

Abstract
Morphine-induced glial proinflammatory responses have been documented to contribute to tolerance to opioid analgesia. Here, we examined whether drugs previously shown to suppress glial proinflammatory responses can alter other clinically relevant opioid effects; namely, withdrawal or acute analgesia. AV411 (ibudilast) and minocycline, drugs with distinct mechanisms of action that result in attenuation of glial proinflammatory responses, each reduced naloxone-precipitated withdrawal. Analysis of brain nuclei associated with opioid withdrawal revealed that morphine altered expression of glial activation markers, cytokines, chemokines, and a neurotrophic factor. AV411 attenuated many of these morphine-induced effects. AV411 also protected against spontaneous withdrawal-induced hyperactivity and weight loss recorded across a 12-day timecourse. Notably, in the spontaneous withdrawal study, AV411 treatment was delayed relative to the start of the morphine regimen so to also test whether AV411 could still be effective in the face of established morphine dependence, which it was. AV411 did not simply attenuate all opioid effects, as co-administering AV411 with morphine or oxycodone caused 3-to-5-fold increases in acute analgesic potency, as revealed by leftward shifts in the analgesic dose response curves. Timecourse analyses revealed that plasma morphine levels were not altered by AV411, suggestive that potentiated analgesia was not simply due to prolongation of morphine exposure or increased plasma concentrations. These data support and extend similar potentiation of acute opioid analgesia by minocycline, again providing converging lines of evidence of glial involvement. Hence, suppression of glial proinflammatory responses can significantly reduce opioid withdrawal, whilst improving analgesia.
Keywords: microglia, astrocyte, neuron, morphine, oxycodone
Introduction
Spinal cord glia are now known to contribute to morphine tolerance (Raghavendra et al., 2004), whereby less and less pain suppression (analgesia) is induced by the same opioid dose, requiring progressive increases in opioid dosing to maintain pain suppression. Astrocytes and microglia respond to repeated opioids in a proinflammatory fashion, with upregulated activation markers and proinflammatory cytokines. Attenuating this response, blocking glial proinflammatory mediators, or genetically disrupting interleukin-1 signaling markedly delays morphine tolerance (Watkins et al. (2007), for review).
Whether microglia and astrocytes broadly influence opioid actions is unknown but clinically important. For example, continued opioid exposure induces physical dependence, wherein continued exposure to opioids becomes required, and without which the organism displays a characteristic behavioral withdrawal syndrome (Fishbain et al., 1992). Treatments that circumvent such aspects of opioid dependence would be greatly beneficial. If drugs known to suppress microglial and astrocytic proinflammatory activation were found to curtail negative consequences of opioid use, this would both provide a novel strategy for increasing the clinical utility of opioids and provide the stimulus for an equally important parallel line of glial-opioid research that would complement the wealth of neuronal-opioid knowledge.
The present studies explore whether different-class drugs that suppress glial proinflammatory responses can suppress the expression of morphine withdrawal. Moreover, association between the glial proinflammatory response in brain nuclei previously implicated in opioid withdrawal and general opioid action, and the concomitant withdrawal behaviors is also addressed. Oxycodone withdrawal was additionally evaluated to determine whether such effects are generalizable beyond the prototypical opioid morphine, a method previously recommended when characterizing novel opioid actions (Sibinga and Goldstein, 1988). Oxycodone was chosen here as it is structurally distinct from morphine but, like morphine, is commonly used clinically for pain control. To explore generality, the effect of a glial attenuator was also assessed on the acute dose-response functions of morphine and oxycodone analgesias. To attenuate systemic opioid-induced glial proinflammatory responses in the central nervous system (CNS), AV411 (ibudilast; 3-isobutyryl-2-isopropylpyrazolo-[1,5-a]pyridine) was employed (Ledeboer et al., 2007). AV411 has been used for many years in the Orient for the treatment of bronchial asthma, post-stroke dizziness, and ocular allergies, based on its inhibition of platelet aggregation, inhibition of tracheal smooth muscle contractility, and improvement of cerebral blood flow (Ledeboer et al., 2007). AV411 was chosen for use here owing to its blood-brain-barrier permeability, documented suppression of proinflammatory responses by microglia in vitro and in vivo, tolerability, and long duration of action (Ledeboer et al., 2007). In addition, AV411 appears to distribute evenly throughout the CNS with no apparent depoting (Ledeboer et al., 2007). AV411 suppresses the production of proinflammatory cytokines and chemokines, nitric oxide, phosphodiesterase activity, and reactive oxygen species; has no known p38 MAP kinase effect; and increases the anti-inflammatory cytokine interleukin-10 and glia-derived neurotrophic factor (GDNF) (Ledeboer et al., 2007; Mizuno et al., 2004). Minocycline (2-Naphthacenecarboxamide, 4,7-bis(dimethylamino)-1,4,4a,5,5a,6,11,12a-octahydro-3,10,12,12a-tetrahydroxy-1,11dioxo-,monohydrochloride,[4S(4a,4aa,5aa,12aa)]-4,7-Bis(dimethylamino)-1,4,4a,5,5a,6,11,12a-octahydro-3,10,12,12a-tetrahydroxy-1,11-dioxo-2-naphthacenecarboxamide monohydrochloride, http://www.newdruginfo.com/pharmacopeia/usp28/v28230/usp28nf23s0_m54170.htm) is structurally distinct from ibudilast and is a compound which inhibits microglial p38 MAP kinase and proinflammatory cytokine production and release but has no known phosphodiesterase activity (Ledeboer et al., 2005; Yrjanheikki et al., 1998), was also assessed for attenuation of morphine withdrawal. Minocycline, a semi-synthetic second-generation tetracycline, was discovered in the early to mid-1990's to have anti-inflammatory properties separate from its actions as an antibiotic, including inhibition of matrix metalloproteinases, depression of oxygen radical release, inhibition of inducible nitric oxide synthase, and peroxynitrite scavenging (Yrjanheikki et al., 1998). These qualities, and its superior blood brain barrier permeability, led minocycline to be discovered as a microglial activation inhibitor in 1998 (Yrjanheikki et al., 1998). Minocycline was used in addition to AV411 in order to provide converging lines of evidence of the potential causal role of the proinflammatory activation of microglia and astrocytes in opioid withdrawal, as each drug shares the ability to attenuate glial proinflammatory responses. If proven efficacious here, it would provide the first evidence that: (a) microglia and astrocytes in brain may be importantly involved in phenomena associated with opioid dependence, and (b) a single manipulation (inhibiting microglial and astrocytic proinflammatory responses) may be able to simultaneously suppress some negative consequences of opioids (withdrawal) and enhance positive opioid actions (analgesia).
Methods
Subjects
Pathogen-free male Sprague–Dawley rats (300-350 g; Harlan Labs, Madison) were housed in pairs in temperature (23±3 °C) and light (12:12 light:dark) controlled rooms with ad libitum chow and water and allowed 1-week acclimatization. All procedures were performed during the light cycle and approved by the Institutional Animal Care and Use Committee. All data collected were obtained by personnel blinded to group assignment.
Drugs
Morphine sulfate (Mallinckrodt), AV411 (ibudilast; Avigen), naloxone HCl (Sigma), minocycline (Sigma), and oxycodone HCl (Sigma) were used. Where applicable, drugs are reported as free base concentrations.
Experiment 1: Effect of AV411 and minocycline on naloxone-precipitated morphine withdrawal when co-administered during the development of morphine dependence
Drug administration and behavior
Habituation (2×60min/day) to handling and the withdrawal environment preceded drug exposure. AV411: Rats received twice-daily i.p. AV411 (7.5mg/kg in 35% polyethylene glycol [PEG; Sigma] in saline; dose volume 2.5ml/kg) or equivolume i.p. vehicle, for 7-days (n=12/group). Minocycline: Rats received a 6-day dosing regimen (n=6/group). The evening prior to the first morphine dose, rats received 50mg/kg minocycline (or vehicle; 5ml/kg) by gavage, followed by twice daily 25mg/kg gavages. Minocycline was administered via gavage to mimic the clinically employed route of delivery and to avoid injection irritation issues experienced with repeated intraperitoneal minocycline injections. The morning injection occurred between 1:45 and 2:15h after lights on, with the afternoon injection occurring between 9:45 and 10:15h after lights on. On day 3 for AV411 and day 2 for minocycline, a 5-day dependence regimen began of subcutaneous morphine or equivolume vehicle (1ml/kg saline). When morphine was administered near AV411 or minocycline, it occurred 45min after the drug administration. The dependence regimen (times relative to lights on) dose escalated from 15 to 22.5mg/kg/day: day 3: 5mg/kg (2h), 5mg/kg (6h), 5mg/kg (10h); day 4: 7.5mg/kg (2h), 12.5mg/kg (10h); day 5: 15mg/kg (2h); day 6: 17.5mg/kg (2h); and day 7: 22.5mg/kg (2h). Such dosing regimens induce reliable dependence and naloxone precipitated withdrawal (Collier, 1972). Body weights were recorded prior to each dosing. Following the final dose, rats were placed in the withdrawal environment, consisting of a cage with bedding with an inverted clear Perspex cage enclosing the rats. Sixty min later, naloxone (10mg/kg in 1ml/kg) was administered subcutaneously to precipitate withdrawal. Withdrawal behaviors were scored by 2 investigators (3 rats/investigator) blinded to treatment for 6×10min blocks, with investigators rotating between the 2 sets of 3 rats every 10min to control for tester biases. Behaviors scored have previously been described as standardly associated with opioid withdrawal and are the set of behaviors routinely measured in studies of precipitated morphine withdrawal (Fdez Espejo et al., 1995; Gellert and Holtzman, 1978): jumping, rearing, exploration (movement greater than one body length), teeth chattering, wet dog shakes, abnormal posture, ptosis, diarrhea, penis licking, pica (oral stimulation by filling of mouth with bedding), paw chewing, cleaning, salivation, vocalization, chewing (large jaw movements including masseter muscle contraction) and fidgeting (a writhing type of behavior involving small shifts in body position). Counts of each were made upon their presentation. In cases where the response was prolonged, for example ptosis, counts were made every 30s. Each behavior was analyzed separately and as a combined score to provide an overall assessment of elicited withdrawal behavior, as is standard in the field (Fdez Espejo et al., 1995; Gellert and Holtzman, 1978).
Tissue collection
Following withdrawal assessment, rats were sacrificed (overdosed sodium pentobarbital; Abbott Laboratories) and then transcardially perfused. Six to nine rats of each group (randomly assigned) were perfused with saline only (for mRNA and protein quantification) and the remaining four with saline followed by 4% paraformaldehyde (for immunohistochemistry) and brains collected. Saline perfusion of tissues for mRNA and protein quantification ensured that measured analytes would not be confounded by any potential changes in circulating levels of these immune products by either morphine or AV411. Samples from saline perfused rats were flash frozen in liquid nitrogen and stored at -80°C. Paraformaldehyde perfused samples were stored in 4% paraformaldehyde for 48h and then transferred to 30% sucrose (0.1% azide) until sagittal sectioning. Brain nuclei from frozen, saline perfused brains were collected into chilled tubes and stored at -80°C for mRNA and protein analysis. The brain nuclei associated with the behavioral sequalae of opioid withdrawal (Bozarth, 1994) that were analyzed were the dentate gyrus and cornu ammonis of the hippocampus (Tremblay and Charton, 1981; Trujillo, 2000), dorsal periaqueductal gray (Maldonado et al., 1992; Punch et al., 1997), cingulate cortex (Trafton and Marques, 1971), substantia nigra (Baumeister et al., 1989; Baumeister et al., 1992), ventral tegmental area (Wang et al., 2004), central nucleus of the amygdala (Calvino et al., 1979; Tremblay and Charton, 1981) and mediodorsal thalamic nuclei (Tremblay and Charton, 1981). The selection of these, versus other withdrawal-associated regions, was based on the structures being of sufficient size to allow the analyses under study. In addition, to define if morphine-induced glial proinflammatory changes are restricted to brain regions associated with withdrawal versus brain regions more generally, the following brain nuclei were also analyzed: ventral periaqueductal gray, the bed nucleus of the stria terminalis, nucleus accumbens and medial prefrontal cortex.
Xb61
Immunohistochemistry
Immunoreactivity for the opioid withdrawal related brain nuclei (ventral tegmental area, nucleus accumbens, dentate gyrus, cornu ammonis of the hippocampus, dorsal periaqueductal gray and substantia nigra) and additional brain nuclei not associated with withdrawal (ventral periaqueductal gray, medial prefrontal cortex, caudate putamen, dorsal raphe nucleus, rostral ventromedial medulla and trigeminal nucleus) were assessed for markers of microglia (CD11b; OX42 labeling) and astrocytes (glial fibrillary acidic protein; GFAP) which are cell type specific markers known to upregulate in response to cellular activation (Milligan et al., 2001). As their use here is to detect increases in GFAP and CD11b expression commonly related to activation, for simplicity these are referred to here as activation markers. Densitometry of immunohistochemical staining was evaluated using an Olympus XB61 light microscope, NIH Image, and a macro that enables the area and intensity of the signal +3.5 SD above background to be determined automatically, as reported previously (Foley et al., 2006).
RNA isolation and enrichment
Total RNA was isolated based on the method of Chomczynski and Sacchi (1987), as previously described (Johnston et al., 2004). UV spectrophotometry was used to assess purity and concentration. Samples were DNase treated (DNA-free kit; Ambion), followed by requantitation before cDNA synthesis.
mRNA quantification
Amplification of cDNA was performed using the QuantiTect SYBR Green PCR Kit (Qiagen) in iCycler iQ 96 well PCR plates (Bio-Rad) on a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad), as previously described (Johnston et al., 2004). Quality controls were also as previously detailed (Johnston et al., 2004). SYBR Green 1 fluorescence (PCR product formation) was monitored in real time using the MyiQ Single Color Real-Time PCR Detection System (Bio-Rad). Threshold for detection of PCR product was set in the log–linear phase of amplification and the threshold cycle (CT, the number of cycles to reach threshold of detection) was determined for each reaction. The levels of the target mRNAs were quantified, using blinded procedures, relative to the level of the housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) using the comparative CT (ΔCT) method (Livak and Schmittgen, 2001). Expression of the housekeeping gene was not significantly altered by experimental treatment. The following targets were investigated: interleukin-1β and interleukin-10 (Ledeboer et al., 2005); and glial derived neurotrophic factor (NM_019139; forward: GGCTAACAAGTGACAAGGTA; reverse: AGGGTCAGATACATCCACA). Primers were purchased from Proligo.
Cytokine, chemokine and GDNF quantification
Tissue processing for protein quantification was as described previously (Johnston et al., 2004). As the balance of proinflammatory to anti-inflammatory cytokines/chemokines is increasingly recognized as important for their final effect, multiplex protein quantification (Thermo Scientific SearchLight) was utilized to quantify the following from single micropunches. Cytokine, chemokine and neurotrophic factors previously reported to be modifiable by at least some opioid regimens in some tissues were included: tumor necrosis factor-α(TNF-α) (Andjelkov et al., 2005; Liang et al., 2008), granulocyte macrophage colony stimulating factor (GMCSF) (Liang et al., 2008), interferon -γ(IFN-γ) (Carrigan et al., 2004; Roy et al., 2001), interleukin (IL)-10 (Limiroli et al., 2002; Messmer et al., 2006), IL-1β(Liang et al., 2008; Pourpak et al., 2004), IL-2 (Wang et al., 2007), and IL-6 (El-Hage et al., 2005; Liang et al., 2008), GRO/KC (CXCL1) (Liang et al., 2008), monocyte chemotatic protein-1 (MCP-1, CCL2) (El-Hage et al., 2005; Rock et al., 2006), macrophage inflammatory protein (MIP) -2 (Wang et al., 2005), Regulated upon Activation Normal T-cell Expressed and Secreted (RANTES; CCL5) (El-Hage et al., 2005; Happel et al., 2008), and fractalkine (Johnston et al., 2004). Glial derived neurotrophic factor (GDNF) was measured as elevation of GDNF has previously been documented to inhibit behavioral responses to drugs of abuse and decrease morphine-induced sensitization and opioid reward (Niwa et al., 2007; Niwa et al., 2007). Il-1α, MIP-1α and MIP-3α were also quantified as part of the broad screen of proinflammatory products but have not previously been reported to be modified by opioids. Each of these analytes are produced by astrocytes and/or microglia. However, a number of these have now been reported to be expressed by other cell types such as neurons. For example, there is expression of TNF in vitro by primary hippocampal neurons (Renauld and Spengler, 2002), GMCSF in neurons in human fetus (Dame et al., 1999), MIP-1alpha in cortical neurons of 1 of 13 neuropsychiatric patients (Ishizuka 97), and IFNgamma in a few neurons of hypothalamus and midbrain and in motoneurons following axotomy (Kiefer and Kreutzberg, 1990 although its relationship to immune-derived interferon-gamma has been questioned {Kiefer, 1991 #89; Olsson et al., 1989). MCP-1 likewise can be induced in neurons in response to injury (Zhang and De Koninck, 2006). IL-1beta has been observed in human hypothalamic neurons (Huitinga et al., 2000) and IL-6 can be induced in neurons in culture by S100beta (Li et al., 2000). Of the analytes investigated, GDNF has the broadest evidence of neuronal expression in widespread brain regions in adult rat (Pochon et al., 1997).
Hence, there is a potential for at least some of the results obtained to reflect changes in neuronal production as well as glial. All molecular protein endpoints were corrected for total protein concentration (Bradford assay).
Experiment 2: Effect of AV411 on spontaneous opioid withdrawal when administration begins after development of opioid dependence
Given the success of AV411 in Experiment 1, the influence AV411 has on spontaneous opioid withdrawal was also investigated. This protracted response mimics the clinical condition where opioid users and abusers experience pharmacokinetically induced removal of opioid action when drug administration is stopped rather than as a result of pharmacological blockade of opioid action. In addition, the experiment was designed to test whether AV411 could be effective when treatment began after morphine dependence was established; that is, several days of morphine dosing preceded the start of AV411. This is an important question as the literature supports that some treatments can suppress opioid withdrawal only when drug treatment begins prior to the establishment of dependence, but cannot affect withdrawal if begun after dependence exists (Shoemaker et al., 1997; Trujillo, 2000).
Rats were individually housed in the telemetry room and acclimatized 1 week. Five handling sessions (60min each group; n=6-10) were performed this week. Rats were anesthetized (isoflurane; Phoenix Pharmaceuticals) and emitters for measuring core body temperature and activity (MiniMitter) were implanted in the peritoneal cavity per manufacturer's instructions. The emitter had to move for activity to be counted. Activity counts and core body temperature were measured every min and a moving average over 120min was calculated for data smoothing. Computer-controlled recording of telemetry data occurred automatically throughout the entire experiment. Times when experimenters entered the room were eliminated from analyses due to spuriously increased activity. In the same surgery, rats were also implanted with 2 subcutaneous lumbar osmotic minipumps (model 2ML2, Alzet), which each pumped ∼5μ/h for 14 days (combined total: 10μ/h). One of the two pumps/rat had a lead length of polyethylene-60 tubing pre-loaded with saline to delay the morphine or oxycodone (or vehicle) delivery for 2 days. Therefore, the pumps delivered a combined dose of 6.25mg of morphine or oxycodone/day on days 1 and 2, and then 12.5mg/day from then onwards. On day 12-post opioid dosing, rats began a 7-day twice daily i.p. AV411 regimen (7.5mg/kg in 35% PEG in saline, dose volume 2.5 ml/kg) or equivolume i.p. vehicle, completing the final dose on the afternoon of day 18, hence ending 4 days after morphine or oxycodone. The morning injection of AV411 or vehicle occurred between 1:45 and 2:15h after lights on, with the afternoon injection occurring between 9:45 and 10:15h after lights on. On day 14, the pumps were removed under brief isoflurane anesthesia to begin spontaneous opioid withdrawal in rats receiving morphine and oxycodone. Body weights were recorded daily, including prior to each dosing session. In accordance with prior literature (Azar et al., 2004), spontaneous opioid withdrawal was quantified as decreases in body weight and changes in activity.
Experiment 3: Effect of AV411 on morphine and oxycodone analgesia
Rats received at least four 60min habituations to the test environment prior to behavioral testing. Thresholds for behavioral response to heat stimuli applied to the tail were assessed using a modified Hargreaves test (Hargreaves et al., 1988). All testing was conducted blinded. Briefly, baseline withdrawal values were calculated from an average of 2 consecutive withdrawal latencies of the tail, measured at 15-min intervals. Latencies for the thermal stimulus at baseline ranged from 2 to 3s, and a cut-off time of 10s was imposed to avoid tissue damage. Baseline withdrawal latency assessments were performed 45min prior to opioid administration, and 30min prior to opioid dosing, rats received i.p. AV411 or vehicle. Withdrawal latencies were again tested 10min prior to opioid administration. At time 0, rats received morphine or oxycodone subcutaneously (0.1, 0.4, 1, or 4mg/ml/kg, n=6/group) or vehicle and were tested every 10min for 230min.
Plasma morphine quantification
Plasma morphine concentrations were quantified to determine whether the pharmacodynamic differences in analgesia could potentially be explained by altered morphine pharmacokinetics with AV411 co-administration. The same dosing regimen as employed in the Hargreaves experiment (4mg/kg morphine, 7.5mg/kg AV411) was used here. Whole blood from tail vein bleeds (∼500μ) were collected 0, 5, 30, 60 and 180min following morphine administration, and plasma collected and frozen until analyzed. Morphine levels were quantified by a modification of a high-performance liquid chromatographic electrochemical detection method previously described (Doverty et al., 2001; Van Crugten et al., 1997). While morphine-6-glucuronide is the major metabolite of morphine, levels are low to undetectable in response to relatively low dose acute morphine administrations and, furthermore, any significant modulation of morphine-6-glucuronide levels would be reflected by alterations in morphine concentrations (Van Crugten et al. 1997).
The system consisted of an ESA 5600A Coularray detector with an ESA 5014B analytical cell and an ESA 5020 guard cell. The column was an ESA MD-150 (C-18, 3μm, 150×3.2mm), and the mobile phase was ESA buffer MD-TM. The analytical cell potentials were kept at -100mV and +250mV and the guard cell at +300mV. Plasma (100μ) was diluted in water for a total volume of 1ml. Samples were then alkalinized with 500μ of sodium bicarbonate buffer (500mM; pH=9.6) and extracted with chloroform (6ml) for 120s on vortex followed by centrifugation (1700×g; 10min). The upper aqueous layer was aspirated to waste followed by a further addition of the sodium bicarbonate buffer. Samples were then vortexed (10s) and centrifuged (1700×g; 10min). After aspirating the aqueous layer to waste, morphine was back extracted from 5 ml of chloroform into 300μ NaH2PO4 (50mM; pH=2) by vortexing for 120s. After centrifugation, an aliquot (100μ) of the aqueous phase was injected onto the system. Calibration standards ranged from 0.25ng/ml to 400ng/ml, and samples above this were diluted with water and reanalyzed. High (300ng/ml) and low (1ng/ml) quality control samples were assayed with each assay, and were within 10% of the nominal concentrations. The limit of quantification was 0.25ng/ml with inter-assay variability of the bottom standard 7.4%. The slope and intercept of the 1/Y weighted line of best-fit varied 4.3% and 3.4%, respectively. There were no interfering peaks in the chromatography.
Statistics
Statistical significance was assessed using a one-way repeated measure ANOVA with Bonferroni posthoc test when comparing the individual behaviors and densitometry values between treatment groups. For protein and mRNA analysis a two way ANOVA with Bonferroni posthoc test was used to compare the treatment effect of each molecular endpoint across each of the brain nuclei (n=6-9/group). The analgesic responses were calculated as the % of maximal possible effect (%MPE) using the following equation (Carmody, 1995). The area under the response curve was calculated using Prism 5.0 for individual rats and analyzed between groups with a Students t-test. All analyses and calculations were conducted with Excel 2003 SP2 (Microsoft), R Project version 2.6.1, SPSS 14.0.1 (SPSS) and Prism 5.0 (GraphPad). Significance was set at P<0.05.
Results
Experiment 1: AV411 and minocycline protect against naloxone-precipitated morphine withdrawal when co-administered during the development of morphine dependence
Morphine injections were escalated over 7 days (s.c. 15 to 22 mg/kg/day). This produced dependence as vehicle+morphine produced marked withdrawal behaviors across the 60min post-naloxone observation period (area under the withdrawal score curve: vehicle+morphine 2,358±260 vs. AV411+saline 332±73; Figure 1). Rats receiving AV411+morphine or minocycline+morphine exhibited significantly reduced overall withdrawal behavior scores compared to controls. AV411 and minocycline reliably reduced jumping (AV411: P<0.05; minocycline: P<0.05), teeth chattering (AV411: P<0.001; minocycline: P<0.05), abnormal posture (AV411: P<0.001; minocycline: P<0.05), ptosis (AV411: P<0.001; minocycline: P<0.05), diarrhea (AV411: P<0.001; minocycline: P<0.01), penis licking (AV411: P<0.05; minocycline: P<0.05), pica (AV411: P<0.05; minocycline: P<0.01), paw chewing (AV411: P<0.05; minocycline: P<0.05), cleaning (AV411: P<0.05; minocycline: P<0.05), salivation (AV411: P<0.05; minocycline: P<0.05), chewing (AV411: P<0.001; minocycline: P<0.01), and fidgeting (AV411: P<0.001; minocycline: P<0.01). No changes were observed, for either drug, for rearing, exploration, or wet dog shakes, compared to vehicle+morphine treated rats (P>0.05). Thus consistent effects were observed using the 2 mechanistically distinct inhibitors of glial proinflammatory responses. To facilitate comparison of data across glial attenuators, Figure 1 is expressed as the percent of maximal observed area under the naloxone precipitated opioid withdrawal timecourse. This was done since the PEG vehicle used for AV411 delivery caused a substantial enhancement in the morphine-induced withdrawal behaviors observed (P<0.001, ###).
Figure 1. AV411 and minocycline significantly reduced naloxone precipitated morphine withdrawal.
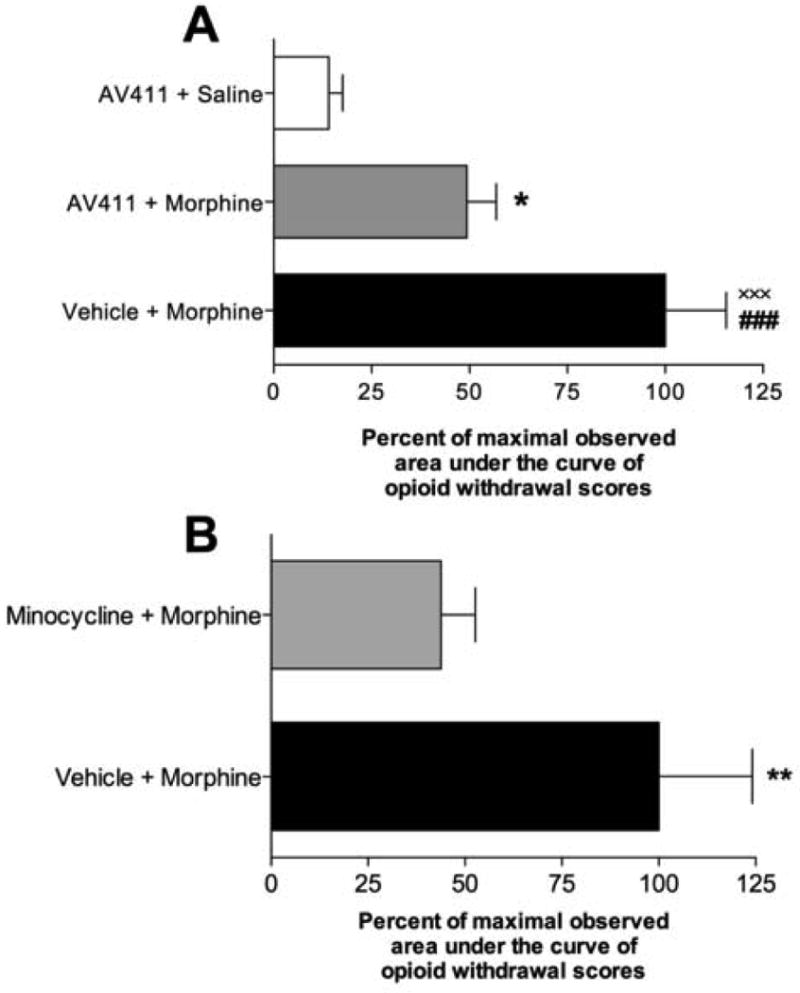
A: Co-administration of AV411 (7.5mg/kg twice daily) with morphine significantly reduced the area under the naloxone precipitated opioid withdrawal behaviors across a 60min post-naloxone timecourse, compared to vehicle+morphine treated rats. To enable of comparison of data, data are expressed as percent of maximal observed withdrawal score as the vehicle (PEG) caused a substantial modification in the behaviors observed (P<0.001, ###). Behaviors included in this analysis included: jumping, rearing, exploration (movement greater than one body length), teeth chattering, wet dog shakes, abnormal posture, ptosis, diarrhea, penis licking, pica (oral stimulation by filling of mouth with bedding), paw chewing, cleaning, salivation, vocalization, chewing (large jaw movements including masseter muscle contraction) and fidgeting (a writhing type of behavior involving small shifts in body position). Counts of each of these behaviors were made upon their presentation. In cases where the response was prolonged, for example ptosis, counts were made every 30s. As seen in the figure, some behaviors still persisted compared to AV411+saline treated rats (P<0.05, *). Vehicle+morphine rats displayed significant withdrawal behaviors compared to AV411+vehicle controls (P<0.001; ×××), n=10-11/group. B: Co-administration of minocycline (25mg/kg gavage twice daily) with morphine significantly reduced the sum of all scored naloxone precipitated opioid withdrawal behaviors compared to vehicle+morphine treated rats (P<0.01, **). n=6/group. Scored behavior and presentation are as in A, above.
Tissues were collected from these animals to enable analyses of where microglial and/or astrocytic activation would be observed in brain, by immunohistochemistry. The intent was to define whether microglia and astrocyte would both be globally affected, independent of brain site, versus revealing site-specificity and/or cell type specificity in the activation patterns observed. Immunohistochemical analyses of the microglial activation marker CD11b and astrocyte activation marker GFAP were thus conducted on the AV411+morphine brains and related control tissues (Table 1). For simplicity and space considerations, representative examples of the drug-induced changes in immunohistochemically-detected microglia and astrocyte activation markers are illustrated in Figure 2 from one brain region, chosen at random, but representative of the effects observed across nuclei. Vehicle+morphine significantly upregulated both microglial and astrocytic activation markers compared to AV411+saline in both opioid withdrawal related and other brain nuclei (Table 1). Morphine reliably increased the expression of GFAP in astrocytes in 5 of the 6 brain nuclei examined in both brain nuclei related to opioid withdrawal and general brain nuclei. In contrast morphine significantly increased the expression microglial CD11b in 3 opioid withdrawal related brain nuclei, and significantly decreased CD11b expression in the other 3 regions (Table 1). In the general brain nuclei examined, microglial CD11b expression was increased in 4 of 6 regions (Table 1). Of these opioid withdrawal related and general brain nuclei, AV411 reliably attenuated increases in GFAP expression in 4 of 6 and 3 of 6 brain nuclei, respectively. For morphine-induced CD11b expression changes, AV411 attenuated 5 of 6 morphine-induced changes in opioid withdrawal brain nuclei. AV411 attenuated morphine-induced CD11b changes in the general brain nuclei in 3 of 4 regions and caused a significant decrease in CD11b expression in 2 others (Table 1).
Table 1. AV411 reduced morphine-induced elevations of astrocytic GFAP and microglial CD11b activation markers as assessed by densitometry.
Densitometry analysis of individual brain nuclei for expression of GFAP or CD11b in several brain regions. A one-way ANOVA with Bonferroni post hoc analysis revealed significant reductions in glial activation markers when compared to saline+AV411 (* P<0.05, ** P<0.01, *** P<0.001) or morphine+AV411 (# P<0.05, ## P<0.05, ### P<0.001). Dark shading of the table cells indicates an increase in staining, whilst lighter cell shading signifies a decrease. n=4/group.
| Astrocyte activation (GFAP) | Microglial activation (CD11b) | |||||
|---|---|---|---|---|---|---|
| Saline + AV411 | AV411 + Morphine | Vehicle + Morphine | Saline + AV411 | AV411 + Morphine | Vehicle + Morphine | |
| OPIOID WITHDRAWAL ASSOCIATED BRAIN NUCLEI | ||||||
| Ventral tegmental area | 453.8±59.1 | 535.3±44.8 |
1041±82.0 *** ### |
54.4±20.10 | 64.0±19.1 |
185.2±39.6 * # |
| Nucleus accumbens | 120.4±15.6 |
313.3±23.0 *** |
340.5±31.6 *** |
226.3±22.1 | 245.8±76.1 |
58.8±15.5 * # |
| Dentate gyrus | 184.3±10.4 | 162.9±13.9 |
260.2±33.4 * ## |
21.2±3.5 | 22.2±3.0 |
49.3±2.9 *** ### |
| Cornu ammonis of the hippocampus | 190.7±22.6 | 232.2±18.7 |
406.3±27.7 *** ### |
132.4±22.7 | 102.6±7.0 |
59.3±6.6 * |
| Dorsal periaqueductal gray | 387.9±41.1 | 383.0±24.9 |
756.4±35.4 *** ### |
69.0±25.9 | 50.7±13.9 |
140.2±30.7 # |
| Substantia nigra | 291.0±31.2 |
133.1±30.7 * |
111.1±37.4 * |
340.6±55.3 |
141.5±19.8 * |
147.6±23.1 * |
| GENERAL BRAIN NUCLEI OF INTEREST | ||||||
| Medial prefrontal cortex | 42.2±4.3 |
242.7±35.4 ** ## |
253.7±26.5 ** ## |
90.6±63 |
31.2±6.8 ** |
61.9±5.7 |
| Ventral periaqueductal gray | 406.7±48.0 | 455.0±19.3 |
776.0±53.9 *** ### |
87.1±24.0 | 72.1±18.8 |
261.9±18.0 *** ### |
| Caudate Putamen | 227.2±32.2 | 243.5±18.5 |
442.0±26.4 *** ## |
25.8±2.0 | 24.4±3.5 |
64.0±3.6 *** ### |
| Dorsal raphe nucleus | 831.1±129.3 | 752.9±217.5 | 873.0±127.0 | 290.2±114.4 | 257.2±53.0 | 280.5±38.0 |
| Rostral ventromedial medulla | 71.8±5.9 | 168.8±22.3 |
443.0±2.5 *** ## |
30.1±6.2 | 22.7±2.0 |
75.3±7.7 * # |
| Trigeminal Nucleus | 85.3±17.7 |
188.4±14.8 * |
429.7±10.8 *** ## |
53.2±3.0 |
23.02±3.0 * |
83.5±7.6 ** # |
Figure 2. Morphine-induced increases in microglial (CD11b expression) and astrocytic (GFAP expression) activation were significantly reduced by AV411 treatment.
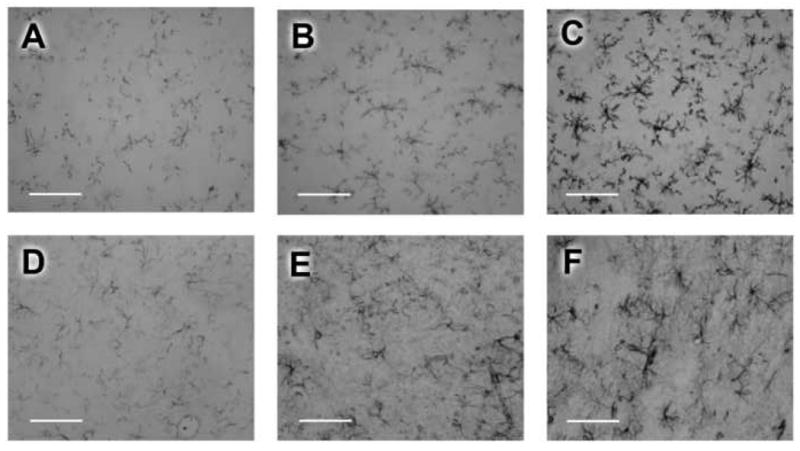
For simplicity, representative examples of the drug-induced changes in immunohistochemically-detected glial activation markers are illustrated from one brain region, chosen at random. Microglial CD11b expression from the dorsal periaqueductal gray of representative rats (A, B and C) following naloxone precipitated withdrawal in rats that received AV411+saline (A), AV411+morphine (B) and vehicle+morphine (C). Astrocytic GFAP expression from the dorsal periaqueductal gray of representative rats (D, E and F) following naloxone precipitated withdrawal in rats that received AV411+saline (D), AV411+morphine (E) and vehicle+morphine (F). Image acquisition was done using an Olympus XB61 microscope and NIH image. Scale bar=100μm.
Brains from separate rats from the behavioral study were used to analyze mRNA and protein of potential mediators of interest (for citations below, see Experiment 1 Methods). A broad analysis was done, in part, given increasing recognition that the relative balance of proinflammatory and anti-inflammatory mediators can impact brain function and behavior. The choices of analytes were also governed by prior literature reporting that TNF-α, GMCSF, IFN-γ, IL-10, IL-1β, IL-2, IL-6, GRO/KC, MCP-1 (CCL2), MIP-2, RANTES (CCL5), and fractalkine are modifiable by at least some opioid regimens in some tissues. GDNF was measured as GDNF elevation has previously been reported to suppress behavioral responses to drugs of abuse, decrease morphine-induced sensitization, and decrease morphine reward. Related analytes (IL-1α, MIP-1α and MIP-3α) were included as part of the proinflammatory screen but not previously studied with regard to modifiability by opioids. These analyses were done on both opioid withdrawal related nuclei and other nuclei not previously related to opioid withdrawal in order to define whether observed changes across sites were selective for sites linked to opioid withdrawal versus pervasive and nonspecific in their distribution. In addition, a broad site analysis allowed definition of whether specific changes in specific analytes were always observed in response to morphine and/or blocked by AV411, versus displaying site specificity in the results obtained.
Analyses of mRNA and protein in micropunched brain nuclei revealed numerous reliable treatment effects on cytokine, chemokine and GDNF protein and mRNA expression that occurred 7 of the 8 opioid withdrawal related nuclei examined, but in only 1 of the 4 general brain nuclei (Table 2). In addition, IL-1βmRNA was significantly reduced in the medial prefrontal cortex in AV411+morphine treated animals and significantly elevated in the dorsal periaqueductal gray in vehicle+morphine animals. The majority of the significant protein changes quantified for vehicle+morphine treated animals were predominantly proinflammatory (9 of 12) whilst for AV411+morphine treated animals significant changes were mostly reductions in the expression of proinflammatory mediators (6 of 11). The mRNA and protein changes documented here cannot be accounted for by alterations in circulating levels of these analytes as a result of morphine or AV411 treatment, as all animals were transcardially perfused with saline prior to tissue collections to avoid this potential confound.
Table 2. Protein analyses following morphine withdrawal.
Summary of the significant cytokine, chemokine and GDNF changes in brain nuclei resulting from morphine withdrawal and the effect of AV411 treatment. Micropunches from individual brain nuclei were analyzed for cytokine, chemokine and GDNF protein. Significant changes were assessed using a two-way ANOVA with Bonferroni posthoc test comparing each molecular endpoint across the brain nuclei. P < 0.05; Significances are classified as to which treatment group the difference refers to, represented by the use of the column are identifier. Hence a superscripted A following a table value means that this value is significantly different from the parallel value shown in column A (AV411 + Saline), a superscripted B following a table value means that this value is significantly different from the parallel value shown in column B (AV411 + Morphine), and a superscripted C following a table value means that this value is significantly different from the parallel value shown in column C (Vehicle + Morphine). n=6-7/group.
Cytokine, chemokine and GDNF analyses
| Brain nuclei | Chemokine/Cytokine | AV411 + Saline (pg/μg protein) A |
AV411 + Morphine (pg/μg protein) B |
Vehicle + Morphine (pg/μg protein) C |
|---|---|---|---|---|
| Ventral tegmental area | IL-1β | 0.24±0.06 | 0.12±0.03 A | 0.18±0.05 |
| IL-6 | 1.56±0.89 | 0.19±0.06 A | 0.38±0.04 | |
| MIP-1α | 0.08±0.03 | 0.02±0.01 A | 0.03±0.01 | |
| RANTES | 0.16±0.02 | 0.11±0.02 C | 0.18±0.04 | |
| GDNF | 0.56±0.09 | 0.29±0.08 A, C | 0.53±0.07 | |
| Nucleus accumbens | IFN-γ | 0.18±0.06 | 0.44±0.09 | 0.60±0.06 A |
| RANTES | 0.06±0.02 | 0.08±0.01 | 0.15±0.02 A | |
| Dentate gyrus | GDNF | 0.07±0.02 | 0.13±0.02 | 0.01±0.01 B |
| MIP-3α | 2.07±0.69 | 1.28±0.13 | 0.52±0.17 A, B | |
| Cornu ammonis of the hippocampus | IL-1β | 0.08±0.01 | 0.05±0.01 | 0.16±0.04 B |
| Fractalkine | 4.73±0.56 | 2.73±0.27 A, C | 4.85±0.89 | |
| GRO/KC | 0.12±0.02 | 0.09±0.01 | 0.19±0.05 A, B | |
| MCP-1 | 0.22±0.03 | 0.15±0.02 | 0.38±0.13 B | |
| Dorsal periaqueductal gray | IL-1β | 0.09±0.02 | 0.09±0.03 | 0.16±0.03 B |
| MCP-1 | 0.27±0.07 | 0.17±0.04 | 0.40±0.09 B | |
| MIP-3α | 0.86±0.29 | 0.55±0.22 | 1.35±0.16 B | |
| Substantia nigra | IL-1β | 0.06±0.02 | 0.13±0.02 A, C | 0.05±0.02 |
| IL-6 | 1.8±0.10 | 0.33±0.08 A | 0.32±0.08 A | |
| GDNF | 0.56±0.09 | 0.32±0.08 A | 0.45±0.08 | |
| Central Nucleus of the Amygdala | MIP-2 | 0.01±0.01 | 0.04±0.01 A | 0.04±0.01 A |
| Medial prefrontal cortex | GDNF | 0.13±0.02 | 0.25±0.01 A, C | 0.12±0.02 |
Experiment 2: AV411 decreases spontaneous opioid withdrawal when administration begins after development of opioid dependence
Subcutaneous osmotic minipumps delivered 6.25mg of morphine or oxycodone per day on days 1 and 2, and then 12.5mg/day from then onwards through the end of the experiment. A weeklong AV411 (vs. vehicle) treatment was delayed until 12 days after the start of the opioid regimen to allow dependence to develop prior to AV411 administration. This delay was included to test whether AV411 could be effective after morphine dependence was established, as prior literature documents other drugs that fail to affect withdrawal if drug treatment begins after the establishment of opioid dependence (Shoemaker et al., 1997; Trujillo, 2000). AV411 administered after the establishment of morphine dependence significantly protected rats from both morphine and oxycodone spontaneous withdrawal-induced weight loss that occurred following removal of drug pumps (Figure 3). Within one day after spontaneous withdrawal initiation, vehicle+morphine treated rats lost 53.8±4.0g and vehicle+oxycodone treated rats lost 55.7±5.0g whilst AV411+morphine rats (36.0±5.1g) and AV411+oxycodone rats (28.2±2.5g) lost significantly less (P<0.05).
Figure 3. AV411 prevents spontaneous morphine and oxycodone withdrawal induced weight loss.
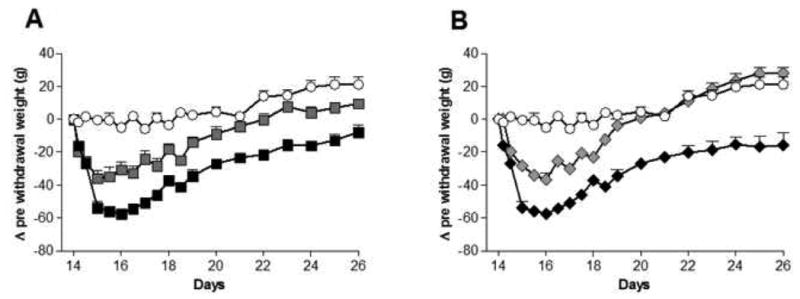
AV411 (7.5mg/kg twice daily) protects against morphine (A; ■) and oxycodone (B; ◆) induced weight loss when compared to morphine (■) or oxycodone (◆) plus vehicle treated rats. It appears that these weight loss effects are likely due to the opioid withdrawal, since AV411+saline rats do not show such weight loss (○). Data are expressed as the change in weight after the start of spontaneous withdrawal, compared to weight just prior to the initiation of spontaneous withdrawal. A two-way ANOVA with Bonferroni post hoc analysis revealed significant differences between AV411 and vehicle treated rats just one day after withdrawal that continued thought the rest of the observation (day 15 to day 26; P<0.05). n=6-9/group.
Telemetry monitoring of core body temperature and activity during morphine treatment revealed significant morphine-induced hyperactivity and associated dysregulation of diurnal core body temperature pattern. Upon commencement of AV411, brief, small yet significant hypothermic responses were observed following AV411 administration, which tolerated by the fourth administration. Analysis of core body temperature data suggests that once AV411 treatment terminated, no other significant systematic differences exist between treatment groups (P>0.05). However, activity data following completion of AV411 and vehicle treatment revealed substantial differences between groups with significant and extended hyperactivity during the active dark phase in vehicle+morphine treated rats (total activity count for the dark phase of day 20: 2361±611, day 21: 2262±599) compared to AV411+saline (day 20: 1122±91, day 21:1083±21; P<0.01) which is not apparent following AV411+morphine (day 20: 1346±77, day 21:1330±90; P<0.05). Given the severity of oxycodone withdrawal, analyses were restricted to the morphine-treated groups.
Experiment 3: AV411 potentiates morphine and oxycodone analgesia
Rats were acutely administered AV411 or vehicle, plus one of four doses of either morphine or oxycodone. AV411 alone did not produce any change in hind paw or tail flick latencies over a 100min timecourse (data not shown). However, when combined with morphine or oxycodone, a significant potentiation and prolongation of analgesia was observed (Figure 4) resulting in significant leftward shifts in the dose response curve and a corresponding reduction in the ED50. Importantly, like the generalized effects observed for opioid withdrawal, the effects of AV411 were not restricted to simply morphine but rather extended to another clinically relevant opioid (oxycodone) as well. As there were no significant changes in morphine plasma concentrations observed when morphine was administered in combination with AV411 as compared to morphine-alone animals (30min post morphine maximal concentration morphine+vehicle 97.5±3.3 ng/ml versus 93.7±9.3ng/ml and no change in area under the curve (AUC); P>0.05), this is suggestive that the results are not readily accounted for by higher morphine exposure or elevated plasma concentrations. This potentiation of opioid analgesia is paralleled by our recent observations that minocycline markedly potentiates morphine analgesia as well (Hutchinson et al., 2008).
Figure 4. AV411 potentiates both morphine and oxycodone analgesia.
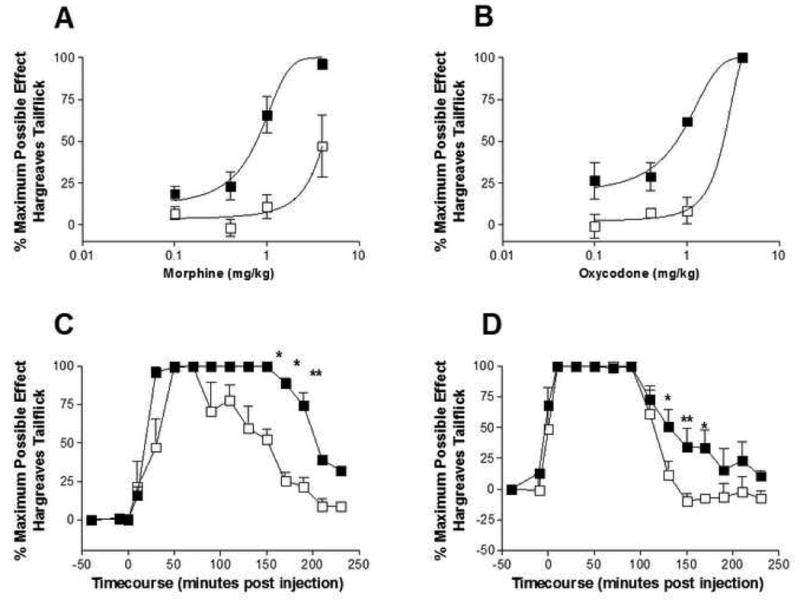
AV411 (7.5mg/kg; ■) significantly potentiated morphine (A and C: 4mg/kg) and oxycodone (B and D: 4mg/kg) analgesia compared to vehicle treated rats (□). The EC50 of morphine+vehicle dose response (30min after opioid administration) was 4.1mg/kg whilst AV411 co-administration reduced this significantly to 0.77mg/kg (P<0.0001). The EC50 of oxycodone+vehicle dose response was 2.6mg/kg whilst AV411 co-administration reduced this significantly to 0.78mg/kg (P<0.001). n=6/group.
Discussion
These data provide the first characterization of attenuated opioid withdrawal by AV411 and minocycline. These studies support involvement of opioid-induced proinflammatory responses in withdrawal, as minocycline and AV411 attenuate glial proinflammation via distinct mechanisms. Precipitated and spontaneous opioid withdrawals were reduced by AV411, a drug candidate in clinical development for treating neuropathic pain. Both morphine and oxycodone were tested as structurally distinct opioids commonly used clinically for pain control. As such, this comparison provides a test of generality that the effects observed are not restricted to simply morphine. Notably, in the spontaneous withdrawal study, AV411 was delayed relative to initiating morphine treatment to test whether AV411 could effect established morphine dependence. This is important if one were to consider a drug such as AV411 for treating dependence clinically. Minocycline also reduced precipitated withdrawal, providing converging lines of evidence suggestive of microglia and/or astrocyte involvement. AV411 did not reduce all opioid actions, as morphine and oxycodone analgesias were potentiated. This caused a leftward shift in the dose response curves, significantly reducing analgesia ED50s. AV411 also enhanced the magnitude and duration of opioid analgesia. These AV411 effects parallel our observation that minocycline potentiates morphine analgesia (Hutchinson et al., 2008) and are consistent with potentiated analgesia by suppressing actions of opioid-induced pronociceptive glial products (Shavit et al., 2005). Potentiation of analgesia by AV411 and minocycline is likely indirect, via suppressing release of pain-enhancing glial products that oppose, or counter-regulate, analgesia. For neurochemical analyses, brain nuclei were chosen to test specificity of effects to sites relevant to opioid withdrawal (see Methods) versus generality of morphine-induced changes in glial activation markers, chemokines, cytokines, and GDNF. Repeated morphine administration induced significant changes in these analytes that, by-and-large, were prevented by AV411. Despite many nuclei displaying changes in microglial and/or astrocyte activation marker expression, changes in proinflammatory mediators were primarily observed in brain nuclei previously associated with morphine withdrawal.
AV411 and minocycline attenuated most morphine withdrawal behaviors. Rearing and exploration were unaffected and rats treated with AV411 or minocycline appeared qualitatively more alert. These latter observations are consistent with effects unrelated to overt anesthesia or sedation. AV411 also suppressed spontaneous withdrawal. These effects also suggest that the common denominator across the glial modulators is not enhancement of cAMP. While AV411 does enhance cAMP (Ledeboer et al, 2007), minocycline has the opposite effect so to reduce cAMP (Mork and Geisler, 1995).
Repeated morphine induced an intriguing pattern of glial activation markers and proinflammatory mediator expression. Firstly, morphine-induced changes in glial activation markers were similarly distributed between many brain regions, regardless of whether or not the nuclei were linked to withdrawal phenomena. The brain regions examined here incorporate a much wider range of sites than investigated previously. Song et al. (2001) documented heterogeneous GFAP expression following repeated morphine in posterior cingulate, hippocampus and thalamus. Other studies of GFAP expression following repeated morphine demonstrated increases in locus coeruleus and solitary nucleus (Alonso et al., 2007), ventral tegmental area (VTA), nucleus accumbens (NAcc) shell and frontal cortex (Garrido et al., 2005). The present study is the first survey of morphine-induced microglial activation (CD11b), alone or in combination with astrocyte GFAP. It is clear, based on the site specific results reported, that microglia and astrocytes can be differentially and site-specifically affected by systemic opioids. AV411 attenuated morphine-induced GFAP and CD11b changes in most cases, mirroring reductions in withdrawal behaviors. If AV411 reduces opioid withdrawal by attenuating opioid-induced glial activation, these glial activation marker expression patterns provide anatomical targets for future investigations. Moreover, the results suggest a diversity of mechanisms for opioid-induced glial regulation in various brain regions.
However, glial activation markers do not necessarily predict molecular mediator expression. Therefore, proteins were examined utilizing multiplex technology. Morphine induced proinflammatory changes in NAcc (IFNγ, RANTES), cornu ammonis of hippocampus (IL-1β, GRO/KC, MCP-1), dorsal periaqueductal gray (dPAG) (IL-1β, MCP-1, MIP-3α) and central nucleus of the amygdala (MIP-2), but decreased MIP-3α in dentate gyrus (DG). This pattern occurred only in opioid withdrawal-associated brain nuclei and not in other brain regions. AV411 attenuated the protein changes in NAcc, DG, cornu ammonis of the hippocampus and dPAG. AV411+morphine also induced specific changes in VTA (broadly reducing proinflammatory mediators), substantia nigra (increased IL-1β, decreased GDNF) and medial prefrontal cortex (increased GDNF). This pattern of changes is the first of their kind reported. In contrast to our findings of both proinflammation and attenuation proinflammation in the hippocampus, Patel et al. (1996) reported decreased IL-1β throughout hippocampus, using a much higher morphine regimen.
Little other data exist relating chronic morphine, morphine dependence, and changes in brain cytokines, chemokines and GDNF. However, elevations of IL-β in cat dPAG induce defensive rage (Bhatt et al., 2008; Zalcman and Siegel, 2006). This provides an interesting parallel with the current data, given elevations of several proinflammatory cytokines and chemokines occurred in this region and several behaviors displayed during opioid withdrawal are rage-like in nature. Further, involvement of immunocompetent cells in opioid withdrawal was suggested 25 years ago by Dafny and coworkers (Dafny, 1983; Dougherty et al., 1990), who demonstrated that whole body immunosuppression inhibited morphine withdrawal. Our data suggest that Dafny may have non-selectively inhibited glial proinflammatory responses, thereby reducing morphine withdrawal. Such proinflammatory mediators can exert direct actions on neurons (Zhang et al., 2007) as well as induce various downstream changes that alter neuronal function, such as downregulation of glial glutamate transporters (Nakagawa et al., 2005; Nakagawa and Satoh, 2004; Ozawa et al., 2004; Ozawa et al., 2001) potentially contributing to opioid withdrawal (Nakagawa and Satoh, 2004). Collectively these data are intriguing as they suggest possible site specificity of morphine action and microglial and/or astrocytic response to morphine, mediated in such sites or indirectly via projections received from other regions directly affected by opioids. This latter possibility would be parsimonious with upregulated glial activation markers in projection regions of activated neurons (Holguin et al., 2007). Alternatively, variations in kinetic access of morphine to specific brain regions may account for the differences observed.
It is recognized that it is a very complex undertaking to understand the different interactions of various cytokines and markers seen in opioid withdrawal-associated brain nuclei with behavioral responses. The present study provides the first data toward an understanding of these complexities. The analysis of multiple brain sites identify a number of specific nuclei that are versus are not affected by the procedures under study and provide information as to what potential mediators are altered in specific sites. The present data lay the groundwork for future studies aimed at integrating the observed glial and proinflammatory changes in withdrawal-associated nuclei with withdrawal behaviors. First, while the present study identified specific mRNA/protein changes in micropunches from specific nuclei, their cellular origin(s) remains to be defined. Secondly, the significance of the patterns of cytokine and chemokine changes observed will be important to explore, given that multiple cytokines and chemokines can work in concert to produce behavioral responses (Hutchinson et al., 2008). Finally, the downstream neuronal consequences of these proinflammatory changes that potentially contribute to withdrawal behaviors can now be addressed, given the documented site-specific chemokine/cytokine changes. Such studies will likely advance the understanding of mechanisms underlying opioid withdrawal.
It is also noted that the tissues for mRNA, protein, and glial activation marker analyses were all collected after repeated morphine and a one hour precipitated withdrawal, so that what the values were pre-withdrawal for morphine-treated rats is at present unknown. Given the size and complexity of the study, adding these additional groups were not possible in the current work. This is a consideration primarily, if not exclusively, for the mRNA and multiplex protein data rather than for the glial activation marker expression by immunohistochemistry. That is because alteration in CD11b and GFAP levels is a far slower process than for the other mRNA/protein analytes under study. For example, even upon strong stimulation of spinal glial activation by intrathecal HIV-1 gp120, it was 8-18 hr later that GFAP expression became elevated and 4 hr later for elevated CD11b expression (Milligan et al., 2001). Such delayed timings are common in the glial activation literature. Hence it is likely that the immunohistochemistry results reported here reflect the effect of repeated morphine exposure rather than the relatively brief survival after precipitation of morphine withdrawal.
The present data would seem to present a dilemma, since it could be hypothesized that by blocking proinflammatory responses one may globally inhibit opioid activity. However, AV411 potentiated morphine and oxycodone analgesia 3-to-5 fold, with no change in plasma morphine levels. Potentiation of analgesia is consistent with previous reports of potentiated analgesia caused by suppressing the action of opioid-induced pronociceptive glial products, which oppose opioid analgesia (Hutchinson et al., 2008; Shavit et al., 2005). It may prove parsimonious to view enhancement of opioid analgesia by AV411 not as enhancing pain suppressive mechanisms, per se, but rather removing opioid-induced pain enhancement acting as an opponent process in parallel. We have also demonstrated that AV411 and minocycline reduce the behavioral (Hutchinson et al., 2007) and neurochemical (Ledeboer et al., 2007) signs of morphine reward. Likewise, morphine induced respiratory depression is reduced by minocycline (Hutchinson, 2007). Therefore, drugs like AV411 and minocycline enhance analgesia while attenuating reward and dependence and improving the safety profile.
Mechanistically, minocycline possesses neuroprotective and anti-inflammatory properties, independent of its antibiotic activity. Its anti-inflammatory actions include p38 MAPK inhibition (Cui et al., 2008; Stirling et al., 2005), reduced cell motility, and downregulation of β1-integrin and Kv1.3 channels (Nutile-McMenemy et al., 2007). AV411 attenuates microglia proinflammatory responses in vitro and in vivo (Ledeboer et al., 2007; Suzumura et al., 1999). AV411 does not interact with a number of neuronal targets at least linked to neuropathic pain regulation, if not opioid analgesia and withdrawal (Ledeboer et al., 2007; Ledeboer et al., 2007). cAMP elevation related to phosphodiesterase inhibition cannot be excluded as a component of glial regulation by AV411, and at least one phosphodiesterase inhibitor, rolipram, has been reported to regulate glial activation (Zhang et al., 2002). However, phosphodiesterase inhibition appears insufficient since a closely-related AV411 analog with little-to-no PDE inhibition attenuates glia and mimics AV411 pharmacology in vivo, including attenuation of opioid withdrawal (Ledeboer et al., 2007). Notably, minocycline has no known PDE inhibitory activity, again suggestive that PDE inhibition is not required for effects reported here. A proinflammatory cytokine target for AV411, which has not been previously recognized and which may regulate glial proinflammatory responses, has been preliminarily identified and is currently under investigation (K. Johnson et al., unpublished data).
In summary, we demonstrate that two structurally and mechanistically distinct glial modulatory drugs, AV411 and minocycline, each suppress the expression of opioid withdrawal and enhance acute opioid analgesia. These effects are generalized, as common effects were observed for both morphine and oxycodone. Together with our recent demonstration that AV411, as well as minocycline, reduces morphine reward (Hutchinson et al., 2007), this novel indication suggests that human clinical trials may be warranted to determine if these findings can be extrapolated to humans or whether other drugs of abuse may be similarly modified by glial modulatory compounds.
Acknowledgments
Funding for these studies was provided by Avigen Inc., NIH grants DA015642, DA017670 and DE017782, National Health and Medical Research Council of Australia CJ Martin Fellowship (ID 465423), International Association for the Study of Pain International Collaborative grant and American Australian Association Merck Company Foundation Fellowship. Special thanks to the Pierce technical support team for the development of multiplex assays suitable for rat central nervous system tissue and to Mallinkrodt for kindly gifting the morphine. We would like to thank Lindsey Chao, Jeffrey Kearney and Paige Sholar for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso E, Garrido E, Diez-Fernandez C, Perez-Garcia C, Herradon G, Ezquerra L, Deuel TF, Alguacil LF. Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha(2)-adrenoceptor gene expression in hippocampus. Neuroscience Letters. 2007;412:163–7. doi: 10.1016/j.neulet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Andjelkov N, Elvenes J, Martin J, Johansen O. Opiate regulation of IL-1beta and TNF-alpha in cultured human articular chondrocytes. Biochem Biophys Res Commun. 2005;333:1295–9. doi: 10.1016/j.bbrc.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Azar MR, Ahmed SH, Lintz R, Gutierrez T, Stinus L, Koob GF. A non-invasive gating device for continuous drug delivery that allows control over the timing and duration of spontaneous opiate withdrawal. Journal of Neuroscience Methods. 2004;135:129–35. doi: 10.1016/j.jneumeth.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Baumeister AA, Anticich TG, Hebert G, Hawkins MF, Nagy M. Evidence that physical dependence on morphine is mediated by the ventral midbrain. Neuropharmacology. 1989;28:1151–7. doi: 10.1016/0028-3908(89)90204-9. [DOI] [PubMed] [Google Scholar]
- Baumeister AA, Richard AL, Richmond-Landeche L, Hurry MJ, Waguespack AM. Further studies of the role of opioid receptors in the nigra in the morphine withdrawal syndrome. Neuropharmacology. 1992;31:835–41. doi: 10.1016/0028-3908(92)90119-a. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Bhatt R, Zalcman SS, Siegel A. Role of IL-1 beta and 5-HT2 receptors in midbrain periaqueductal gray (PAG) in potentiating defensive rage behavior in cat. Brain Behav Immun. 2008;22:224–33. doi: 10.1016/j.bbi.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozarth MA. Physical dependence produced by central morphine infusions: an anatomical mapping study. Neuroscience and Biobehavioral Reviews. 1994;18:373–83. doi: 10.1016/0149-7634(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Calvino B, Lagowska J, Ben-Ari Y. Morphine withdrawal syndrome: differential participation of structures located within the amygdaloid complex and striatum of the rat. Brain Res. 1979;177:19–34. doi: 10.1016/0006-8993(79)90915-6. [DOI] [PubMed] [Google Scholar]
- Carmody J. Avoiding fallacies in nociceptive measurements. Pain. 1995;63:136. doi: 10.1016/0304-3959(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Carrigan KA, Saurer TB, Ijames SG, Lysle DT. Buprenorphine produces naltrexone reversible alterations of immune status. Int Immunopharmacol. 2004;4:419–28. doi: 10.1016/j.intimp.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Colloier HO. Drug dependence: a pharmacological analysis. Brit J Addict Alcohol Other Drugs. 1972;67:277–286. [PubMed] [Google Scholar]
- Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, Chen PX, Feng JQ. A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain, Behavior, and Immunity. 2008;22:114–23. doi: 10.1016/j.bbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Dafny N. Interferon modifies morphine withdrawal phenomena in rodents. Neuropharmacology. 1983;22:647–51. doi: 10.1016/0028-3908(83)90157-0. [DOI] [PubMed] [Google Scholar]
- Dame JB, Christensen RD, Juul SE. The distribution of granulocyte-macrophage colony-stimulating factor and its receptor in the developing human fetus. Pediatr Res. 1999;46:358–66. doi: 10.1203/00006450-199910000-00002. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Pellis NR, Dafny N. The brain and the immune system: an intact immune system is essential for the manifestation of withdrawal in opiate addicted rats. Neuroscience. 1990;36:285–9. doi: 10.1016/0306-4522(90)90293-d. [DOI] [PubMed] [Google Scholar]
- Doverty M, Somogyi AA, White JM, Bochner F, Beare CH, Menelaou A, Ling W. Methadone maintenance patients are cross-tolerant to the antinociceptive effects of morphine. Pain. 2001;93:155–63. doi: 10.1016/S0304-3959(01)00306-2. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fdez Espejo E, Cador M, Stinus L. Ethopharmacological analysis of naloxone-precipitated morphine withdrawal syndrome in rats: a newly-developed “etho-score”. Psychopharmacology. 1995;122:122–30. doi: 10.1007/BF02246086. [DOI] [PubMed] [Google Scholar]
- Fishbain DA, Rosomoff HL, Rosomoff RS. Drug abuse, dependence, and addiction in chronic pain patients. Clinical Journal of Pain. 1992;8:77–85. doi: 10.1097/00002508-199206000-00003. [DOI] [PubMed] [Google Scholar]
- Foley TE, Greenwood BN, Day HE, Koch LG, Britton SL, Fleshner M. Elevated central monoamine receptor mRNA in rats bred for high endurance capacity: implications for central fatigue. Behavioural Brain Research. 2006;174:132–42. doi: 10.1016/j.bbr.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Garrido E, Perez-Garcia C, Alguacil LF, Diez-Fernandez C. The alpha2-adrenoceptor antagonist yohimbine reduces glial fibrillary acidic protein upregulation induced by chronic morphine administration. Neuroscience Letters. 2005;383:141–4. doi: 10.1016/j.neulet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Gellert VF, Holtzman SG. Development and maintenance of morphine tolerance and dependence in the rat by scheduled access to morphine drinking solutions. Journal of Pharmacology and Experimental Therapeutics. 1978;205:536–46. [PubMed] [Google Scholar]
- Happel C, Steele AD, Finley MJ, Kutzler MA, Rogers TJ. DAMGO-induced expression of chemokines and chemokine receptors: the role of TGF-beta1. J Leukoc Biol. 2008;83:956–63. doi: 10.1189/jlb.1007685. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Holguin A, Frank MG, Biedenkapp JC, Nelson K, Lippert D, Watkins LR, Rudy JW, Maier SF. Characterization of the temporo-spatial effects of chronic bilateral intrahippocampal cannulae on interleukin-1beta. Journal of Neuroscience Methods. 2007;161:265–72. doi: 10.1016/j.jneumeth.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitinga I, van der Cammen M, Salm L, Erkut Z, van Dam A, Tilders F, Swaab D. IL-1beta immunoreactive neurons in the human hypothalamus: reduced numbers in multiple sclerosis. J Neuroimmunol. 2000;107:8–20. doi: 10.1016/s0165-5728(00)00248-4. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. TheScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid induced acute and chronic analgesia. Brain, Behavior, and Immunity. 2008 doi: 10.1016/j.bbi.2008.05.004. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. Journal of Neuroscience. 2004;24:7353–65. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer R, Kreutzberg GW. Gamma interferon-like immunoreactivity in the rat nervous system. Neuroscience. 1990;37:725–34. doi: 10.1016/0306-4522(90)90103-b. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs. 2007a;16:935–50. doi: 10.1517/13543784.16.7.935. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vijay S, Gross MI, Vargas JA, Sultzbaugh L, Claypool MD, Sanftner LM, Watkins LR, Johnson KW. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biology. 2007b;21:279–291. doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Li Y, Barger SW, Liu L, Mrak RE, Griffin WS. S100beta induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000;74:143–50. doi: 10.1046/j.1471-4159.2000.0740143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Shi X, Qiao Y, Angst MS, Yeomans DC, Clark JD. Chronic morphine administration enhances nociceptive sensitivity and local cytokine production after incision. Mol Pain. 2008;4:7. doi: 10.1186/1744-8069-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limiroli E, Gaspani L, Panerai AE, Sacerdote P. Differential morphine tolerance development in the modulation of macrophage cytokine production in mice. J Leukoc Biol. 2002;72:43–8. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. 1992;261:669–77. [PubMed] [Google Scholar]
- Messmer D, Hatsukari I, Hitosugi N, Schmidt-Wolf IG, Singhal PC. Morphine reciprocally regulates IL-10 and IL-12 production by monocyte-derived human dendritic cells and enhances T cell activation. Mol Med. 2006;12:284–90. doi: 10.2119/2006-00043.Messmer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci. 2001;21:2808–19. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46:404–11. doi: 10.1016/j.neuropharm.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Mork A, Geisler A. A comparative study on the effects of tetracyclines and ltium on the cyclic AMP second messenger system in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19:157–160. doi: 10.1016/0278-5846(94)00112-u. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Fujio M, Ozawa T, Minami M, Satoh M. Effect of MS-153, a glutamate transporter activator, on the conditioned rewarding effects of morphine, methamphetamine and cocaine in mice. Behavioural Brain Research. 2005;156:233–9. doi: 10.1016/j.bbr.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Satoh M. Involvement of glial glutamate transporters in morphine dependence. Ann N Y Acad Sci. 2004;1025:383–8. doi: 10.1196/annals.1307.047. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Yamada K, Nabeshima T. The roles of glial cell line-derived neurotrophic factor, tumor necrosis factor-alpha, and an inducer of these factors in drug dependence. J Pharmacol Sci. 2007;104:116–21. doi: 10.1254/jphs.cp0070017. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Yamada Y, Nakajima A, Saito K, Seishima M, Noda Y, Nabeshima T. Tumor necrosis factor-alpha and its inducer inhibit morphine-induced rewarding effects and sensitization. Biol Psychiatry. 2007;62:658–68. doi: 10.1016/j.biopsych.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Nutile-McMenemy N, Elfenbein A, Deleo JA. Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. Journal of Neurochemistry. 2007;103:2035–46. doi: 10.1111/j.1471-4159.2007.04889.x. [DOI] [PubMed] [Google Scholar]
- Olsson T, Kristensson K, Ljungdahl A, Maehlen J, Holmdahl R, Klareskog L. Gamma-interferon-like immunoreactivity in axotomized rat motor neurons. J Neurosci. 1989;9:3870–5. doi: 10.1523/JNEUROSCI.09-11-03870.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Sekiya Y, Minami M, Satoh M. Effect of gene transfer of GLT-1, a glutamate transporter, into the locus coeruleus by recombinant adenoviruses on morphine physical dependence in rats. Eur J Neurosci. 2004;19:221–6. doi: 10.1111/j.1460-9568.2004.03101.x. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001;905:254–8. doi: 10.1016/s0006-8993(01)02536-7. [DOI] [PubMed] [Google Scholar]
- Patel NA, Romero AA, Zadina JE, Chang SL. Chronic exposure to morphine attenuates expression of interleukin-1 beta in the rat hippocampus. Brain Res. 1996;712:340–4. doi: 10.1016/0006-8993(95)01575-2. [DOI] [PubMed] [Google Scholar]
- Pochon NA, Menoud A, Tseng JL, Zurn AD, Aebischer P. Neuronal GDNF expression in the adult rat nervous system identified by in situ hybridization. Eur J Neurosci. 1997;9:463–71. doi: 10.1111/j.1460-9568.1997.tb01623.x. [DOI] [PubMed] [Google Scholar]
- Pourpak Z, Ahmadiani A, Alebouyeh M. Involvement of interleukin-1beta in systemic morphine effects on paw oedema in a mouse model of acute inflammation. Scand J Immunol. 2004;59:273–7. doi: 10.1111/j.0300-9475.2004.01396.x. [DOI] [PubMed] [Google Scholar]
- Punch LJ, Self DW, Nestler EJ, Taylor JR. Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci. 1997;17:8520–7. doi: 10.1523/JNEUROSCI.17-21-08520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–34. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Renauld AE, Spengler RN. Tumor necrosis factor expressed by primary hippocampal neurons and SH-SY5Y cells is regulated by alpha(2)-adrenergic receptor activation. J Neurosci Res. 2002;67:264–74. doi: 10.1002/jnr.10101. [DOI] [PubMed] [Google Scholar]
- Rock RB, Hu S, Sheng WS, Peterson PK. Morphine stimulates CCL2 production by human neurons. J Neuroinflammation. 2006;3:32. doi: 10.1186/1742-2094-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Balasubramanian S, Sumandeep S, Charboneau R, Wang J, Melnyk D, Beilman GJ, Vatassery R, Barke RA. Morphine directs T cells toward T(H2) differentiation. Surgery. 2001;130:304–9. doi: 10.1067/msy.2001.116033. [DOI] [PubMed] [Google Scholar]
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Shoemaker WJ, Kosten TA, Muly SM. Ethanol attenuation of morphine dependence: comparison to dizocilpine. Psychopharmacology (Berl) 1997;134:83–7. doi: 10.1007/s002130050428. [DOI] [PubMed] [Google Scholar]
- Sibinga NE, Goldstein A. Opioid peptides and opioid receptors in cells of the immune system. Annu Rev Immunol. 1988;6:219–49. doi: 10.1146/annurev.iy.06.040188.001251. [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neuroscience Research. 2001;39:281–6. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist. 2005;11:308–22. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Research. 1999;837:203–12. doi: 10.1016/s0006-8993(99)01666-2. [DOI] [PubMed] [Google Scholar]
- Trafton CL, Marques PR. Effects of septal area and cingulate cortex lesions on opiate addiction behavior in rats. J Comp Physiol Psychol. 1971;75:277–85. doi: 10.1037/h0030810. [DOI] [PubMed] [Google Scholar]
- Tremblay EC, Charton G. Anatomical correlates of morphine-withdrawal syndrome: differential participation of structures located within the limbic system and striatum. Neurosci Lett. 1981;23:137–42. doi: 10.1016/0304-3940(81)90030-6. [DOI] [PubMed] [Google Scholar]
- Trujillo KA. Are NMDA receptors involved in opiate-induced neural and behavioral plasticity? A review of preclinical studies. Psychopharmacology (Berl) 2000;151:121–41. doi: 10.1007/s002130000416. [DOI] [PubMed] [Google Scholar]
- Van Crugten JT, Somogyi AA, Nation RL, Reynolds G. The effect of old age on the disposition and antinociceptive response of morphine and morphine-6 beta-glucuronide in the rat. Pain. 1997;71:199–205. doi: 10.1016/s0304-3959(97)03363-0. [DOI] [PubMed] [Google Scholar]
- Wang HL, Zhao Y, Xiang XH, Wang HS, Wu WR. Blockade of ionotropic glutamatergic transmission in the ventral tegmental area attenuates the physical signs of morphine withdrawal in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:1079–87. doi: 10.1016/j.pnpbp.2004.05.043. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Charboneau R, Roy S. Morphine impairs host innate immune response and increases susceptibility to Streptococcus pneumoniae lung infection. J Immunol. 2005;174:426–34. doi: 10.4049/jimmunol.174.1.426. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Roy S. Transcriptional and epigenetic regulation of interleukin-2 gene in activated T cells by morphine. J Biol Chem. 2007;282:7164–71. doi: 10.1074/jbc.M604367200. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–69. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15769–74. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalcman SS, Siegel A. The neurobiology of aggression and rage: role of cytokines. Brain Behav Immun. 2006;20:507–14. doi: 10.1016/j.bbi.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Zhang B, Yang L, Konishi Y, Maeda N, Sakanaka M, Tanaka J. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: comparison with other types of cAMP-elevating agents. Neuropharmacology. 2002;42:262–9. doi: 10.1016/s0028-3908(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2007 doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]