides (e.g. 9(s)-HODE and 13(s)-HODE), and in linoleic acid (9, 10). The efficacy of these compounds as anti-cancer agents has been examined in a variety of cancers including colon, breast and prostate, and they have been found to inhibit cancer cell growth in vitro and in vivo (11).
In view of the above, it has been suggested that the anti-cancer properties of fish oil are dependent on activation of PPARγ; however, the downstream events involved in this process remain unclear. One of the potential targets for PPARγ ligands is integrin-linked kinase (ILK), which links cell-adhesion receptors, integrins, and growth factors to the actin cytoskeleton and to a range of signaling pathways that are implicated in the regulation of anchorage-dependent cell growth/survival, cell cycle progression, invasion and migration, and tumor angiogenesis (12). Moreover, overexpression of ILK results in oncogenic transformation and progression to invasive and metastatic phenotypes (13, 14). Thus, we explored the effects of fish oil on ILK expression. This work revealed that fish oil inhibits NSCLC proliferation by suppressing ILK expression through activation of PPARγ. This results in the activation of P38 mitogen activated protein kinase (p38 MAPK) and induction of AP-2α, which in turn inhibits ILK gene expression. To our knowledge, this is the first report linking fish oil to ILK expression.
MATERIALS AND METHODS
Culture, Chemicals and Fish Oil Treatment
The human NSCLC cell lines (H522, H1792, H1838 and A549) and normal bronchial epithelial cell lines (BEAS-2B and 16-HBE), and NIH3T3 cells were obtained from the American Type Culture Collection (American Type Culture Collection, Rockville, MD) and routinely grown in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, HEPES buffer, 50 IU/ml penicillin/streptomycin, and 1 µg amphotericin (complete medium) as previously described (15). Fish oil was obtained from Sigma (St. Louis, MO). As described in detail elsewhere (16), the fish oil was obtained from Mendaden fish and is typically composed of the following fatty acids:14:0 Myristic acid, 6–9%; 16:0 Palmitic acid, 15–20%; 16:1 Palmitoleic acid, 9–14%; 18:0 Stearic acid, 3–4%; 18:1 Oleic acid, 5–12%; 18:2 Linoleic acid, <3%; 18:3 Linolenic acid, <3%; 18:4 Octadecatetraenoic acid, 2–4%; 20:4 Arachidonic acid, <3%; 20:5 Eicosapentaenoic acid (EPA), 10–15%; 22:6 Docosahexaenoic Acid (DHA), 8–15%. These sum components added up to 80% (the remaining 20% represents other unidentified fatty acids). Note that EPA and DHA omega-3 fatty acids, known to act as PPARγ activators, are the major components for fish oil. The fish oil was emulsified with 5% (w/w) egg phosphatidylcholine (Sigma) and 0.03% (w/w) butylated hydroxytoluene (as anti-oxidant) in phosphate buffered saline (PBS) at a final oil concentration of 15 mg/ml. For treatment, cells were plated in regular growth medium for 24 h, after which the medium was replaced with fresh oil-enriched medium at indicated concentrations of fish oil. The control group received egg phosphatidylcholine alone at an equal volume. The oil-enriched medium was changed daily in order to decrease the reactions of fatty acid oxidation on cells. Polyclonal antibodies specific for ILK, p38 MAPK, Phospho-p38 MAPK (Thr180/Tyr182), PD98059 and SB239023 were purchased from Cell Signaling (Beverly, MA). GW9662, rosiglitazone, and WY14643 were purchased from Cayman Chemical Co. (Ann Arbor, Michigan). Polyclonal antibodies against AP-2α, β and γ were purchased from Santa Cruz Biotechnology, Inc. The inactive (ILK-S343A) and superactive ILK cDNAs (ILKS-343D) in pUSEamp vectors under the control of the cytomegalovirus promoter were purchased from Upstate USA, Inc. (Charlottesville, VA). Linoleic acid, antibodies against PPARγ, and other chemicals were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise indicated.
Cell viability assay
NSCLC cells (105 cells/well) were cultured with increasing concentrations of fresh oil-enriched medium for up to 48 h, or were transfected with control or AP-2α siRNA for 30 h before exposure of the cells to fresh oil-enriched medium for an additional 48 h in 96-well plates. In a separate experiment, cells were transfected with inactive (ILK-S343A) or superactive ILK (ILKS-343D) cDNA, or with control or AP-2 expression reporter construct SP (RSV)AP-2 (#12100) (purchased from Addgene, Inc.; Cambridge, MA) (17) using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h of incubation, cells were treated with or without fresh oil-enriched medium for 48 h. Afterwards, the number of viable cells in culture was determined using the CellTiter-Glo Luminescent Cell Viability Assay kit, which is based on quantitation of the ATP present according to the manufacturer’s instructions (Promega).
Western Blot analysis
The procedure was performed as previously described (15). Protein concentrations were determined by the Bio-Rad protein assay. Equal amounts of protein from whole cell lysates were solubilized in 2× SDS-sample buffer and separated on SDS-8–10% polyacrylamide gels. Blots were incubated with antibodies against PPARγ, ILK, p38 MAPK, and their phosphorylated forms (1:1000), as well as AP-2α, β, and δ (1:500). The blots were washed and followed by incubation with a secondary goat antibody raised against rabbit IgG conjugated to horseradish peroxidase (1:2000, Cell Signaling, Beverly, MA). The blots were washed, transferred to freshly made ECL solution (Amersham, Arlington, IL) for 1 min, and exposed to X-ray film. In controls, the primary antibodies were omitted or replaced with a control rabbit IgG.
Treatment with AP-2α small interfering RNA (siRNA)
The AP-2α siRNA (Cat No. sc-29200) and control nonspecific siRNA oligonucleotides (Cat No. sc-37007) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, California). For the transfection procedure, cells were grown to 60% confluence, and AP-2α and control siRNAs were transfected using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, oligofectamine reagent was incubated with serum–free medium for 10 min. Subsequently, a mixture of respective siRNA was added. After incubation for 15 min at room temperature, the mixture was diluted with medium and added to each well. The final concentration of siRNAs in each well was 100 nM. After culturing for 30 h, cells were washed, and resuspended in fresh oil-enriched medium culture media for an additional 24 or 48 h for luciferase assays or cell growth assays.
Immunoprecipitation assays (IP)
Protein lysates were prepared from cells treated with fish oil for 24 h by extraction in modified RIPA buffer [50 mM Tris (pH 7.4), 0.5% Nonidet P-40, 0.25% Na-deoxycholate, 125 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4 and protease inhibitor cocktail] on ice for 1 h. Cells were sonicated for 10 sec, lysates centrifuged at 12,000 × g for 15 min at 4 C, and the supernatant removed for IP. Samples containing 200 µg proteins were precleared for 30 min with 30 µl of Protein A/G Plus-agarose (sc-2003, Santa Cruz Biotechnology) and incubated for 1 h at 4 C with the appropriate antibodies (anti-PPARγ) or normal IgG preabsorbed to Protein A/G Plus-agarose. Immune complexes were collected after incubation overnight at 4 C, and washed once with lysis buffer and three times with PBS. Protein was eluted by boiling in 25 µl 2× SDS sample buffer [125 mM Tris (pH 6.8), 10% 2-mercaptoethanol, 4% SDS, 20% glycerol], and eluted proteins were analyzed by SDS-PAGE and Western blotting using the appropriate antibodies described above.
Transient transfection assays
The wild-type human ILK (pILK-Pr) and deletion ILK promoter constructs (dILK-pr) ligated to the luciferase reporter gene were a gift from Drs. Michalik and Desvergne at The University of Lausanne and have been reported previously (18). The ILK promoter construct contains approximately 730 base pairs (bp) of the 5’ flanking region of the human ILK gene connected to the pGL3 Basic Luciferase reporter vector (Promega). Briefly, NSCLC cells were seeded at a density of 5 ×105 cells/well in 6-well dishes and grown to 50 –60% confluence. For each well, 2 µg of the above ILK plasmid DNA constructs, with or without 0.2 µg of the internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector, were cotransfected into the cells using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions as described in our earlier work (19). After 24 h of incubation, cells were treated with or without rosiglitazone, linoleic acid, WY14643, or fish oil for 24 h, or with SB239023 for 2 h before exposure of the cells to fresh oil-enriched medium for an additional 24 h. The preparation of cell extracts and measurement of luciferase activities were carried out using the Dual-Luciferase Reporter Kit according to recommendations by the manufacturer (Promega). The assays for firefly luciferase activity and Renilla luciferase activity were performed sequentially in a Labsystems Luminoskan Ascent luminometer equipped with dual injectors. Changes in firefly luciferase activity were calculated and plotted after normalization with changes in Renilla luciferase activity within the same sample.
Electrophoretic mobility shift assays (EMSA)
EMSA experiments were performed as described before (16). The oligonucleotides used as probes were : Wild-type AP-2 (5’-TCCTCCCCGCCTCCGC-3’), Mutant AP-2 (5’-TCCTCtttGCCTCCGC -3’); Wild type Sp1 (5’-GGCCCCCACGGGCGGG-3’), Mutant Sp1 (5’-GGCCCCCACGGttGGG -3’); Wild type NF-κB (5’-ACGGGAGTTCCCCG-3’), Mutant NF-κB (5’- ACGttAGTTCCCCG-3’), which were based on the ILK promoter sequences (20) and consensus AP-2 binding motifs (5’-GATCGAACTGACCGCCCGCGGCCCGT-3’). The complimentary oligonucleotides were annealed and purified following the manufacturer’s protocol. The AP-2, Sp1, and NF-κB oligonucleotides were end-labeled with [γ-32P] ATP using T4 polynucleotide kinase as recommended by the manufacturer. Nuclear proteins (5 µg) were first incubated under binding conditions [10 mM HEPES, Tris-HCL (pH 7.9), 50 mM KCl, 0.1 mM EDTA, 1 mM DTT, 12% (vol/vol) glycerol, and 2 µg poly (dI-dC)] for 10 min, then [γ-32P] ATP probe was added for another 20 min at room temperature in a final volume of 20 µl. For cold competition, a 100-fold excess of the respective unlabeled consensus oligonucleotides was incubated for 15 min before adding the probe. The same amount of mutated oligonucleotides mixed with the probe was used as another control. All of these were performed in the same binding conditions as described before. Afterwards, the protein-DNA complexes were electrophoresed on a native 4.5% polyacrylamide gel at 150 volts using 1× Tris-Glycine buffer. Each gel was then dried and subjected to autoradiography at −80 C.
RESULTS
Fish oil decreases NSCLC cell proliferation which is associated with inhibition of ILK protein expression through PPARγ activation
We began by examining the effects of fish oil on NSCLC cell proliferation. As shown in Figure 1A, fish oil resulted in a significant decrease in cell proliferation of two NSCLC cell lines (H1838 and H1792) as compared to the control cells (no fish oil) with maximal effects noted at 15 µg/ml. Note that two human bronchial epithelial cell lines (BEAS-2B and 16-HBE) showed no inhibition in response to fish oil (Fig. 1A).
Figure 1. Fish oil decreases NSCLC cell growth and ILK protein expression through PPARγ.

A, NSCLC cells (H1838 and H1792) and normal bronchial epithelial cells (BEAS-2B and 16-HBE) were plated in different concentrations of fish oil enriched medium for up to 48 h. Afterwards, viable cells were detected using Cell Titer-Glo Luminescent Cell Viability Assay Kit according to the protocol of the manufacturer. Actin was used as internal control for loading purpose. Con, indicates untreated control cells. B, Cellular protein was isolated from H1838 cells that were cultured with increased concentrations of fish oil as indicated for 24 h followed by western blot analysis with antibodies against ILK protein. Actin served as internal control for normalization purposes. C, Cellular protein was isolated from several NSCLC cell lines (H1838, A549, H522 and H1792) that were cultured with fish oil (10 µg/ml) for up to 24, followed by Western Blot analysis with antibodies against ILK protein. D, Cellular protein was isolated from normal epithelial cells (BEAS-2B and 16-HBE) and NIH-3T3 cells that were cultured with fish oil (10 µg/ml) for up to 24, followed by Western Blot analysis with antibodies against ILK protein. E, Cellular protein was isolated from H1838 cells treated with GW9662 (20 µM) for 2 h before exposure of the cells to linoleic acid (10 µM) or fish oil (10 µg/ml) for an additional 24 h, then subjected to Western Blot analysis with antibodies against ILK. F, Cellular protein was isolated from H1838 cells transfected with control or PPARγ siRNA (100 nM each) for 30 h before exposure of the cells to fish oil (10 µg/ml) enriched medium for an additional 24 h. Afterwards, Western blot analysis was performed using polyclonal antibodies against PPARγ and ILK. Actin served as internal control for normalization purposes. Con, indicates untreated control cells.
The ILK signal pathway has been implicated in the regulation of anchorage-dependent cell growth/survival, cell cycle progression, invasion and migration, and tumor angiogenesis (21, 22). We found that fish oil inhibited ILK protein levels in a dose-dependent manner in H1838 NSCLC cells (Fig. 1B). Similar results were also observed in several other NSCLC cell lines (Fig. 1C); while fish oil had little effect on ILK protein expression in BEAS-2B, 16-HBE and NIH3T3 fibroblast cells (Fig. 1D).
Since fish oil can activate nuclear receptors such as PPARγ, we examined if inhibition of ILK expression by fish oil was mediated by PPARγ. To this end, we tested the cells with GW9662, a specific PPARγ antagonist. As shown in Fig. 1E, the effects of fish oil on ILK expression were eliminated in the presence of GW9662, and this was confirmed by PPARγ siRNA (Fig. 1F). Note that fish oil induced PPARγ protein expression (Fig. 1F); this effect was due to upregulation of PPARγ gene transcription since fish oil stimulated PPARγ gene promoter activity in separate experiments (not shown). Similar results were also observed with another PPARγ natural activator, linoleic acid (Fig. 1E).
Overexpression of ILK overcomes, while silencing of ILK enhances the inhibitory effect of fish oil on cell growth
Having established that fish oil may act via affecting expression of ILK, we tested the role of ILK further. Specifically, we tested if overexpression of ILK blocked the effect of fish oil on cell growth. We were transiently transfected cells with ILK kinase-inactive (ILK-S343A) and ILK kinase-hyperactive (ILK-S343D) cDNAs. Exogenous high ILK protein expression was confirmed by Western blot with anti-ILK antibody in ILK-S343A and ILK-S343D treatment groups (Figure 2A, upper panel). Cell viability assays showed that ILK kinase-hyperactive (ILK-S343D) cDNA opposed the inhibitory effect of fish oil on cell growth (Figure 2A, lower panel), whereas the inactive ILK slightly enhanced the effect of fish oil. Knockdown of ILK with siRNA reduced ILK protein production (Fig. 2B, upper panel) and enhanced the inhibitory effect of fish oil on cell growth as determined by CellTiter-Glo Luminescent Cell Viability Assay (Fig. 2B, lower panel).
Figure 2. The role of ILK in mediating the inhibitory effect of fish oil on cell growth.
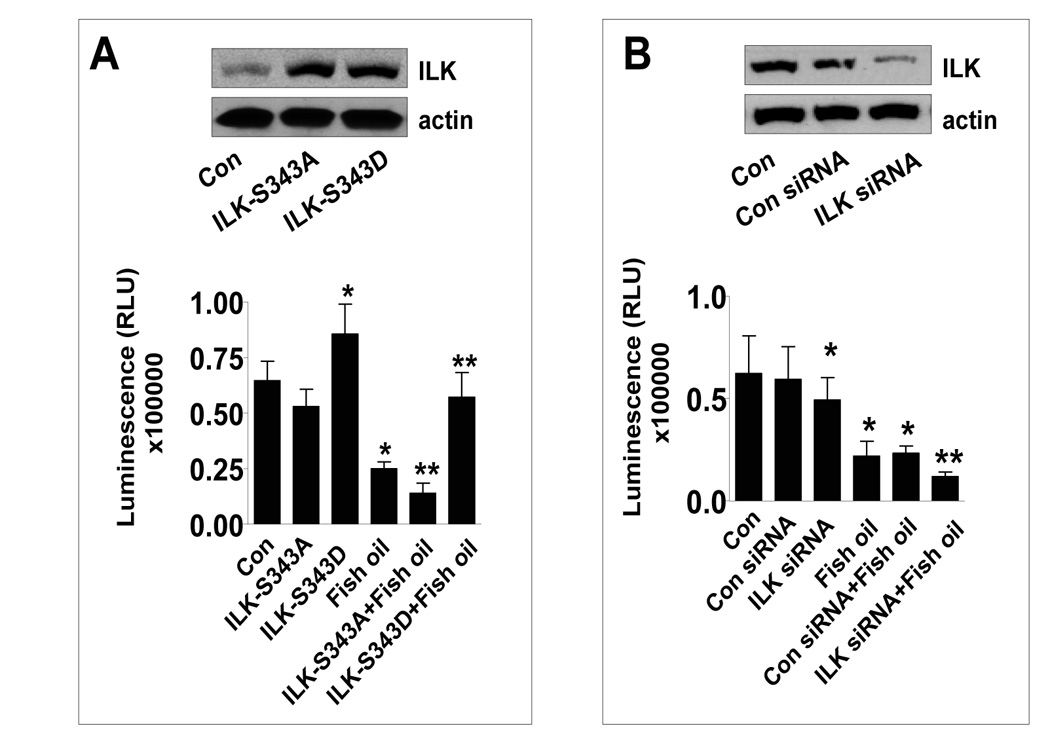
A, H1838 cells were transfected with the inactive (ILK-S343A) and superactive ILK cDNA (ILK-S343D) using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h of incubation, cells were treated with or without (15 µg/ml) for an additional 24 h. Afterwards, viable cells were detected using Cell Titer-Glo Luminescent Cell Viability Assay Kit according to the protocol of the manufacturer. The insert in upper panel represents Western blot results for ILK protein. Actin was used as internal control for loading purpose. Con, indicates untreated control cells. B, H1838 cells were transfected with control or ILK siRNA (100 nM) for 40 h followed by exposing the cells to fish oil (15 µg/ml) for an additional 24 h. Afterwards, viable cells were detected using Cell Titer-Glo Luminescent Cell Viability Assay Kit according to the protocol of the manufacturer. The insert in upper panel represents Western blot results for ILK protein. Actin was used as internal control for loading purpose. Con, indicates untreated control cells.
The inhibitor of p38 MAPK, SB239023, abrogates the effect of fish oil on ILK, whereas the inhibitor of ERK, PD98059, has no effect
We and others have shown that PPARγ ligands activate kinase signaling related to p38 MAPK and extracellular signal-regulated kinase½ (ERK1/2) in several cell systems including lung cancer (23–25). Therefore, we tested whether regulation of ILK expression by fish oil was related to p38 MAPK and ERK activation. We found that fish oil increased the phosphorylation of p38 MAPK (Fig. 3A) and ERK (Fig. 3B). However, the inhibitors of p38 MAPK, SB239023 and SB203580 (not shown), blocked the effect of fish oil on ILK protein expression (Fig. 3C) and cell growth (not shown). The ERK1/2 inhibitor, PD98059 had no effect (Fig. 3D). This indicated that activation of p38 MAPK, but not that of ERK, was involved in the regulation of ILK by fish oil.
Figure 3. The role of p38 MAPK and ERK signaling in ILK protein expression in the setting of fish oil treatment.

A, Cellular protein was isolated from H1838 cells treated with fish oil in the indicated time period. Afterwards, Western blot analysis were performed with antibodies against phosphor-p38 MAPK and total p38 MAPK. Actin was used as internal control for loading purposes. Con, indicates untreated control cells. B, Cellular protein was isolated from H1838 cells treated with fish oil in the indicated time period. Afterwards, Western blot analysis was performed with antibodies against phosphor-ERK1/2 and total ERK1/2. Actin was used as internal control for loading purposes. Con, indicates untreated control cells. C, Cellular protein was isolated from H1838 cells treated with SB239023 (10 µM) for 2 h before exposure of the cells to fish oil enriched medium (10 µg/ml) for an additional 24 h, then subjected to Western Blot analysis for ILK protein. D, Cellular protein was isolated from H1838 cells treated with PD98059 (25 µM) for 2 h before exposure of the cells to fish oil enriched medium (10 µg/ml) for an additional 24 h, then subjected to Western Blot analysis for ILK protein. Actin was used as internal control for loading purposes. Con, indicates untreated control cells.
Fish oil reduces ILK promoter activity and this is abolished by the p38 MAPK inhibitor
We next examined whether the effects of fish oil on ILK expression occurred at the transcriptional level. As shown in Figure 4A, the ILK promoter contains multiple transcription factor binding sites including AP-2, NF-κB, and Sp1, among others. We found that H1838 cells transfected with a wild-type ILK promoter luciferase reporter construct showed decreased promoter activity when exposed to fish oil, linoleic acid, and rosiglitazone, a synthetic PPARγ ligand, while little effect was observed in cells exposed to WY14643, a PPARα ligand (Fig. 4B). The inhibitory effect of fish oil on ILK promoter activity was abolished by the inhibitor of p38 MAPK, SB239023 (Fig. 4C).
Figure 4. Fish oil decreases ILK promoter activity in human lung carcinoma cells.

A, The human ILK wild type and deletion promoter constructs schematics are presented. These regions contain several transcription factor binding sites including AP-2, Sp1 and NF-κB. B. H1838 cells (1×105 cells) were cotransfected with a wild type human ILK promoter reporter constructs (shown in A) ligated to a luciferase reporter gene and an internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector as described in Materials and Methods for 24 h using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h of incubation, cells were treated with vehicle control (Con), rosiglitazone (10 µM), fish oil (10 µg/ml), linoleic acid (10 µM) and WY146463 (15 µM) for an additional 24 h. Con, indicates untreated control cells. C, H1838 cells (1×105 cells) were cotransfected with a wild type human ILK promoter reporter construct (shown in A) ligated to luciferase reporter gene and an internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector as described in Materials and Methods for 24 h using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h of incubation, cells were treated with SB239023 (10 µM) for 2 h before exposure of the cells to fish oil (10 µg/ml) for an additional 24 h. The ratio of firefly luciferase to renilla luciferase activity was quantified as described in Materials and Methods. The bars represent the mean ± SD of at least three independent experiments for each condition. * indicates significant increase of activity as compared to controls. ** indicates significance of combination treatment as compared with fish oil alone (P < 0.05). Con, indicates untreated control cells.
Fish oil induces AP-2 DNA binding activity in the ILK promoter through activation of PPARγ
Having established the importance of ILK, we turned our attention to transcription factors affecting its expression. Transcription factors AP-2 and NF-κB have been inversely correlated with tumor cell growth. PPARγ ligand inhibition of cancer cell proliferation has been associated with inactivation of NF-κB and activation of AP-2 in several studies (26, 27). We found that H1838 cells treated with fish oil (10 µg/ml) for 24 h show increased AP-2 nuclear protein binding activity as compared to the solvent controls (Fig. 5A) In contrast, fish oil slightly decreased Sp1 and NF-κB DNA binding activity (Fig. 5B and C). The specific bands for AP-2, Sp1, and NF-κB were attenuated by a 100-fold molar excess of unlabeled respective oligonucleotides, but were not inhibited by a mutated unlabeled AP-2, Sp1, or NF-κB oligonucleotides (mAP-2, mSp1, mNF-κB). Consistent with the role of PPARγ, we found that GW9662 blocked the stimulatory effect of fish oil on AP-2 DNA binding activity (Fig. 5D). Moreover, consistent with the stimulatory effect of fish oil on PPARγ protein expression (Fig. 1F); we showed that fish oil enhanced protein complex formation between AP-2α and PPARγ (Fig. 5E).
Figure 5. Effect of fish oil on AP-2, Sp1 and NF-κB binding activities.
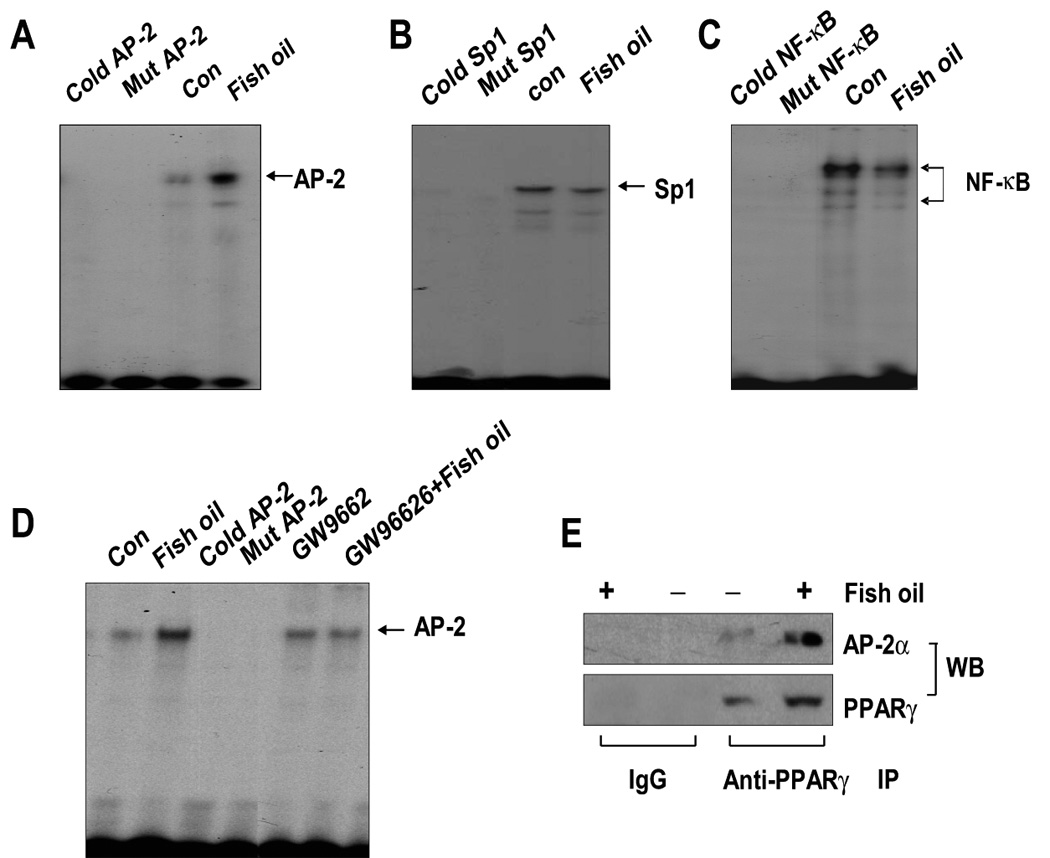
A, Oligonucleotides containing the AP-2 (A), Sp1 (B) and NF-κB (C) sites were end-labeled with [γ-32P]-ATP and incubated with nuclear extracts (5 µg) from H1838 cells treated with fish oil (10 µg/ml) for an additional 24 h. For competition assays, a molar excess (×100) of consensus Sp1 (Cold Sp1) or NF-κB (Cold NF-κB) or AP-2 (Cold AP-2) oligonucleotide were added to the binding reaction. Oligonucleotides containing a mutated Sp1 (Mut Sp1) or NF-κB (Mut NF-κB) or AP-2 (Mut AP-2) site that were end-labeled with γ32P-ATP were used to confirm the binding specificity. Con, indicates untreated control cells. D, Oligonucleotides containing the AP-2 site was end-labeled with [γ-32P]-ATP and incubated with nuclear extracts (5 µg) from H1838 cells treated with GW9662 (20 µM) for 2 h before exposure of the cells to fish oil (10 µg/ml) for an additional 24 h. For competition assays, a molar excess (×100) of consensus AP-2 (Cold AP-2) oligonucleotide was added to the binding reaction. Oligonucleotides containing a mutated AP-2 (Mut AP-2) site that was end labeled with γ32P-ATP were used to confirm the binding specificity. Con, indicates untreated control cells. E, H1838 cells were treated with fish oil (10 µg/ml) for 24 h and cell lysates were immunoprecipitated with either IgG, or anti-PPARγ. Immunoprecipitates were separated by SDS-PAGE in 10% Tris-glycine gels, transferred onto nitrocellulose and Western blotting (WB) was carried out with either anti-PPARγ or anti-AP-2α antibody.
Blockade of AP-2α abrogates the effect of fish oil on ILK expression and on cell growth
To further test the role of AP-2 in mediating the effect of fish oil on ILK expression, we examined the effect of fish oil on AP-2 family protein levels. We found that fish oil increased the expression of AP-2α subunits, but had no effect on AP-2γ and AP-2β (Fig. 6A). The stimulatory effect of fish oil on AP-2α protein expression was blocked by GW9662 (Fig. 6B) and by the inhibitor of p38 MAPK (Fig. 6C). In addition, the inhibitory effect of fish oil on ILK promoter activity was abolished in cells with silencing of AP-2α (Fig. 6D), and with a deletion ILK promoter construct most likely lacking AP-2 sites (Fig. 6E). This suggests that inhibition of ILK expression by fish oil occurs at the transcriptional level through activation of p38 MAPK and induction of AP-2α in NSCLC cells.
Figure 6. The role of transcription factor AP-2α in inhibition of ILK expression by fish oil.

A, Cellular proteins were isolated from H1838 cells treated with fish oil (10 µg/ml) for the indicated time period. Afterwards, Western Blot analyses were performed using polyclonal antibodies against AP-2α, β, γ. B, H1838 cells were treated with GW9662 (20 µM) for 2 h before exposure of the cells to fish oil (10 µg/ml) for an additional 24 h followed by Western blot analysis for AP-2α protein. C, H1838 cells were treated with SB239023 (10 µM) for 2 h before exposure of the cells to fish oil (10 µg/ml) for an additional 24 h followed by Western blot analysis for AP-2α protein. D, H1838 cells were transfected with control or AP-2α siRNA (100 nM) together with a wild type ILK promoter constructs (−267/+463 bp) for 30 h, followed by exposing the cells to fish oil (10 µg/ml) for an additional 24 h. E, H1838 cells were transfected with a truncated human ILK promoter reporter construct (shown in Fig.4A) ligated to luciferase reporter gene and an internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector as described in Materials and Methods for 24 h using the oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h of incubation, cells were treated with vehicle control (Con) and fish oil (10 µg/ml) for an additional 24 h. Con, indicates untreated control cells. F, H1838 cells were transfected with control or AP-2α siRNA (100 nM) for 30 h before exposure of the cells to fish oil (10 µg/ml) for an additional 48 h. Afterwards, viable cells were detected using Cell Titer-Glo Luminescent Cell Viability Assay Kit according to the protocol of the manufacturer. Actin was used as internal control for loading purpose. Con, indicates untreated control cells. The insert on the top showed the Western blot result for AP-2α protein production. G, H1838 cells were transfected with control and AP-2 expression reporter constructs [SP(RSV)AP-2], and an internal control phRL-TK Synthetic Renilla Luciferase Reporter Vector as described in Material and Methods section for 24 h before exposure of the cells to fish oil (10 µg/ml) for an additional 48 h. The insert in upper panel represents Western blot results for AP-2α protein. Con, indicates untreated cells. The ratio of firefly luciferase to renilla luciferase activity was quantified as described in Materials and Methods. The bars represent the mean ± SD of at least four independent experiments for each condition. * indicates significant increase of activity as compared to controls. ** indicates significance of combination treatment as compared to the nicotine alone (p<0.05). Con, indicates untreated control cells.
Next, we assessed the role of AP-2α in mediating the inhibitory effect of fish oil on lung cancer cell growth. We showed that cells transfected with AP-2α siRNA diminished the inhibitory effects of fish oil on cell growth as determined CellTiter-Glo Luminescent Cell Viability Assay (Fig. 6F, lower panel). The control siRNA had no effect. Note that AP-2α siRNA blocked protein production of AP-2α (Fig. 6F, upper panel). On the contrary, cells transfected with AP-2 expression vector [SP(RSV)AP-2] showed enhanced inhibition of the effect of fish oil on cell proliferation (Fig. 6G, lower panel). Note that cells transfected with [SP(RSV)AP-2] showed increased AP-2 protein levels, while the control vector had no effect (Fig. 6G, upper panel).
DISCUSSION
The expression of PPARγ and the effects of dietary PPARγ ligands such as fish oil on cell growth have been extensively studied in many carcinoma cell types including lung cancer (3, 7). However, the exact mechanisms mediating the effects of dietary PPARγ ligands on cell growth inhibition are not fully understood. Fish oil, which is an activator of PPARγ, contains polyunsaturated fatty acids (PUFAs) of the ω3 and ω6 family, and is believed to have anti-cancer properties (4–6). In this study, we show that fish oil inhibits human lung carcinoma cell growth through suppression of ILK gene expression suggesting that ILK represents a target for PPARγ ligands. Interestingly, fish oil had little effect on the growth of normal cells and this seems to correlate with the relative lower PPARγ protein levels found in normal cells.
The concentrations of fish oil used in this study are similar or even lower than those reported by others showing inhibition of cell growth and invasion (28, 29). One study found that fish oil at a dose range between 1 to 100 µg/ml significantly inhibited growth of human hepatoma cells (28). The maximal dose of fish oil tolerated by patients seems to be around 6 g per day (30). Therefore, the concentrations of fish oil used here are much lower than the 1.2 mg/ml found in human blood in those studies.
Alterations in integrin function and ILK expression enhance the aggressive capabilities of several tumor cells (31). Furthermore, ILK overexpression has been correlated with tumor progression (13, 32). ILK is a ubiquitously expressed serine-threonine protein kinase capable of interacting with the cytoplasmic domain of the integrin β1 and plays a critical role in integrin-dependent cell adhesion, spreading, and cell shape change (12). ILK has been implicated in the regulation of cell growth/survival, cell cycle progression, invasion and tumor angiogenesis (21, 22). Overexpression ILK results in oncogenic transformation and progression to invasive and metastatic phenotypes (13, 14). ILK and the activated form of PI3-K down stream signal Akt are mutually associated with poor prognosis in NSCLC, and the simultaneous overexpression of these proteins is an independent prognostic factor (33). Thus, ILK-pAkt signaling may provide a novel prognostic marker and therapeutic target for NSCLC (33). We found that knockdown of ILK resulted in further inhibition of cell growth, whereas exogenous expression of ILK blocked the inhibitory effect of fish oil on cell growth. Interestingly, silencing of ILK was not as effective as fish oil in inhibiting tumor cell growth. This is probably due to the fact that other kinases such as Akt, ERK, tumor promoters such as PGE2/COX-2, and extracellular matrixes like fibronectin may also drive NSCLC cell growth (34, 35). These effects may be unrelated to ILK, but affected by fish oil.
Consistent with other reports (7, 36), we found that the effect of fish oil on the expression of ILK was dependent on activation of PPARγ. In other work, eicosapentaenoic acid (EPA), a component of fish oil, induced a PPAR response element reporter assay and stimulated cell growth in colon cancer cells, which was inhibited by GW9662, a PPARγ antagonist. In contrast, overexpression of PPARγ enhanced the effect of EPA (36). However, PPARγ-independent mechanisms have also been reported (37). These discrepancies may be due to the different cells studied and the concentrations used for these reagents.
ILK has been linked to p38 MAPK and ERK signaling pathways in many cell systems (31, 38). We demonstrated that inhibition of p38 MAPK, but not ERK, blocked the inhibitory effect of fish oil on ILK. Consistent with this, another study showed that ILK played an important role in interleukin-1α-induced adhesion and invasion of pancreatic cancer cells through p38 MAPK signaling (31). Thus, our results suggest that ILK is downstream of p38 MAPK.
Having demonstrated the important role of ILK, we investigated whether downregulation of ILK by fish oil reflected inhibition of transactivation of the ILK gene. Our results show that fish oil decreased ILK promoter activity through activation of p38 MAPK. Several transcription factor binding sites within regions of the ILK promoter have been characterized including regulatory elements for AP-2, NF-κB, Sp1 and others (20). As such, we evaluated the possibility that these sites might play a role in inhibition of ILK expression by fish oil. The effect of fish oil on ILK promoter activity was blocked when the upstream portion of the ILK gene promoter containing AP-2 sites was deleted, and in cells transfected with AP-2α siRNA suggesting that AP-2α plays a critical role in mediating the effect of fish oil on ILK gene expression. Furthermore, increased AP-2α expression and AP-2α DNA binding activity were responsible for the inhibitory effect of fish oil on ILK expression. On the contrary, silencing AP-2α blocked the inhibitory effect of fish oil on NSCLC cell proliferation further indicating the tumor suppressive property of this transcription factor. Similarly, another study demonstrated that targeting constitutive expression of AP-2α in colon cancer cells with siRNA resulted in an increase in their invasive potential, downregulation of E-cadherin, and increased expression of matrix metalloproteinase-9 (39). The AP-2 transcription factor is involved in the regulation of cell proliferation, differentiation, apoptosis and carcinogenesis. AP-2α functions as a tumor suppressor in several cancers (40). Overexpression of AP-2α significantly decreased cell proliferation and invasion concomitant to the up-regulation of p27 expression in pancreatic cancer cells (41). Also, transient expression of AP-2α resulted in the induction of PTEN transcription in AP-2α-negative colon cancer cells (42).
Taken together, our results demonstrate that fish oil inhibits ILK expression through PPARγ signaling that results in activation of p38 MAPK and induction of AP-2α protein expression, followed by AP-2 DNA binding activity. In turn, the inhibition of ILK expression results in inhibition of NSCLC cell proliferation. This study unveils a novel mechanism by which fish oil inhibits human lung cancer cell growth.
ACKNOWLEDGEMENT
We are grateful to Drs. Michalik and Desvergne, Center for Intergrative Genomics at University of Lausanne in Switzerland, for providing the ILK promoter constructs; and Dr. ShiYong Sun (Whinship Cancer Institutes, Emory University) for providing H1792 and H522 cells. This work was supported by American Thoracic Society (ATS)/LUNGevity Foundation Partnership Grant LC-06-004, by Emory University Research Fund 2-55016, and by the NIH/NCI CA123104 (S. W. H), and by a Merit Review Grant from the Department of Veterans Affairs and NIH/NCI CA116812 (J. R.).
REFERENCE
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: diagnosis and management. Am Fam Physician. 2007;75:56–63. [PubMed] [Google Scholar]
- 3.Reddy BS, Patlolla JM, Simi B, Wang SH, Rao CV. Prevention of colon cancer by low doses of celecoxib, a cyclooxygenase inhibitor, administered in diet rich in omega-3 polyunsaturated fatty acids. Cancer Res. 2005;65:8022–8027. doi: 10.1158/0008-5472.CAN-05-0212. [DOI] [PubMed] [Google Scholar]
- 4.Nowak J, Weylandt KH, Habbel P, et al. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis. 2007;28:1991–1995. doi: 10.1093/carcin/bgm166. [DOI] [PubMed] [Google Scholar]
- 5.Hardman WE. Dietary canola oil suppressed growth of implanted MDA-MB 231 human breast tumors in nude mice. Nutr Cancer. 2007;57:177–183. doi: 10.1080/01635580701277445. [DOI] [PubMed] [Google Scholar]
- 6.Sauer LA, Blask DE, Dauchy RT. Dietary factors and growth and metabolism in experimental tumors. J Nutr Biochem. 2007;18:637–649. doi: 10.1016/j.jnutbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Trombetta A, Maggiora M, Martinasso G, Cotogni P, Canuto RA, Muzio G. Arachidonic and docosahexaenoic acids reduce the growth of A549 human lung-tumor cells increasing lipid peroxidation and PPARs. Chem Biol Interact. 2007;165:239–250. doi: 10.1016/j.cbi.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 8.Kliewer SA, Willson TM. The nuclear receptor PPARgamma - bigger than fat. Curr Opin Genet Dev. 1998;8:576–581. doi: 10.1016/s0959-437x(98)80014-2. [DOI] [PubMed] [Google Scholar]
- 9.Chene G, Dubourdeau M, Balard P, et al. n-3 and n-6 polyunsaturated fatty acids induce the expression of COX-2 via PPARgamma activation in human keratinocyte HaCaT cells. Biochim Biophys Acta. 2007;1771:576–589. doi: 10.1016/j.bbalip.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Schopfer FJ, Lin Y, Baker PR, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome vfproliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 12.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Costello PC, Pham NA, et al. Integrin-linked kinase inhibitor KP-392 demonstrates clinical benefitsin an orthotopic human non-small cell lung cancer model. J Thorac Oncol. 2006;1:771–779. [PubMed] [Google Scholar]
- 14.Cordes N. Overexpression of hyperactive integrin-linked kinase leads to increased cellular radiosensitivity. Cancer Res. 2004;64:5683–5692. doi: 10.1158/0008-5472.CAN-04-1056. [DOI] [PubMed] [Google Scholar]
- 15.Han S, Ritzenthaler JD, Wingerd B, Roman J. Activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) increases the expression of prostaglandin E2 receptor subtype EP4. The roles of phosphatidylinositol 3-kinase and CCAAT/enhancer-binding protein beta. J Biol Chem. 2005;280:33240–33249. doi: 10.1074/jbc.M507617200. [DOI] [PubMed] [Google Scholar]
- 16.Fox PL, DiCorleto PE. Fish oils inhibit endothelial cell production of platelet-derived growth factor-like protein. Science. 1988;241:453–456. doi: 10.1126/science.3393911. [DOI] [PubMed] [Google Scholar]
- 17.Williams T, Tjian R. Analysis of the DNA-binding and activation properties of the human transcription factor AP-2. Genes Dev. 1991;5:670–682. doi: 10.1101/gad.5.4.670. [DOI] [PubMed] [Google Scholar]
- 18.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 19.Han S, Sidell N, Fisher PB, Roman J. Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells. Clin Cancer Res. 2004;10:1911–1919. doi: 10.1158/1078-0432.ccr-03-0985. [DOI] [PubMed] [Google Scholar]
- 20.Melchior C, Kreis S, Janji B, Kieffer N. Promoter characterization and genomic organization of the gene encoding integrin-linked kinase 1. Biochim Biophys Acta. 2002;1575:117–122. doi: 10.1016/s0167-4781(02)00247-6. [DOI] [PubMed] [Google Scholar]
- 21.Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- 22.Koul D, Shen R, Bergh S, et al. Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol Cancer Ther. 2005;4:1681–1688. doi: 10.1158/1535-7163.MCT-05-0258. [DOI] [PubMed] [Google Scholar]
- 23.Yano M, Matsumura T, Senokuchi T, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–1451. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 24.Li M, Lee TW, Mok TS, Warner TD, Yim AP, Chen GG. Activation of peroxisome proliferator-activated receptor-gamma by troglitazone (TGZ) inhibits human lung cell growth. J Cell Biochem. 2005;96:760–774. doi: 10.1002/jcb.20474. [DOI] [PubMed] [Google Scholar]
- 25.Han S, Zheng Y, Roman J. Rosiglitazone, an Agonist of PPARgamma, Inhibits Non-Small Cell Carcinoma Cell Proliferation In Part through Activation of Tumor Sclerosis Complex-2. PPAR Res. 2007;2007:29632. doi: 10.1155/2007/29632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa V, Foti D, Paonessa F, et al. The insulin receptor: a new anticancer target for peroxisome proliferator-activated receptor-{gamma} (PPAR{gamma}) and thiazolidinedione-PPAR{gamma} agonists. Endocr Relat Cancer. 2008;15:325–335. doi: 10.1677/ERC-07-0226. [DOI] [PubMed] [Google Scholar]
- 27.Patel KM, Wright KL, Whittaker P, Chakravarty P, Watson ML, Ward SG. Differential modulation of COX-2 expression in A549 airway epithelial cells by structurally distinct PPAR(gamma) agonists: evidence for disparate functional effects which are independent of NF-(kappa)B and PPAR(gamma) Cell Signal. 2005;17:1098–1110. doi: 10.1016/j.cellsig.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Pandey MK, Pant AB, Das M. In vitro cytotoxicity of polycyclic aromatic hydrocarbon residues arising through repeated fish fried oil in human hepatoma Hep G2 cell line. Toxicol In Vitro. 2006;20:308–316. doi: 10.1016/j.tiv.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Istfan NW, Chen ZY, Rex S. Fish oil slows S phase progression and may cause upstream shift of DHFR replication origin ori-beta in CHO cells. Am J Physiol Cell Physiol. 2002;283:C1009–C1024. doi: 10.1152/ajpcell.00614.2001. [DOI] [PubMed] [Google Scholar]
- 30.Cerchietti LC, Navigante AH, Castro MA. Effects of eicosapentaenoic and docosahexaenoic n-3 fatty acids from fish oil and preferential Cox-2 inhibition on systemic syndromes in patients with advanced lung cancer. Nutr Cancer. 2007;59:14–20. doi: 10.1080/01635580701365068. [DOI] [PubMed] [Google Scholar]
- 31.Sawai H, Okada Y, Funahashi H, et al. Integrin-linked kinase activity is associated with interleukin-1 alpha-induced progressive behavior of pancreatic cancer and poor patient survival. Oncogene. 2006;25:3237–3246. doi: 10.1038/sj.onc.1209356. [DOI] [PubMed] [Google Scholar]
- 32.Imanishi Y, Hu B, Jarzynka MJ, et al. Angiopoietin-2 Stimulates Breast Cancer Metastasis through the {alpha}5{beta}1 Integrin-Mediated Pathway. Cancer Res. 2007;67:4254–4263. doi: 10.1158/0008-5472.CAN-06-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamura M, Yamaji S, Nagashima Y, et al. Prognostic value of integrin beta1-ILK-pAkt signaling pathway in non-small cell lung cancer. Hum Pathol. 2007;38:1081–1091. doi: 10.1016/j.humpath.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Han S, Ritzenthaler JD, Sitaraman SV, Roman J. Fibronectin increases matrix metalloproteinase 9 expression through activation of c-Fos via extracellular-regulated kinase and phosphatidylinositol 3-kinase pathways in human lung carcinoma cells. J Biol Chem. 2006;281:29614–29624. doi: 10.1074/jbc.M604013200. [DOI] [PubMed] [Google Scholar]
- 35.Marks JL, Gong Y, Chitale D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–5528. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allred CD, Talbert DR, Southard RC, Wang X, Kilgore MW. PPARgamma1 as a molecular target of eicosapentaenoic acid in human colon cancer (HT-29) cells. J Nutr. 2008;138:250–256. doi: 10.1093/jn/138.2.250. [DOI] [PubMed] [Google Scholar]
- 37.Yee LD, Young DC, Rosol TJ, Vanbuskirk AM, Clinton SK. Dietary (n-3) polyunsaturated fatty acids inhibit HER-2/neu-induced breast cancer in mice independently of the PPARgamma ligand rosiglitazone. J Nutr. 2005;135:983–988. doi: 10.1093/jn/135.5.983. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Ikegami T, Honda A, et al. Involvement of integrin-linked kinase in carbon tetrachloride-induced hepatic fibrosis in rats. Hepatology. 2006;44:612–622. doi: 10.1002/hep.21315. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz B, Melnikova VO, Tellez C, et al. Loss of AP-2alpha results in deregulation of E-cadherin and MMP-9 and an increase in tumorigenicity of colon cancer cells in vivo. Oncogene. 2007;26:4049–4058. doi: 10.1038/sj.onc.1210193. [DOI] [PubMed] [Google Scholar]
- 40.Pellikainen JM, Kosma VM. Activator protein-2 in carcinogenesis with a special reference to breast cancer--a mini review. Int J Cancer. 2007;120:2061–2067. doi: 10.1002/ijc.22648. [DOI] [PubMed] [Google Scholar]
- 41.Fauquette V, Aubert S, Groux-Degroote S, et al. Transcription factor AP-2alpha represses both the mucin MUC4 expression and pancreatic cancer cell proliferation. Carcinogenesis. 2007;28:2305–2312. doi: 10.1093/carcin/bgm158. [DOI] [PubMed] [Google Scholar]
- 42.Choi HJ, Chung TW, Kim SJ, et al. The AP-2alpha transcription factor is required for the ganglioside GM3-stimulated transcriptional regulation of a PTEN gene. Glycobiology. 2008;18:395–407. doi: 10.1093/glycob/cwn016. [DOI] [PubMed] [Google Scholar]