

Abstract
A simple and efficient methodology for the preparation of N-chlorinated hydantoins is presented. These versatile chlorenium sources were isolated in high yield after a simple recrystallization. Among the ten examples are the first chiral N-chlorohydantoins.
N-chlorinated hydantoins (e.g., 1) are important and versatile chlorinating agents that have found use in a range of synthetic operations. The prototypical example, 1,3-dichloro-5,5-dimethylhydantoin (DCDMH 3), has been employed as a chlorine source for the α-chlorination of acetophenones1 and 1-aryl-2-pyrazolin-5-ones,2 for the preparation of chlorohydrin derivatives of corticosteroids, 3 for the benzylic chlorination of 2-methylpyrazine,4 and for the selective chlorination of a heavily functionalized quinolone derivative en route to the antibiotic ABT-492.5 In the latter case, DCDMH was found to be a milder, more selective reagent than sulfuryl chloride. Researchers at Schering Plough exploited the enhanced reactivity of DCDMH relative to NCS to improve upon the large-scale preparation of Davis’ oxaziridine.6,7 Moreover, DCDMH was employed to trap an enolate resulting from the attack of dimethylzinc onto an α,β-unsaturated ketone, thus generating an α-chloro-β-methyl ketone functionality en route to a building block of Amphotericin B.8
DCDMH has been employed as an oxidant for a number of transformations including the halodeboronation of aryl boronic acids,9 the oxidation of urazoles to provide triazolinediones,10 the microwave-assisted cleavage of oximes,11 the preparation of dialkyl chlorophosphates,12 and as a terminal oxidant in the Sharpless asymmetric aminohydroxylation reaction.13,14 The compound can also serve as a mild oxidant in the presence of wet silica gel in the sodium nitrite-mediated nitration of phenols15 and in the preparation of N-nitrosoamines.16
Furthermore, DCDMH has emerged as the reagent of choice for the activation, prior to loading, of silyl linkers for solid-phase organic synthesis.17–20 Finally, DCDMH has been used as an oxidative activator of an iridium catalyst employed for the asymmetric hydrogenation of substituted quinolines.21 Other uses of DCDMH include employment as redox titrants in non-aqueous media,22 and as a cheap yet safe disinfectant and bactericide for municipal water sources.23
N-chlorinated hydantoins have also received attention from the pharmacological community. The myriad of adverse side-effects associated with hydantoin-based drugs such as the anti-convulsant Dilantin and the aldose reductase inhibitor Sorbinil have been attributed to N-chloro metabolites generated in vivo via chlorination by myeloperoxidase and hydrogen peroxide.24–26
There are two classical methods for the preparation of N-chlorinated hydantoins. One approach involves passing chlorine gas through an aqueous alkaline solution of hydantoin.27 The toxicity and reactivity of elemental chlorine make this approach somewhat undesirable. The second method involves treatment of the hydantoin with an aqueous sodium hypochlorite solution (Chlorox®) followed by extraction.28,24 In our hands, the latter approach was met with difficulties when less substituted, more water-soluble hydantoins were employed.
Due to our need in a number of projects, we sought a simple and high-yielding methodology for the preparation of several new di-and mono-chlorinated hydantoin derivatives. We were particularly interested in the possibility of generating chiral N-chlorohydantoins, for asymmetric applications. Our interest was piqued by a report by Nagao and Katagiri published in 1991.29 These authors correctly predicted that compounds possessing N–Cl bonds with smaller 35Cl nuclear quadrupole resonance (NQR) frequencies than that of trichloroisocyanuric acid30,31 (TCCA 2, 58.902 MHz29,32) ought to suffer chlorination by action of the latter. In their report, DCDMH 3 (35Cl NQR ν = 55.960 and 53.639 MHz) was prepared from the corresponding dimethylhydantoin by action of TCCA.29 TCCA has also been employed previously to chlorinate various amides and carbamates, although hydantoins were not represented in these studies.33,34 We sought to explore the versatility of 2 as a reagent for the preparation of a number of these useful chlorinating agents.
In this Letter, we present a modified and improved procedure for the TCCA-mediated preparation of a series of N-chlorinated hydantoins, including three of the first known chiral examples. In addition, we have assembled a compendium of hitherto scarce physical data for these compounds in the Supplementary data. Whereas historically, these compounds have been characterized only by melting point, elemental analysis, and sometimes 13C NMR28,24, we decided to fully characterize these compounds, collecting 1H and 13C NMR, IR, elemental analysis, melting point, and optical rotation data (where appropriate) for all of the products isolated in this study. Importantly, we were also able to secure X-ray crystal structures for nine of the ten products presented herein.
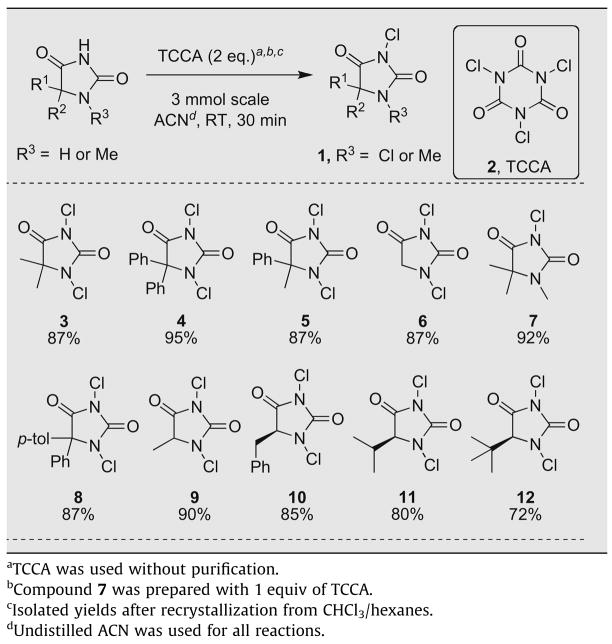
At the outset of our investigation, the synthetic operation was simplified by shortening the reaction time and by employing unpurified TCCA and undistilled acetonitrile. In addition, cognizant of the crystalline nature of N-chlorohydantoins,24 the goal was set to purify the products via recrystallization. As indicated in Table 1, compound 3 was isolated in 87% yield by adding TCCA to a stirred slurry of 5,5-dimethylhydantoin in acetonitrile, followed by stirring for 30 min. The reduced byproducts of TCCA were readily removed on trituration of the isolate in chloroform followed by filtration of the filtrate through a 1 cm-thick pad of silica gel. On concentration, this crude isolate was easily purified by recrystallization from chloroform and hexanes. This methodology was general, returning the desired chlorinated hydantoin products (1) with various substitutions in excellent yields. All of the products disclosed in Table 1 were rigorously characterized by 1H and 13C NMR, IR, X-ray diffraction (except 10), elemental analysis, and melting point (see Supplementary data). To the best of our knowledge, the X-ray structures of these compounds were previously unknown, save that of compound 4.24
Table 1.
TCCA-mediated chlorination of hydantonis
|
In addition to several examples of 5,5-disubstituted hydantoins (3–5, 7 and 8), 5-monosubstituted hydantoins (9–12) and nonsubstituted hydantoin 6 were chlorinated without incident. The preparation of trialkylated compound 7 confirms the ability to affect a mono-chlorination by employing one equivalent of TCCA. Of note is the fact that the relatively activated C5 of nonsubstituted hydantoin 6 as well as the benzylic positions of compounds 8 and 10 were not chlorinated under the reaction conditions.
Moreover, this methodology is compatible with chiral hydantoin starting materials, returning the desired chiral N,N-dichlorohydantoins 10–12. The parent chiral hydantoins were prepared in high enantiomeric purity (confirmed by optical rotation) from the corresponding chiral α-amino acids by known methodology.35–37 Importantly, reduction of the chiral N,N-dichlorohydantoins with aqueous sodium sulfite returned enantiomerically pure parent hydantoins as judged by optical rotation (see Supplementary data for details). The ready preparation of chiral hydantoin-based chlorenium sources may facilitate the discovery of new asymmetric halogenation reactions. Such studies are ongoing in our laboratories.
Realizing that one molar equivalent of TCCA harbors three chlorenium equivalents (Scheme 1), the net three-fold excess of chlorenium source relative to the parent hydantoin when 2 mol equiv of TCCA was employed seemed unnecessary. Hence, the fate of the reaction when substantially less TCCA was employed was evaluated. As indicated in Scheme 1, the amount of TCCA can be reduced to as low as 0.7 mol equiv (a 1:1 ratio of chlorenium relative to the number of hydantoin nitrogen atoms) without substantial loss of yield. Given these results, we are in a position to recommend employing less than 1 mol equiv of TCCA relative to the parent hydantoin.
Scheme 1.

TCCA equivalent study.
Additionally, these reactions can be conducted on large scale, as shown in Scheme 2. Diphenylhydantoin 13 was chlorinated on a 33 mmol scale to return 8.72 g (82%) of 1,3-dichloro-5,5-diphenylhydantoin 4.
Scheme 2.

Scaled preparation of 4.
In conclusion, we have presented an operationally simple method for the preparation of a number of N-chlorinated hydantoins by action of unpurified, commercially available TCCA in undistilled solvents. This methodology returns crystalline products in high yield without recourse to chromatographic purification, save a simple filtration step. We have also presented a catalogue of physical data for N-chlorohydantoins that has been previously undisclosed in the literature. Most important among these data are the X-ray diffraction data for 9 of the 10 compounds presented herein. Current efforts in our laboratory include exploiting these new chlorinating reagents for organic synthesis. Particular attention will be given to asymmetric transformations employing chiral N-chlorinated hydantoins.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2008.11.091.
Acknowledgments
We are grateful to the NIH (Grant No. R01-GM082961) and the ACS-PRF (Grant No. 47272-AC) for the support of this research. D.C.W. thanks Mr. R. Adam Mosey for helpful suggestions.
References and notes
- 1.Xu ZJ, Zhang DY, Zou XZ. Synth Commun. 2006;36:255–258. [Google Scholar]
- 2.Spitulnik MJ. Synthesis. 1985:299–300. [Google Scholar]
- 3.Shapiro EL, Gentles MJ, Tiberi RL, Popper TL, Berkenkopf J, Lutsky B, Watnick AS. J Med Chem. 1987;30:1581–1588. doi: 10.1021/jm00392a010. [DOI] [PubMed] [Google Scholar]
- 4.Karmazin L, Mazzanti M, Bezombes JP, Gateau C, Pecaut J. Inorg Chem. 2004;43:5147–5158. doi: 10.1021/ic049538m. [DOI] [PubMed] [Google Scholar]
- 5.Barnes DM, Christesen AC, Engstrom KM, Haight AR, Hsu MC, Lee EC, Peterson MJ, Plata DJ, Raje PS, Stoner EJ, Tedrow JS, Wagaw S. Org Process Res Dev. 2006;10:803–807. [Google Scholar]
- 6.Chen BC, Murphy CK, Kumar A, Reddy RT, Clark C, Zhou P, Lewis BM, Gala D, Mergelsberg I, Scherer D, Buckley J, DiBenedetto D, Davis FA. Organic Synthesis. Vol. 73. 1996. pp. 159–173. [Google Scholar]
- 7.Mergelsberg I, Gala D, Scherer D, Dibenedetto D, Tanner M. Tetrahedron Lett. 1992;33:161–164. [Google Scholar]
- 8.Miesch L, Gateau C, Morin F, Franck-Neumann M. Tetrahedron Lett. 2002;43:7635–7638. [Google Scholar]
- 9.Szumigala RH, Devine PN, Gauthier DR, Volante RP. J Org Chem. 2004;69:566–569. doi: 10.1021/jo035184p. [DOI] [PubMed] [Google Scholar]
- 10.Zolfigol MA, Nasr-Isfahani H, Mallakpour S, Safaiee M. Synlett. 2005:761–764. [Google Scholar]
- 11.Khazaei A, Manesh AA. Synthesis. 2005:1929–1931. [Google Scholar]
- 12.Shakya PD, Dubey DK, Pardasani D, Palit M, Gupta AK. Org Prep Proced Int. 2005;37:569–574. [Google Scholar]
- 13.Barta NS, Sidler DR, Somerville KB, Weissman SA, Larsen RD, Reider PJ. Org Lett. 2000;2:2821–2824. doi: 10.1021/ol006255z. [DOI] [PubMed] [Google Scholar]
- 14.Harding M, Bodkin JA, Hutton CA, McLeod MD. Synlett. 2005:2829–2831. [Google Scholar]
- 15.Zolfigol MA, Ghaemi E, Madrakian E, Choghamarani AG. Mendeleev Commun. 2006:41–42. [Google Scholar]
- 16.Niknam K, Zolfigol MA. J Iran Chem Soc. 2006;3:59–63. [Google Scholar]
- 17.DiBlasi CM, Macks DE, Tan DS. Org Lett. 2005;7:1777–1780. doi: 10.1021/ol050370y. [DOI] [PubMed] [Google Scholar]
- 18.Grela K, Tryznowski M, Bieniek M. Tetrahedron Lett. 2002;43:9055–9059. [Google Scholar]
- 19.Kobori A, Miyata K, Ushioda M, Seio K, Sekine M. Chem Lett. 2002:16–17. doi: 10.1021/jo010813l. [DOI] [PubMed] [Google Scholar]
- 20.Komatsu M, Okada H, Akaki T, Oderaotoshi Y, Minakata S. Org Lett. 2002;4:3505–3508. doi: 10.1021/ol026634n. [DOI] [PubMed] [Google Scholar]
- 21.Zhou YG. Acc Chem Res. 2007;40:1357–1366. doi: 10.1021/ar700094b. [DOI] [PubMed] [Google Scholar]
- 22.Radhamma MP, Indrasenan P. Talanta. 1983;30:49–50. doi: 10.1016/0039-9140(83)80009-5. [DOI] [PubMed] [Google Scholar]
- 23.Rao ZM, Zhang XR, Baeyens WRG. Talanta. 2002;57:993–998. doi: 10.1016/s0039-9140(02)00143-1. [DOI] [PubMed] [Google Scholar]
- 24.Lipinski CA, Bordner J, Butterfield GC, Marinovic LC. J Heterocycl Chem. 1990;27:1793–1799. [Google Scholar]
- 25.Mays DC, Pawluk LJ, Apseloff G, Davis WB, She ZW, Sagone AL, Gerber N. Biochem Pharmacol. 1995;50:367–380. doi: 10.1016/0006-2952(95)00151-o. [DOI] [PubMed] [Google Scholar]
- 26.Uetrecht J, Zahid N. Chem Res Toxicol. 1988;1:148–151. doi: 10.1021/tx00003a004. [DOI] [PubMed] [Google Scholar]
- 27.Corral RA, Orazi OO. J Org Chem. 1963;28:1100–1104. [Google Scholar]
- 28.Derosa M, Melenski E, Holder AJ. Heterocycles. 1993;36:1059–1064. [Google Scholar]
- 29.Nagao YK. Sci Rep Hirosaki Univ. 1991;38:20–23. [Google Scholar]
- 30.Juenge EC, Beal DA, Duncan WP. J Org Chem. 1970;35:719–722. [Google Scholar]
- 31.Tilstam U, Weinmann H. Org Process Res Dev. 2002;6:384–393. [Google Scholar]
- 32.Poleshchuk OK, Makiej K, Ostafin M, Nogaj B. Magn Reson Chem. 2001;39:329–333. [Google Scholar]
- 33.De Luca L, Giacomelli G, Nieddu G. Synlett. 2005:223–226. [Google Scholar]
- 34.Hiegel GA, Hogenauer TJ, Lewis JC. Synth Commun. 2005;35:2099–2105. [Google Scholar]
- 35.Anteunis MJO, Spiessens L, Dewitte M, Callens R, Reyniers F. Bull Soc Chim Belg. 1987;96:459–465. [Google Scholar]
- 36.Lickefett H, Krohn K, Konig WA, Gehrcke B, Syldatk C. Tetrahedron: Asymmetry. 1993;4:1129–1135. [Google Scholar]
- 37.Reist M, Carrupt PA, Testa B, Lehmann S, Hansen JJ. Helv Chim Acta. 1996;79:767–778. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2008.11.091.