

Abstract
Hexokinase isoforms I and II bind to mitochondrial outer membranes in large part by interacting with the outer membrane Voltage-Dependent Anion Channel (VDAC). This interaction results in a shift in the susceptibility of mitochondria to pro-apoptotic signals that are mediated through Bcl2-family proteins. The upregulation of hexokinase II expression in tumor cells is thought to provide both a metabolic benefit and an apoptosis suppressive capacity that gives the cell a growth advantage and increases its resistance to chemotherapy. However, the mechanisms responsible for the anti-apoptotic effect of hexokinase binding and its regulation remain poorly understood. We hypothesize that hexokinase competes with Bcl2 family proteins for binding to VDAC to influence the balance of pro-and anti-apoptotic proteins that control outer membrane permeabilization. Hexokinase binding to VDAC is regulated by protein kinases, notably glycogen synthase kinase (GSK)-3β and protein kinase C (PKC) -ε. In addition, there is evidence that the cholesterol content of the mitochondrial membranes may contribute to the regulation of hexokinase binding. At the same time, VDAC associated proteins are critically involved in the regulation of cholesterol uptake. A better characterization of these regulatory processes is required to elucidate the role of hexokinases in normal tissue function and to apply these insights for optimizing cancer treatment.
Introduction
The voltage dependent anion channel (VDAC), also known as mitochondrial porin, is the most abundant protein of the outer mitochondrial membrane (OMM) and the major channel for the exchange of metabolites and ions between the mitochondria and other cellular compartments. As a consequence of its location at the boundary between mitochondria and cytosol, VDAC also is a binding partner for proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities. Notably, VDAC has been reported to bind pro- and anti-apoptotic proteins of the Bcl-2 family that regulate the permeability of the outer membrane [1], it may interact with mitochondrial proteins that form contact sites between the inner and outer membranes [2] and that may generate the mitochondrial permeability transition pore complex [3], and it binds to cytosolic proteins, such as hexokinase I and hexokinase II [4, 5], which influences not only its metabolic function, but also its capacity to respond to Bcl-2 family proteins and its role in apoptosis [6]. How these different functions of VDAC are regulated remains a matter of much debate, but there is evidence that multiple protein kinases are involved in its control, resulting from the phosphorylation of VDAC itself or its binding partners. In addition, the functional state of VDAC and its capacity to interact with proteins such as hexokinase isoforms may depend on the constituents of the membrane in which it is embedded. In this brief review, we will discuss the regulation of VDAC function with an emphasis on the regulation of its interaction with hexokinase and Bcl2-family proteins.
Interactions between hexokinase and VDAC
Early studies on VDAC characterized it as the outer membrane hexokinase-binding protein [7, 8]. Hexokinase isoforms I and II both bind to the outer mitochondrial membrane (OMM) and there is an abundance of evidence that this binding is greatly enhanced by their interaction with VDAC, more specifically the most abundant isoform, VDAC-1. Hexokinase I is highly expressed in brain and hexokinase II is prevalent in cardiac muscle [4]. In addition, hexokinase II is highly overexpressed in many tumor cells where it contributes to the tumor’s enhanced capacity for oxidative glycolysis, commonly referred to as the Warburg effect [5, 6]. However, hexokinase binds equally well to heterologous mitochondria from cells or tissues such as liver that do not normally express these isoforms. Moreover, hexokinase binds effectively to VDAC-1 reconstituted in lipid membranes and affects its ion permeability characteristics, promoting a closed state of the channel [1]. Both hexokinase I and II possess a hydrophobic N-terminal sequence of 15 amino acids that has features compatible with an amphipathic α-helix and may be at least partly inserted into the membrane [9]. The N-terminal domain is necessary and sufficient for mitochondrial binding and hexokinase I or II can be displaced from mitochondria by a peptide corresponding to the N-terminal domain [10]. Moreover, the interaction of hexokinase I with the reconstituted VDAC-1 also requires the presence of the N-terminal domain [1]. Hexokinase isoforms, such as hexokinase III and hexokinase IV (glucokinase), which lack the N-terminal domain, do not bind directly to mitochondria, although indirect interaction with mitochondria mediated by other proteins may occur [11, 12]. However, despite the recognition that the N-terminal α-helix of hexokinase I and II is required for binding to VDAC, it is not clear how the interaction is enhanced by this domain. There is evidence that the hydrophobic tail of the N-terminal domain is inserted into the membrane bilayer [9], where it may interact with one or more of the transmembrane domains of VDAC-1. It is likely that other regions of hexokinase come in close apposition to the mitochondria and may help to stabilize the interaction of hexokinase with VDAC or other proteins bound to VDAC. This is supported both by antibody interference studies [13] and by the observation that effective binding of hexokinase to mitochondria depends on the conformation of the protein. A high concentration of its metabolite glucose-6-phosphate (G6P), which induces a conformational change in the protein, displaces it from the mitochondria [1, 4, 10]. However, no other specific domains required for mitochondrial binding have yet been identified. Divalent cations such as Mg2+ enhance the binding of hexokinase I and II to mitochondria [4, 14]. This may involve surface charge shielding or a more specific bridging effect to facilitate the apposition of negatively charged amino acids residues on the enzyme and anionic phospholipids or charged residues of interacting proteins in the OMM. It should be noted that Mg2+ also enhances the binding of hexokinase to yeast mitochondria, which do not express a VDAC isoform capable of binding the protein [14].
As with hexokinase I and II, several regions of VDAC appear to be required for hexokinase binding. Computer modeling of the structure of membrane-embedded VDAC suggests a β-barrel structure containing 13 to 16 antiparallel transbilayer β-strands connected by peptide loops, in addition to an N-terminal α-helical domain presumably localized to the intermembrane space. N,N-dicyclohexylcarbodiimide (DCCD) abolishes hexokinase binding to mitochondria through modification of glutamate 72, presumed to be located in the middle of the first cytoplasmic loop [15, 16]. Indeed, recent mutational studies by Shoshan-Barmatz and coworkers [17] who assessed both hexokinase binding to intact yeast mitochondria expressing VDAC-1 and its effect on VDAC conductance in a reconstituted lipid membrane, confirmed that glutamate 72 is essential for hexokinase binding to VDAC. The E72Q mutation also prevents protection by hexokinase I of VDAC-mediated apoptotic cell death [17]. Several other charged residues in VDAC-1 were identified in subsequent studies by the same group as being required for hexokinase I protection against VDAC-1-mediated apotosis and for channel closure in the reconstituted membrane system. These include glutamate 65, aspartate 77 and lysine 73, all localized within the putative first cytoplasmic loop [18]. In addition, mutations in glutamate 202 and 188, charged residues in the putative fourth cytoplasmic loop, while not essential for binding, were found to stabilize the interaction of hexokinase I with VDAC and to provide additional protection against VDAC-mediated apoptosis [18]. However, the nature of that stabilizing interaction remains poorly defined.
It is interesting to note that the majority of the VDAC-1 residues that have been identified as being required for hexokinase I binding are charged, and are located in the cytoplasm-exposed loops of VDAC-1. This finding lends further support to the notion that the hydrophobic portion of the N-terminal domain, while required for mitochondrial binding, is not directly engaging these essential residues on VDAC-1. Further characterization of the hexokinase domains involved in these interactions would provide better insight into the nature of the binding interaction.
Recent crosslinking studies [19, 20] and atomic force microscopy analyses of OMM [21, 22] demonstrated that VDAC exists as dimeric, trimeric and tetrameric structures and may be assembled in higher order aggregates in the OMM [20]. The dynamic nature of these oligomerization reactions was suggested by the finding that crosslinking of VDAC oligomers prevented its role in the permeabilization of the OMM and the release of cytochrome c, although transport of ions was not impeded [19]. Similarly, hexokinase I assumes a tetrameric structure when bound to rat brain mitochondria [23], which may be facilitated by the underlying structure of the VDAC oligomers. This is reminiscent of the higher order oligomerization of creatine kinase when bound to VDAC from the interior of the mitochondrial intermembrane space [24, 25]. The mechanisms and signaling pathways regulating the formation of VDAC and hexokinase oligomers and their consequences to mitochondrial function and susceptibility to damage largely remain a matter of speculation.
Although the precise nature of the mechanisms by which hexokinase binding protects against apoptosis still remains unclear, other VDAC interacting proteins may contribute to this effect. VDAC has been reported to interact with multiple other proteins that contribute to its role as the mediator of mitochondria-cytosolic interactions. The classical paradigm of VDAC as an essential partner in the permeability transition involves its ability to interact with inner membrane complexes, such as the adenine nucleotide translocator (ANT) [26, 27]. In cells that express mitochondrial creatine kinase, an octameric form of this protein has been suggested to mediate these interactions [24, 25]. The effects of Bcl2 family proteins on VDAC has also been suggested to involve direct binding interactions [28]. A recent NMR study identified residues involved in binding of VDAC-1 to Bcl-XL [29]. This model predicted that the VDAC binding of Bcl-XL depends primarily on the helices 5 and 6 that are inserted into the membrane. The BH4 domain of Bcl-XL, which is essential for its anti-apoptotic function, appeared to have only a more superficial interaction with the cytosolic face of VDAC. However, the BH4 domain is essential for its inhibitory effect on VDAC, i.e., the functionally effective domain was not primarily responsible for its binding [29].
In these respects, therefore, Bcl-XL binding resembles the nature of the hexokinase interaction with VDAC-1, where the hydrophobic N-terminal domain is thought to insert into the membrane and other interactions may mediate functional effects. Also, hexokinase binding, similar to Bcl-XL, inhibits VDAC conductance [1]. In this context, it is of interest to point out that we identified a BH4 homology region in hexokinase II, which we postulated to play a role in the anti-apoptotic effects that depend on its binding to VDAC [6]. It is conceivable that hexokinase binding displaces Bcl-XL from VDAC and thereby facilitates its availability for interaction anti-apoptotic Bcl2 family proteins, such as Bax/Bak that can contribute to outer membrane permeabilization (Fig. 1A).
Fig. 1. Proposed mechanism of hexokinase-II-mediated protection against outer mitochondrial membrane (OMM) permeabilization.
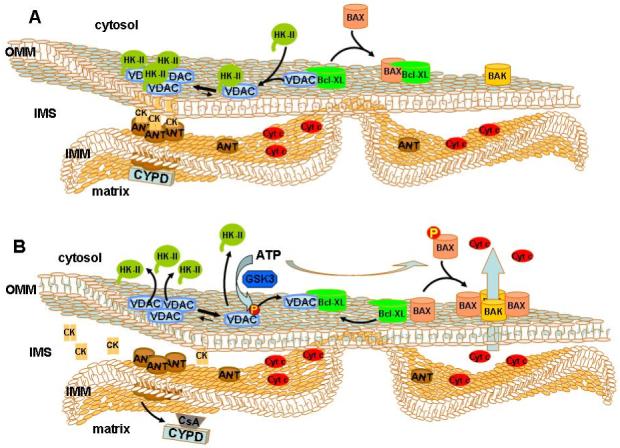
(A) Hexokinase II (HKII) binds to VDAC through both its N-terminal membrane binding domain and its putative BH4 domain [6], which is proposed to compete with VDAC-bound BCL-XL. Release of BCL-XL facilitates BAX binding, which prevents BAX-BAK interaction and protects against OMM permeabilization. Hexokinase II binding promotes VDAC oligomerization and the formation of contact sites between the inner and outer membranes in which the adenine nucleotide translocator (ANT) and octameric mitochondrial creatine kinase (CK) are presumed to play a role. Cyclosporin D (CYPD) may further enhance ANT binding to VDAC-HKII oligomeric complexes. (B) Phosphorylation of VDAC by GSK3 displaces hexokinase II from VDAC, thereby promoting binding of BCL-XL to VDAC. Disruption of CYPD interaction with ANT through cyclosporin A or through CYPD knockout would also promote hexokinase II displacement from VDAC, disrupting VDAC oligomers and making monomeric VDAC available for Bcl-XL binding. BAX is now free to interact with BAK to form pore structures that promote the release of cytochrome c (cyt c) and other intermembrane space (IMS) pro-apoptotic proteins.
Interestingly, Malia and Wagner [29] found that the VDAC-Bcl-XL interaction shifts the oligomerization equilibrium from a predominantly oligomeric towards a monomeric state of VDAC. This contrasts with the effect of hexokinase binding, which promotes the formation of oligomeric VDAC structures. This observation further suggests that the binding of hexokinase and Bcl-XL may be mutually exclusive. To what extent this transition plays a role in the protective effects of hexokinase remains to be established. It also remains unclear what role VDAC oligomerization plays in membrane permeabilization.
Signaling Pathways that Regulate the Interaction of Hexokinases I and II with VDAC
There is an abundance of evidence that the interaction of hexokinase with mitochondria is regulated to promote its protective anti-apoptotic effects when needed and to facilitate its removal when apoptosis is required. In addition, the metabolic function of hexokinase binding requires integration with other regulatory devices that control metabolic flux and energy supply to meet the demands of the cell [6, 30]. Several mechanisms have been identified that affect the hexokinase-VDAC interaction. As pointed out above, hexokinase I and II binding is decreased by high concentrations of its reaction product glucose-6-phosphate, which induces a conformational change in the protein that causes its release from VDAC [1, 4, 13]. Also, divalent cations are required for hexokinase binding to VDAC-1 [4, 14], although it is not known whether these contribute to the regulation of the interaction in intact cells. In addition, there is evidence that the binding of hexokinase I or II to VDAC is regulated by protein kinases.
Akt (also known as protein kinase B) is an oncogene protein kinase that is activated in response to stimulation of phosphatidylinositol 3-kinase (PI3K) and exhibits dysregulated activity in a variety of malignancies [30, 31]. Akt hyperactivity in cancer cells is associated with increased glycolysis, at least in part by enhancing the rate of glucose uptake through increased expression of the glucose carriers GLUT1 and GLUT4. Elstrom et al [32] demonstrated that Akt activation also enhances binding of hexokinase to the mitochondria. The Akt mediated binding of hexokinase to mitochondria was dependent on the availability of glucose, suggesting that the enzymatic activity of hexokinase is necessary for the interaction with VDAC [33, 34]. These authors suggested that Akt would cause an increased binding of hexokinase to the mitochondria by augmenting the uptake and metabolism of glucose. However, despite evidence that enzymatic activity of hexokinase II is required for its effects on apoptosis [34, 35], the mechanism by which increased glycolytic flux increased mitochondrial binding of hexokinase remained unclear in these studies. Other studies discussed below indicate that Akt also has more direct effects on the binding of hexokinase to VDAC by affecting the phosphorylation state of VDAC and/or hexokinase.
The ability of Akt to directly impact mitochondrial function is illustrated by the finding that activated Akt localizes to the mitochondria following activation of the upstream PI3K [36, 37]. Indeed, Akt can directly phosphorylate hexokinase-II [36, 37]. Activation of Akt in cardiomyocytes was shown to stimulate association between Akt and hexokinase II, with Akt subsequently phosphorylating hexokinase II at threonine 473, positioned in an Akt phosphorylation consensus sequence. The phosphorylation of hexokinase II by Akt was accompanied by an increased binding of the enzyme to the mitochondria and also was found to contribute to the anti-apoptotic effects of Akt against ischemia/reperfusion injury. These observations illustrate the ability of Akt to positively regulate the binding of hexokinase II to VDAC.
In addition, work in our laboratory has shown that Akt can also impact the binding of hexokinase II to mitochondria by negatively regulating the activity of glycogen synthase kinase 3β (GSK3β) and its phosphorylation of VDAC [38]. Like Akt, GSK3β can localize to the mitochondria. However unlike Akt, activation of GSK3β is associated with mitochondrial dysfunction and cell injury [39-45]. Indeed, it has been demonstrated that many signaling pathways that promote cardioprotection converge at inhibition of GSK3β activation. Part of the protective effect of GSK3β inhibition in the heart may be through the preservation of mitochondrial binding of hexokinase II. Similar mechanisms may apply to other cells and tissues that express hexokinase isoforms that can bind to mitochondria, including tumor cells [38].
GSK3β can phosphorylate VDAC-1 on threonine 51 resulting in the detachment of hexokinase II from VDAC-1 and a sensitization of cancer cells to chemotherapeutic agents such as paclitaxel and doxorubicin [38]. The location of this threonine residue is not entirely clear. Depending on the structural model of VDAC, threonine 51 would either be located in the putative first cytoplasmic loop or in one of the transmembrane β-sheets [18, 46-48] and it is conceivable that phosphorylation of this residue influences the conformation of VDAC-1 or its ability to form oligomeric structures (Fig. 1B). Future mutational studies focusing on the functional consequences of this phosphorylation would be of interest.
The enhanced vulnerability of the mitochondria to pro-apoptotic conditions resulting from this phosphorylation is mediated in part by a heightened sensitivity to the pro-apoptotic protein Bax, [10]. How Bax sensitivity is increased has not been well characterized. One model would be that Bax is able to gain direct access to VDAC when hexokinase is not bound. Alternatively, if hexokinase detachment promotes binding of Bcl-XL to VDAC, this may prevent it from binding Bax, thereby leaving Bax free to interact with Bak and form pore structures in the outer membrane that promote the release of cytochrome c and other apoptosis enhancing proteins (Fig. 1B). This model is also supported by the finding that overexpression of VDAC-1 itself enhances the susceptibility of cells to apoptosis [49]. Additionally, activation of GSK3β has been shown to phosphorylate Bax directly, promoting its translocation from the cytosol to the mitochondria [50]. This would suggest that GSK3β activation has the twofold impact of making mitochondria more vulnerable to Bax by detaching hexokinase and tying up the anti-apoptotic Bax binding partner Bcl-XL and then promoting the translocation of Bax to the mitochondrial membrane where it can interact with pro-apoptotic partners, such as Bak (Fig. 1B).
In addition to promoting the lethality of pro-apoptotic proteins such as Bax, activation of GSK3β decreases the half-life of anti-apoptotic proteins [51, 52]. GSK3β phosphorylates the anti-apoptotic protein MCL-1 on serine 159 leading to its increased ubiquitinylation and degradation. A phosphorylation site mutant of MCL-1 demonstrated increased stability and conferred protection against growth factor withdrawal. Stimulation of glucose catabolism brought on by growth factors was shown to prevent MCL-1 degradation by inhibiting GSK3β activation [52]. However, in this instance the signaling pathway identified as mediating the growth factor dependent inhibitory phosphorylation of GSK3β involved protein kinase C. Whether GSK3β was a direct or indirect target of protein kinase C was not established in these studies, nor was it determined which isoform of protein kinase C mediated this effect. Nevertheless, this finding exemplifies that different protein kinases can result in phosphorylation and inhibition of GSK3β activity, thus illustrating the critical node that this enzyme occupies in mediating the vulnerability of the mitochondria to injury.
In contrast to the detachment of hexokinase from VDAC brought about by GSK3β mediated phosphorylation, phosphorylation of VDAC by PKC-ε promotes hexokinase binding [53, 54]. Studies in cardiac mitochondria demonstrated an interaction of PKC-ε with VDAC-1. Moreover, in vitro studies revealed that PKC-ε directly binds to and phosphorylates VDAC-1 [53]. Intriguingly, over-expression of hexokinase I or II in HEK293 cells caused a 5-10 fold increase in VDAC phosphorylation that was prevented by an inhibitor of PKC-ε. Such results suggest that the binding of hexokinase to VDAC may help to trigger VDAC phosphorylation by PKC-ε, which then further enhances the affinity of hexokinase for VDAC, thus establishing a positive feedback cycle.
Although these studies did not identify the target site for PKC-ε-dependent phosphorylation, a recent mass spectrometric analysis of covalent modifications in OMM proteins detected multiple phosphorylation sites (as well as other covalent modifications) on all three VDAC isoforms in rat liver mitochondria [55]. Interestingly, VDAC-1 showed evidence of phosphorylation on Serine 12 and Serine 136, the first being a PKC consensus site located in the N-terminal domain, the second a CAMII/GSK3 consensus site, presumably located on the side of the VDAC structure facing the intermembrane space. Recent evidence suggests that a fraction of cellular PKC-ε is present in mitochondria, both in the intermembrane space and in the matrix, although the mechanism by which it is translocated to these sites remains to be established [56].
Other protein kinases have been reported to affect mitochondrial function. C-Raf kinase, a member of the MAP kinase cascade that can be targeted to mitochondria, forms a complex with VDAC both in vivo and in reconstituted membranes, resulting in its phosphorylation and subsequent closure [57]. However, it was not determined if C-Raf-mediated phosphorylation of VDAC exhibited any effects on hexokinase binding. Activation of p38 MAP kinase (MAPK) has also been reported to cause phosphorylation of VDAC during myocardial ischemia and reperfusion, with inhibition of p38 activity exerting a cardioprotective effect [58]. However, this study provided evidence that activation of p38 MAPK resulted in tyrosine phosphorylation of the protein, suggesting that this effect is indirect and involves an unknown tyrosine kinase or tyrosine phosphatase that may act downstream of p38 MAPK. The mass spectrometric analysis of Distler et al [55] identified a tyrosine phosphorylation site on VDAC-2. However, there is evidence that VDAC-2 does not directly interact with hexokinase I or II [59].
Regulation of hexokinase binding to VDAC by Cyclophilin D
Several recent studies have reported that mitochondrial binding of hexokinase II is affected by the activity of cyclophilin D [59, 60]. Cyclophilin D is an immunophilin that exhibits peptidyl-prolyl cis-trans isomerase (PPIase) activity and is localized to the mitochondrial matrix. The ability of cyclosporine A to prevent onset of the mitochondrial permeability transition was the first indication that cyclophilin D regulates the permeability transition pore (PTP) [61]. These findings have been confirmed and extended with the utilization of cyclophilin D knock-out mice [62-64]. In the absence of cyclophilin D, the PTP still formed and opened in response to some challenges, but was not responsive to Ca2+ or inhibition by cyclosporine A. Additionally, the effects of knockout of cyclophilin D established that opening of the PTP was essential for mitochondrial injury in necrotic cell death but dispensable for mitochondrial mediation of apoptosis. By contrast, several studies have provided evidence that cyclophilin D suppresses mitochondrial injury and can function as an apoptosis repressor [65, 66]. Moreover, cyclophilin D is up-regulated in a number of human tumors. Overexpression of cyclophilin D in HEK293 cells or rat glioma C6 cells caused desensitization to induction of apoptosis. Importantly, in these studies the PPIase activity of cyclophilin D was found to be necessary for apoptosis suppression.
Recent studies have shown that the anti-apoptotic effects of cyclophilin D may be exerted by stabilization of hexokinase II binding to mitochondria [59, 60]. Inactivation of cyclophilin D with cyclosporine A or knock-down of its expression utilizing siRNA caused a release of mitochondrially bound hexokinase II. Moreover, the anti-apoptotic effects of cyclophilin D were abrogated by the forced detachment of hexokinase II from the mitochondria. Such observations are in agreement with the concept that hexokinase II prevents access of pro-apoptotic proteins such as Bax to the mitochondria or inhibits its pore-forming abilities. However, it is presently unclear how cyclophilin D activity regulates the binding of hexokinase II to the mitochondria. At first sight, it would seem likely that this occurs through modification of the hexokinase II-VDAC interaction, e.g., by inducing a conformational change in VDAC through a mitochondrial constitutent that can interact with both proteins. Since cyclophilin D is thought to be localized exclusively to the mitochondrial matrix, the modulation of hexokinase II binding to VDAC by cyclophilin D must occur through an intermediate located in the mitochondrial inner membrane. A promising candidate for this intermediation role is the adenine nucleotide translocator (ANT). Classically, the ANT was thought to be a component of the PTP and to exert its pore-forming function by interacting with VDAC at mitochondrial contact sites. However, this model has come under doubt recently as a result of studies demonstrating that cyclosporin-sensitive PTP formation could occur in the absence of ANT [67]. Nevertheless, the ANT-2 isoform has been shown to be expressed in cancer cells that display enhanced glycolysis and in such cells a large proportion of hexokinase II is bound to the mitochondria [68]. A hypothesis that could account for these effects is illustrated in Fig. 1, where it is proposed that cyclophilin D stabilizes the interaction between ANT and the oligomeric form of VDAC that is enhanced by hexokinase II binding.
However, a recent study by Chiara et al [59] suggests that the story may be more complex. These authors showed that detachment of hexokinase II from the mitochondria triggers apoptosis through opening of the PTP and this was associated with a disruption of the association of cyclophilin D with ANT. Moreover, inhibition of the ANT or cyclophilin D prevented the onset of the PTP and cytotoxicity elicited by hexokinase II detachment and, vice versa, inhibition of cyclophilin D or its knockout promoted detachment of hexokinase II from the mitochondria. Remarkably, however, in this study, VDAC was found to be dispensable not only for induction of the PTP and cell death, but also for the hexokinase II detachment induced by inhibition or knockout of cyclophilin D [59]. Also, these studies suggested that the PPIase activity of cyclophilin D was not required for its effects on hexokinase II binding. These observations suggest that the role of VDAC and the VDAC-hexokinase interaction are not irreplaceable constituents of the mitochondrial permeabilization process. It may be that other, yet to be identified proteins (or other membrane constituents) can act as surrogate binding partners for hexokinase II at the OMM in the absence of VDAC and also influence onset of the PTP. It will be of interest to establish if such alternate binding partners also interact with anti-apoptotic proteins, such as Bcl-XL. If so, the essential features of the mechanism by which hexokinase binding protects against Bax-mediated membrane permeabilization may extend to such other binding partners.
Cholesterol enhancement of hexokinase binding to mitochondria
The mitochondrial outer and inner membranes contain lower levels of cholesterol than most other cellular membranes, such as the plasma membrane, the Golgi membrane, or the ER membrane. However, cancer cells have been shown to a 2-10 fold increase in mitochondrial cholesterol content per mg mitochondrial protein compared to liver mitochondria [69]. Most of the increase occurred in the OMM as free (i.e., non-esterified) cholesterol. For instance, the OMM fraction isolated from Ehrlich or AS30-D tumor cell mitochondria was found to contain 3.5-5 times the free cholesterol content found in PMM fractions from rat liver mitochondria, with a much smaller (< 2-fold) increase occurring in the inner membrane fraction [69]. Both of these tumor cell lines are highly glycolytic and exhibit an increased expression and mitochondrial binding of hexokinase II. It was suggested that the increased content of cholesterol in the mitochondria of cancer cells could impact the binding of hexokinase to VDAC [69]. Conversely the increased binding of hexokinase to the mitochondria of cancer cells may play a role in mediating an increased synthesis and uptake of cholesterol into the mitochondria of cancer cells. Indeed, an increase in the content of mitochondrial cholesterol has been shown to affect the levels and activity of a number of mitochondrial membrane proteins.
Mitochondria from cancer cells export citrate at up to a four-fold higher rate than liver mitochondria [70]. This is probably due in part to an increased activity of the tricarboxylate carrier (TCC) responsible for the transport of citrate and isocitrate across the inner mitochondrial membrane. Citrate export is required for the supply of cytosolic acetyl-CoA, the building block of cholesterol, to HMG-CoA reductase, the first committed step of the cholesterol biosynthesis pathway. Intriguingly, this augmented activity of the tricarboxylate carrier in the mitochondria of cancer cells may be associated with their increased content of cholesterol (Fig. 2). Enrichment of liver mitochondria with exogenous cholesterol resulted in a two-fold stimulation in the activity of the mitochondrial citrate carrier. Conversely, an increased content of mitochondrial cholesterol can inhibit the activity of other inner membrane proteins. Baggetto et al [69] reported that the inner membrane proton permeability (responsible for the basal rate of oxygen uptake in mitochondria at high membrane potential) was decreased as a function of the inner membrane cholesterol content. Elevated levels of mitochondrial cholesterol were found to diminish the ability of the ANT to assist in the onset of the mitochondrial permeability transition [71]. Additionally, and possibly related to this effect on ANT, the accumulation of cholesterol in mitochondrial membranes inhibited the ability of Bax to permeabilize the mitochondrial membranes and thus prevented stress induced apoptosis [72]. It is not clear whether these effects of cholesterol loading on the activity of mitochondrial constituents are due to the cholesterol content of the inner membrane or whether the OMM, which contains the majority of this cholesterol, contributes to the control of these metabolite transport activities. VDAC is the major gateway for exit of all such metabolites from the mitochondrial intermembrane space to the cytosol and changes in its activity may also contribute to the metabolic consequences of cholesterol loading.
Fig. 2. Mitochondrial cholesterol accumulation promotes hexokinase II binding and protects against OMM permeabilization.
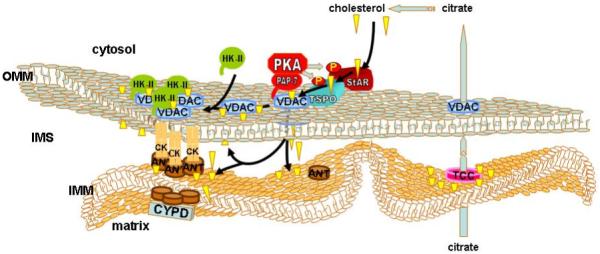
The interaction of VDAC with TSPO (also known as the peripheral benzodiazepine receptor) and StAR enhances cholesterol uptake through VDAC. StAR and TSPO are activated by protein kinase A (PKA)-dependent phosphorylation, which is promoted by VDAC binding of the anchoring protein PAP-7. Increased cholesterol content of the mitochondrial membrane promotes binding of HKII to VDAC, possibly by enhancing the formation of oligomeric complexes involving ANT, resulting in increased protection against OMM permeabilization. Enhanced mitochondrial cholesterol content also promotes citrate export to increase the supply of cholesterol precursor in the cytosol by activation of the tricarboxylate carrier (TCC) located in the inner membrane. The resulting positive feedback loop may cause a switch-like enhancement of mitochondrial cholesterol uptake and hexokinase binding in tumor cells.
Whereas the increased content of cholesterol in cancer cells may be driven in part by the amplified export rate of citrate from the mitochondria into the cytosol, where cholesterol is synthesized, VDAC has also been implicated in the subsequent import of the resultant cholesterol from the cytosol into the mitochondria [73, 74]. As mentioned above, glutamate 72 of VDAC is essential for hexokinase binding. Intriguingly, an E72Q mutant that inhibited hexokinase binding to VDAC and mitochondria, also caused a reduction in the mitochondrial cholesterol content of Morris hepatoma cells [75]. These observations suggest that the binding of hexokinase to VDAC by some means helps to stimulate the uptake of cholesterol into the mitochondria. The converse may also arise, in that the elevated cholesterol content of the mitochondria of cancer cells may facilitate the enhanced binding of hexokinase to VDAC. In this way, a positive feedback loop is initiated where hexokinase binding to mitochondria enhances the uptake of cholesterol by VDAC. In turn, the elevated cholesterol content amplifies the binding of hexokinase to VDAC (Fig. 2). These observations, together with the effect of increased mitochondrial cholesterol on the ability of Bax and the ANT to initiate mitochondrial dysfunction, suggest that the loading of cholesterol characteristic of the mitochondria of cancer cells may make them refractory to injury and hence provide the cancer cells with a resistance to induction of apoptosis and a survival advantage.
Importantly, VDAC may be only one component of a complex that mediates the uptake of cholesterol into the mitochondria and is regulated by a number of signaling pathways. The translocator protein 18kDa (TSPO), formerly know as the peripheral benzodiazepine receptor, is a transmembrane protein located in the OMM [76] that associates with VDAC [77-81]. Importantly, TSPO helps to mediate the uptake of cholesterol into the mitochondria [82]. As such, TSPO is found in great abundance in steroidogenic tissues. Additionally, some cancer types such as gliomas with a high glycolytic capacity have been shown to have an amplified expression of TSPO [83]. TSPO contains a cholesterol recognition/interaction amino acid consensus sequence (CRAC domain) at the carboxyl-terminus located on the cytosolic side of the OMM [84]. Once the cholesterol is bound to the CRAC domain, how it transits though the OMM and IMM is currently unknown. TSPO-associated VDAC may be involved in this process.
Another member of this putative complex is StAR (steroidogenic acute regulatory protein), a 37-kDa protein located in the cytoplasm. Like the TSPO, StAR possesses a cholesterol binding domain (START) [85-88]. Interestingly, MLN64 (metastatic lymph node 64), a homologue of StAR, is over-expressed in a number of cancers [89-91]. StAR is activated by phosphorylation of serine 195 and is then imported into the mitochondria as a 32-kDa intermediate, which, in turn, is cleaved to a 30-kDa protein once it is imported into the mitochondrial matrix [92]. StAR has been shown to interact with the cytoplasmically exposed domains of VDAC-1 and to require VDAC expression to exert its ability to mediate cholesterol uptake. However StAR has also been shown to associate with TSPO and TSPO knock-down cells display a loss of StAR activity [93]. Interestingly, TSPO binds the anchoring protein, PAP-7 (peripheral benzodiazepine receptor-associated protein-7) [94-96], a member of the A-kinase anchor protein family, which promotes the interaction of the VDAC-TSPO-StAR complex with the PKA regulatory subunit-1α (PKAR1α). PKA is thereby thought to mediate the phosphorylation and consequent activation of StAR. PKA has also been shown to phosphorylate VDAC, however the consequences to cholesterol uptake and hexokinase binding, if any, are unknown [97]. PAP-7 possesses a 15 residue targeting element homologous to the N-terminal targeting sequence of hexokinase I and II, suggesting that PAP-7 may interact with VDAC through a mechanism that is similar to that involved in hexokinase II binding [98-100]. Whether PAP-7 directly influences hexokinase binding and contributes to the interregulation between cholesterol uptake and synthesis in tumor cells remains to be established. Fig. 2 presents a model of the interaction of hexokinase binding to VDAC in the context of the complex of VDAC-interacting proteins that regulate cholesterol transport.
In summary, there are several lines of evidence that suggest an intricate role for the mitochondrial cholesterol content in the binding and anti-apoptotic effects of hexokinase binding. The potential for a positive feedback loop involving hexokinase II binding to VDAC, the mitochondrial cholesterol uptake system, and the mitochondrial tricarboxylate carrier may point to the existence of a bistable system in which enhanced hexokinase binding and cholesterol loading of the mitochondria are mutually reinforcing to promote the special conditions prevailing in tumor cells.
Conclusions and further outlook
The past decade has seen a remarkable shift in our understanding of the nature and implications of the binding interactions of hexokinase I and II to the mitochondrial outer membrane and to VDAC in particular. The functional associations of this interaction have shifted from a predominantly metabolic role to the recognition of its major impact on the regulation of apoptotic responsiveness of the cell. Several recent studies have highlighted the potential application of this understanding for therapeutic purposes [38, 101]. As we have pointed out before [6], these findings demonstrate the need for a careful regulation of the binding process so as to integrate the anti-apoptotic protective effects of hexokinase binding with the basic metabolic functions on which the cell depends and which are deregulated in tumor cells. Current evidence suggests that this regulation is at least in part mediated by protein kinase-dependent phosphorylation of VDAC and other proteins involved in the complex formation, most directly by GSK-3β and PKC-ε. However, the regulatory significance of the sites of phosphorylation and the mechanism by which these phosphorylation events affect hexokinase-VDAC interaction remains largely unknown. We hypothesize that VDAC oligomerization and its interaction with ANT and cyclophilin D in the IMM and matrix of the mitochondria may constitute an integral part of these interactions and may be regulated by VDAC phosphorylation. Also, the recent observation that hexokinase II-mediated protection against apoptotic signals can occur in the absence of VDAC may provide further insight into the regulation of these protective interactions in which hexokinase is involved. It is likely that under such conditions the susceptibility of the cells to protein kinase-mediated regulatory controls would be notably different. In addition, the lipid structure of both the inner and outer mitochondrial membranes may contribute to this regulation as indicated by the consequences of cholesterol loading on the hexokinase binding. How these regulatory processes enable normal cells and tissues to manage the balance between protective effects and the response to pro-apoptotic signals is a topic that deserves much more analysis. Also, hexokinase I and II have a distinct tissue distribution and it is not currently clear what the implications are of the different properties of these isoforms for tissue-specific functions. A better understanding of these functions in normal tissues will greatly contribute to the optimization of therapies for the treatment of tumor cells, where hexokinase II is overexpressed.
References
- 1.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–55. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brdiczka D, Beutner G, Ruck A, Dolder M, Wallimann T. The molecular structure of mitochondrial contact sites. Their role in regulation of energy metabolism and permeability transition. Biofactors. 1998;8:235–42. doi: 10.1002/biof.5520080311. [DOI] [PubMed] [Google Scholar]
- 3.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–52. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 4.Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- 5.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 6.Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10:1535–51. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 7.Linden M, Gellerfors P, Nelson BD. Pore protein and the hexokinase-binding protein from the outer membrane of rat liver mitochondria are identical. FEBS Lett. 1982;141:189–92. doi: 10.1016/0014-5793(82)80044-6. [DOI] [PubMed] [Google Scholar]
- 8.Fiek C, Benz R, Roos N, Brdiczka D. Evidence for identity between the hexokinase-binding protein and the mitochondrial porin in the outer membrane of rat liver mitochondria. Biochim Biophys Acta. 1982;688:429–40. doi: 10.1016/0005-2736(82)90354-6. [DOI] [PubMed] [Google Scholar]
- 9.Xie GC, Wilson JE. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Arch Biochem Biophys. 1988;267:803–10. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- 10.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–8. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 11.Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–6. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 12.Arden C, Baltrusch S, Agius L. Glucokinase regulatory protein is associated with mitochondria in hepatocytes. FEBS Lett. 2006;580:2065–70. doi: 10.1016/j.febslet.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto M, Wilson JE. Membrane potential-dependent conformational changes in mitochondrially bound hexokinase of brain. Arch Biochem Biophys. 2000;384:163–73. doi: 10.1006/abbi.2000.2085. [DOI] [PubMed] [Google Scholar]
- 14.Aflalo C, Azoulay H. Binding of rat brain hexokinase to recombinant yeast mitochondria: effect of environmental factors and the source of porin. J Bioenerg Biomembr. 1998;30:245–55. doi: 10.1023/a:1020544803475. [DOI] [PubMed] [Google Scholar]
- 15.Nakashima RA, Mangan PS, Colombini M, Pedersen PL. Hexokinase receptor complex in hepatoma mitochondria: evidence from N,N’-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry. 1986;25:1015–21. doi: 10.1021/bi00353a010. [DOI] [PubMed] [Google Scholar]
- 16.Al Jamal JA. Involvement of porin N,N-dicyclohexylcarbodiimide-reactive domain in hexokinase binding to the outer mitochondrial membrane. Protein J. 2005;24:1–8. doi: 10.1007/s10930-004-0600-2. [DOI] [PubMed] [Google Scholar]
- 17.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–60. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 18.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase -I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J Biol Chem. 2008 doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 19.Zalk R, Israelson A, Garty ES, Azoulay-Zohar H, Shoshan-Barmatz V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem J. 2005;386:73–83. doi: 10.1042/BJ20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoshan-Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim Biophys Acta. 2004;1657:105–14. doi: 10.1016/j.bbabio.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Goncalves RP, Buzhynskyy N, Prima V, Sturgis JN, Scheuring S. Supramolecular assembly of VDAC in native mitochondrial outer membranes. J Mol Biol. 2007;369:413–8. doi: 10.1016/j.jmb.2007.03.063. [DOI] [PubMed] [Google Scholar]
- 22.Hoogenboom BW, Suda K, Engel A, Fotiadis D. The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J Mol Biol. 2007;370:246–55. doi: 10.1016/j.jmb.2007.04.073. [DOI] [PubMed] [Google Scholar]
- 23.Xie G, Wilson JE. Tetrameric structure of mitochondrially bound rat brain hexokinase: a crosslinking study. Arch Biochem Biophys. 1990;276:285–93. doi: 10.1016/0003-9861(90)90040-6. [DOI] [PubMed] [Google Scholar]
- 24.Brdiczka D, Kaldis P, Wallimann T. In vitro complex formation between the octamer of mitochondrial creatine kinase and porin. J Biol Chem. 1994;269:27640–4. [PubMed] [Google Scholar]
- 25.Stachowiak O, Schlattner U, Dolder M, Wallimann T. Oligomeric state and membrane binding behaviour of creatine kinase isoenzymes: implications for cellular function and mitochondrial structure. Mol Cell Biochem. 1998;184:141–51. [PubMed] [Google Scholar]
- 26.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–31. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 27.Halestrap AP, Brennerb C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–25. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 28.Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000;7:1174–81. doi: 10.1038/sj.cdd.4400780. [DOI] [PubMed] [Google Scholar]
- 29.Malia TJ, Wagner G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry. 2007;46:514–25. doi: 10.1021/bi061577h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–42. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 31.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 33.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–40. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–17. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–35. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–9. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 38.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–54. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 39.Morrison RS, Kinoshita Y, Johnson MD, Ghatan S, Ho JT, Garden G. Neuronal survival and cell death signaling pathways. Adv Exp Med Biol. 2002;513:41–86. doi: 10.1007/978-1-4615-0123-7_2. [DOI] [PubMed] [Google Scholar]
- 40.Murphy E. Inhibit GSK-3beta or there’s heartbreak dead ahead. J Clin Invest. 2004;113:1526–8. doi: 10.1172/JCI21986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macanas-Pirard P, Yaacob NS, Lee PC, Holder JC, Hinton RH, Kass GE. Glycogen synthase kinase-3 mediates acetaminophen-induced apoptosis in human hepatoma cells. J Pharmacol Exp Ther. 2005;313:780–9. doi: 10.1124/jpet.104.081364. [DOI] [PubMed] [Google Scholar]
- 44.Park SS, Zhao H, Mueller RA, Xu Z. Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3beta. J Mol Cell Cardiol. 2006;40:708–16. doi: 10.1016/j.yjmcc.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 45.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–70. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Mannella CA, Forte M, Colombini M. Toward the molecular structure of the mitochondrial channel, VDAC. J Bioenerg Biomembr. 1992;24:7–19. doi: 10.1007/BF00769525. [DOI] [PubMed] [Google Scholar]
- 47.Casadio R, Jacoboni I, Messina A, De Pinto V. A 3D model of the voltage-dependent anion channel (VDAC) FEBS Lett. 2002;520:1–7. doi: 10.1016/s0014-5793(02)02758-8. [DOI] [PubMed] [Google Scholar]
- 48.Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256-257:107–15. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 49.Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci U S A. 2006;103:5787–92. doi: 10.1073/pnas.0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–60. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–39. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92:873–80. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korzick DH, Kostyak JC, Hunter JC, Saupe KW. Local delivery of PKCepsilon-activating peptide mimics ischemic preconditioning in aged hearts through GSK-3beta but not F1-ATPase inactivation. Am J Physiol Heart Circ Physiol. 2007;293:H2056–63. doi: 10.1152/ajpheart.00403.2007. [DOI] [PubMed] [Google Scholar]
- 55.Distler AM, Kerner J, Hoppel CL. Post-translational modifications of rat liver mitochondrial outer membrane proteins identified by mass spectrometry. Biochim Biophys Acta. 2007;1774:628–36. doi: 10.1016/j.bbapap.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD. Mitochondrial PKC epsilon and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res. 2006;99:878–83. doi: 10.1161/01.RES.0000245106.80628.d3. [DOI] [PubMed] [Google Scholar]
- 57.Le Mellay V, Troppmair J, Benz R, Rapp UR. Negative regulation of mitochondrial VDAC channels by C-Raf kinase. BMC Cell Biol. 2002;3:14. doi: 10.1186/1471-2121-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwertz H, Carter JM, Abdudureheman M, Russ M, Buerke U, Schlitt A, Muller-Werdan U, Prondzinsky R, Werdan K, Buerke M. Myocardial ischemia/reperfusion causes VDAC phosphorylation which is reduced by cardioprotection with a p38 MAP kinase inhibitor. Proteomics. 2007;7:4579–88. doi: 10.1002/pmic.200700734. [DOI] [PubMed] [Google Scholar]
- 59.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006;281:14314–20. doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- 61.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–30. [PubMed] [Google Scholar]
- 62.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 63.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 64.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 65.Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J Biol Chem. 2002;277:31134–41. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- 66.Schubert A, Grimm S. Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 2004;64:85–93. doi: 10.1158/0008-5472.can-03-0476. [DOI] [PubMed] [Google Scholar]
- 67.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chevrollier A, Loiseau D, Chabi B, Renier G, Douay O, Malthiery Y, Stepien G. ANT2 isoform required for cancer cell glycolysis. J Bioenerg Biomembr. 2005;37:307–16. doi: 10.1007/s10863-005-8642-5. [DOI] [PubMed] [Google Scholar]
- 69.Baggetto LG, Clottes E, Vial C. Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res. 1992;52:4935–41. [PubMed] [Google Scholar]
- 70.Parlo RA, Coleman PS. Enhanced rate of citrate export from cholesterol-rich hepatoma mitochondria. The truncated Krebs cycle and other metabolic ramifications of mitochondrial membrane cholesterol. J Biol Chem. 1984;259:9997–10003. [PubMed] [Google Scholar]
- 71.Colell A, Garcia-Ruiz C, Lluis JM, Coll O, Mari M, Fernandez-Checa JC. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J Biol Chem. 2003;278:33928–35. doi: 10.1074/jbc.M210943200. [DOI] [PubMed] [Google Scholar]
- 72.Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008;15:484–93. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- 73.Thomson M. Does cholesterol use the mitochondrial contact site as a conduit to the steroidogenic pathway? Bioessays. 2003;25:252–8. doi: 10.1002/bies.10243. [DOI] [PubMed] [Google Scholar]
- 74.Papadopoulos V, Liu J, Culty M. Is there a mitochondrial signaling complex facilitating cholesterol import? Mol Cell Endocrinol. 2007;265-266:59–64. doi: 10.1016/j.mce.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 75.Campbell AM, Chan SH. The voltage dependent anion channel affects mitochondrial cholesterol distribution and function. Arch Biochem Biophys. 2007;466:203–10. doi: 10.1016/j.abb.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 76.Papadopoulos V, Amri H, Boujrad N, Cascio C, Culty M, Garnier M, Hardwick M, Li H, Vidic B, Brown AS, et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids. 1997;62:21–8. doi: 10.1016/s0039-128x(96)00154-7. [DOI] [PubMed] [Google Scholar]
- 77.McEnery MW. The mitochondrial benzodiazepine receptor: evidence for association with the voltage-dependent anion channel (VDAC) J Bioenerg Biomembr. 1992;24:63–9. doi: 10.1007/BF00769532. [DOI] [PubMed] [Google Scholar]
- 78.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A. 1992;89:3170–4. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW, Tedeschi H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci U S A. 1993;90:1374–8. doi: 10.1073/pnas.90.4.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McEnery MW, Dawson TM, Verma A, Gurley D, Colombini M, Snyder SH. Mitochondrial voltage-dependent anion channel. Immunochemical and immunohistochemical characterization in rat brain. J Biol Chem. 1993;268:23289–96. [PubMed] [Google Scholar]
- 81.McEnery MW, Snowman AM, Seagar MJ, Copeland TD, Takahashi M. Immunological characterization of proteins associated with the purified omega-conotoxin GVIA receptor. Ann N Y Acad Sci. 1993;707:386–91. doi: 10.1111/j.1749-6632.1993.tb38078.x. [DOI] [PubMed] [Google Scholar]
- 82.Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–7. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 83.Hardwick M, Fertikh D, Culty M, Li H, Vidic B, Papadopoulos V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999;59:831–42. [PubMed] [Google Scholar]
- 84.Li H, Yao Z, Degenhardt B, Teper G, Papadopoulos V. Cholesterol binding at the cholesterol recognition/ interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc Natl Acad Sci U S A. 2001;98:1267–72. doi: 10.1073/pnas.031461598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsujishita Y, Hurley JH. Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol. 2000;7:408–14. doi: 10.1038/75192. [DOI] [PubMed] [Google Scholar]
- 86.Mathieu AP, Fleury A, Ducharme L, Lavigne P, LeHoux JG. Insights into steroidogenic acute regulatory protein (StAR)-dependent cholesterol transfer in mitochondria: evidence from molecular modeling and structure-based thermodynamics supporting the existence of partially unfolded states of StAR. J Mol Endocrinol. 2002;29:327–45. doi: 10.1677/jme.0.0290327. [DOI] [PubMed] [Google Scholar]
- 87.Mathieu AP, Lavigne P, LeHoux JG. Molecular modeling and structure-based thermodynamic analysis of the StAR protein. Endocr Res. 2002;28:419–23. doi: 10.1081/erc-120016817. [DOI] [PubMed] [Google Scholar]
- 88.Soccio RE, Breslow JL. StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J Biol Chem. 2003;278:22183–6. doi: 10.1074/jbc.R300003200. [DOI] [PubMed] [Google Scholar]
- 89.Watari H, Arakane F, Moog-Lutz C, Kallen CB, Tomasetto C, Gerton GL, Rio MC, Baker ME, Strauss JF., 3rd MLN64 contains a domain with homology to the steroidogenic acute regulatory protein (StAR) that stimulates steroidogenesis. Proc Natl Acad Sci U S A. 1997;94:8462–7. doi: 10.1073/pnas.94.16.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bose HS, Baldwin MA, Miller WL. Evidence that StAR and MLN64 act on the outer mitochondrial membrane as molten globules. Endocr Res. 2000;26:629–37. doi: 10.3109/07435800009048583. [DOI] [PubMed] [Google Scholar]
- 91.Bose HS, Whittal RM, Huang MC, Baldwin MA, Miller WL. N-218 MLN64, a protein with StAR-like steroidogenic activity, is folded and cleaved similarly to StAR. Biochemistry. 2000;39:11722–31. doi: 10.1021/bi000911l. [DOI] [PubMed] [Google Scholar]
- 92.Bose M, Whittal RM, Miller WL, Bose HS. Steroidogenic Activity of StAR Requires Contact with Mitochondrial VDAC1 and Phosphate Carrier Protein. J Biol Chem. 2008;283:8837–45. doi: 10.1074/jbc.M709221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hauet T, Yao ZX, Bose HS, Wall CT, Han Z, Li W, Hales DB, Miller WL, Culty M, Papadopoulos V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol. 2005;19:540–54. doi: 10.1210/me.2004-0307. [DOI] [PubMed] [Google Scholar]
- 94.Li H, Degenhardt B, Tobin D, Yao ZX, Tasken K, Papadopoulos V. Identification, localization, and function in steroidogenesis of PAP7: a peripheral-type benzodiazepine receptor- and PKA (RIalpha)-associated protein. Mol Endocrinol. 2001;15:2211–28. doi: 10.1210/mend.15.12.0736. [DOI] [PubMed] [Google Scholar]
- 95.Liu J, Matyakhina L, Han Z, Sandrini F, Bei T, Stratakis CA, Papadopoulos V. Molecular cloning, chromosomal localization of human peripheral-type benzodiazepine receptor and PKA regulatory subunit type 1A (PRKAR1A)-associated protein PAP7, and studies in PRKAR1A mutant cells and tissues. Faseb J. 2003;17:1189–91. doi: 10.1096/fj.02-1066fje. [DOI] [PubMed] [Google Scholar]
- 96.Liu J, Rone MB, Papadopoulos V. Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J Biol Chem. 2006;281:38879–93. doi: 10.1074/jbc.M608820200. [DOI] [PubMed] [Google Scholar]
- 97.Bera AK, Ghosh S, Das S. Mitochondrial VDAC can be phosphorylated by cyclic AMP-dependent protein kinase. Biochem Biophys Res Commun. 1995;209:213–7. doi: 10.1006/bbrc.1995.1491. [DOI] [PubMed] [Google Scholar]
- 98.Huang LJ, Wang L, Ma Y, Durick K, Perkins G, Deerinck TJ, Ellisman MH, Taylor SS. NH2-Terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J Cell Biol. 1999;145:951–9. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ma Y, Taylor S. A 15-residue bifunctional element in D-AKAP1 is required for both endoplasmic reticulum and mitochondrial targeting. J Biol Chem. 2002;277:27328–36. doi: 10.1074/jbc.M201421200. [DOI] [PubMed] [Google Scholar]
- 100.Affaitati A, Cardone L, de Cristofaro T, Carlucci A, Ginsberg MD, Varrone S, Gottesman ME, Avvedimento EV, Feliciello A. Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J Biol Chem. 2003;278:4286–94. doi: 10.1074/jbc.M209941200. [DOI] [PubMed] [Google Scholar]
- 101.Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, Bronner V, Notcovich A, Shoshan-Barmatz V, Flescher E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008 doi: 10.1038/onc.2008.108. [DOI] [PubMed] [Google Scholar]