

Summary
In situ generated [(PPP)Pt][BF4]2 (PPP = triphos) catalyzes the cycloisomerization of 1,6-enyne-ols by initiative π-activation of the alkyne. This generates an isolable cationic Pt-alkenyl species which subsequently participates in turnover limiting protonolysis with in situ generated acid. This latter reactivity contrasts cationic Pt-alkyls which are more difficult to protonolyze. Mechanistic studies on isolated Pt-alkenyls, and deuterium labeling helped to elucidate the mechanistic details.
We have previously established that (PPP)Pt2+ electrophiles1 can stoichiometrically cyclize 1,5- and 1,6-dienyl alcohols into (PPP)Pt-cycloalkyl cations and that these compounds are resistant to Pt-C protonolysis.2,3 In the (PPP)Pt-CH3+ model complex, the ammonium acid [Ph2NH2][BF4] (pKa = 0.84) could liberate methane, but this reactivity did not extend to the catalysis relevant (PPP)Pt-cycloalkyl cations.5 Since metal-vinyl species can be more susceptible to protonolysis6,7 we believed that such an intermediate, obtainable from an alkyne initiated cascade, would be more easily turned over by a protonolysis mechanism, e.g. Scheme 1.
Scheme 1.

To test this hypothesis enyne 1 was subjected to the (PPP)Pt2+ catalyst (20 mol %) generated by stirring (PPP)PtI2 and AgBF4 in CH3NO2. These conditions generated a nitromethane adduct (JPt-P = 3500 Hz)8 that survives the filtrative removal of the silver salts. The reaction was then initiated by adding Ph2NMe9 and 1 (eq 1). In situ monitoring showed that the catalyst rested as a 1:1 ratio of two Pt complexes (31P NMR) with the reduced coupling constants expected of a Pt-vinyl species (JPt-P ~ 1400 Hz).10 When the conversion of 1 to 2 was complete (16 h), these two peaks disappeared and the (PPP)Pt2+-nitromethane adduct returned.
 |
(1) |
The observation that two seemingly distinct Pt-alkenyl intermediates generated a single organic product (2) was most directly investigated by synthesis.11 The putative Pt-alkenyls were synthesized as a 1:1 ratio using a resin bound piperidinomethyl base and a slight excess of 1; slow evaporation of a mixture of CH2Cl2 and Et2O produced fine white needles. Dissolving and collecting 31P and 1H NMR spectra on these crystals at low temperatures (−78 °C), indicated that a single Pt-species had crystallized and persisted up to −18 °C, where upon the other platinum complex began to grow until the 1:1 ratio was reestablished.12
 |
(2) |
Treating either a single isomer of 3 or a 1:1 mixture with HBF4, cleanly generated 2,13 as did the reaction of the isolated Pt-alkenyls with LiBHEt3 (eq 2). The generation of 2 under both acidic and reductive conditions thus argued against the two Pt-alkenyls being structural isomers. The X-ray structure of 3 provided a possible explanation for the two Pt-alkenyl isomers. As shown in Figure 1 the C1/C14/C16 alkenyl plane orients orthogonal to the square plane. The alkenyl methyl (C16) can therefore orient either syn to the central P-Ph (shown) or anti. If Pt-C bond rotation were slow on the NMR time scale, this could lead to two rotamers.14 Protonolysis or reductive cleavage (H−) of either rotamer would thus produce 2.
Figure 1.

Chem3D representation of 3. BF4− anion removed for clarity. Selective bond lengths (Å): Pt-P1 = 2.283(6), Pt-P2 = 2.275(6), Pt-P3 = 2.284(6), Pt-C1 = 2.10(2), C1-C14 = 1.33(3). Selective bond angles (deg): C1-Pt-P1 = 97.6(5), C1-Pt-P3 = 93.0(5), P1-Pt-P2 = 84.4(2), P2-Pt-P3 = 85.4(2).
Consistent with a hindered rotation argument, the Pt-alkenyl obtained from 5 (eq 3) was observed as a single isomer down to low temperature. Unexpectedly, however, 6 diverged from 3 in its reactions with H+ and H−. For example 7 was obtained upon treating with HBF4 while 8 was obtained on treating with LiBHEt3 (Scheme 2).15
Scheme 2.

 |
(3) |
Under the catalytic conditions described above, 6 was also observed to be the resting state, and like 3, was converted to (PPP)Pt2+-nitromethane upon consumption of 5. Reaction times were considerably lower (4 h) than with 1. Like the stoichiometric protonolysis in Scheme 2, the catalytic product was 7 and not the initially expected olefin isomer 8 (eq 4).16
 |
(4) |
The protonolysis thus coupled the metal liberation/turnover to a clean olefin migration. To probe this isomerization, 6 was treated with DBF4 in CD3OD/CD2Cl2, to yield the doubly labeled 10.17 A scenario consistent with these observations is shown in Scheme 3, wherein D+ first isomerizes the 1,2 to the 2,3 form via a putative α-cation intermediate,18 followed by proto-deplatination at C2. The fact that protonolysis yielded no d3 or d4 products suggested that k−2 (and consequently k−1) was not competitive with k3, while the lack of d1 products showed that the direct protonolysis and the alternative intramolecular rearrangements were not competitive (Scheme 3). Unreconciled as of yet is why D+ addition to C2 is competitive in the 2,3 form but not in 6.19 We presume that this sequence also operates in the catalytic cycloisomerization (eq 4).20
Scheme 3.

Previous studies in our lab indicated that changing the tridentate PPP architecture to a combination of a mono- and bidentate ligand (P2P) made protonolysis much slower. In fact, protonolysis of Pt-Me bonds was found to be >50,000 times slower for complexes containing a P2P ligand array.5 Because the (PPP)Pt-alkenyl bond had proven more reactive, experiments were initiated to determine if the chiral P2P catalysts were also viable. Consistent with a markedly different protonolysis mechanism, catalytic reactions with a family of (P2)(PMe3)Pt2+ catalysts (P2 = dppe, (S)-BINAP, (S)-MeO-BIPHEP, (S)-Me-Soniphos) were equally efficient and yielded racemic 7 with similar reaction times to (PPP)Pt2+.21
In summary, we have discovered a catalyst for the cycloisomerization of 1,6-enyne-ols wherein the turnover limiting step is protonolysis of the Pt-alkenyl bond that results from a cascade electrophilic cyclization. Mechanistic studies revealed that for a terminal alkyne, a slow, irreversible, olefin isomerization preceded a fast protonolysis. When the alkyne was internal, protonolysis was turnover limiting and no olefin migration was detected. These results contrast the protonolysis of Pt-alkyls, which do not proceed under similar conditions.
Supplementary Material
Full experimental procedures and crystallographic data are available. This information is free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health, Institute of General Medicine (GM-60578) for support of this research. We thank Dr. Peter White for X-ray structural determinations; correspondence regarding them should be directed to his attention at UNC Chapel Hill (pwhite@unc.edu).
References
- 1.Chianese AR, Lee SJ, Gagné MR. Angew. Chem., Int. Ed. 2007;46:4042–4059. doi: 10.1002/anie.200603954. [DOI] [PubMed] [Google Scholar]
- 2.(a) Koh JW, Gagné MR. Angew. Chem., Int. Ed. 2004;43:3459–3461. doi: 10.1002/anie.200453913. [DOI] [PubMed] [Google Scholar]; (b) Feducia JA, Gagné MR. J. Am. Chem. Soc. 2008;130:592–599. doi: 10.1021/ja075518i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For references showing the stability of cationic Pt-alkyls to protonolysis see: Heyduk AF, Labinger JA, Bercaw JE. J. Am. Chem. Soc. 2003;125:6366–6367. doi: 10.1021/ja029099v. Thorn DL. Organometallics. 1998;17:348–352. Annibale G, Bergamini P, Cattabriga M. Inorg. Chim. Acta. 2001;316:25–32. Butikofer JL, Hoerter JM, Peters RG, Roddick DM. Organometallics. 2004;23:400–408. Peters RG, White S, Roddick DM. Organometallics. 1998;17:4493–4499.
- 4.Stewart R, Dolman D. Can. J. Chem. 1967;45:925–928. [Google Scholar]
- 5.Feducia JA, Campbell AN, Anthis JW, Gagné MR. Organometallics. 2006;25:3114–3117. [Google Scholar]
- 6.For reviews on transition metal catalyzed enyne cycloisomerizations see: Trost BM, Krische MJ. Synlett. 1998;1(25) Fürstner A, Davies PW. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. Michelet V, oullec PY, Genêt J-P. Angew. Chem., Int. Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589. Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271–2296. and references therein.
- 7.For references where protonolysis of the metal vinyl bond is the turn-over mechanism for catalyst regeneration see, for example: Nevado C, Charruault L, Michelet V, Nieto-Oberhuber C, Muñoz MP, Méndez M, Rager M-N, Genêt J-P, Echavarren AM. Eur. J. Org. Chem. 2003:706–713. Nevado C, Cárdenas DJ, Echavarren AM. Chem. Eur. J. 2003;9:2627–2635. doi: 10.1002/chem.200204646. Zhang L, Kozmin SA. J. Am. Chem. 2004;126:11806–11807. doi: 10.1021/ja046112y. Zhang L, Kozmin SA. J. Am. Chem. 2006;128:9705–9710. doi: 10.1021/ja063384n. Yao T, Zhang X, Larock RC. J. Am. Chem. 2004;126:11164–11165. doi: 10.1021/ja0466964. Toullec PY, Genin E, Leseurre L, Genêt J-P, Michelet V. Angew. Chem., Int. Ed. 2006;45:7427–7430. doi: 10.1002/anie.200601980. Imagawa H, Iyenaga T, Nishizawa M. Org. Lett. 2005;7:451–453. doi: 10.1021/ol047472t. Tarselli MA, Chianese AR, Lee SJ, Gagné MR. Angew. Chem., Int. Ed. 2007;46:6670–6673. doi: 10.1002/anie.200701959. Tarselli MA, Gagné MR. J. Org. Chem. 2008;73:2439–2441. doi: 10.1021/jo7024948.
- 8.JPt-P coupling constants are reported for the central P of the triphos ligand that is trans to the reactive site.
- 9.In the absence of Ph2NMe, the acid byproduct of cyclization can cause Brønsted cyclization processes to initiate.
- 10.(PPP)Pt-alkyl cations typically have coupling constants for the trans P between 1300 Hz - 1500 Hz, see footnotes 2b and 5.
-
11.A priori we considered the 5-exo addition product 4 to be a second potential isomer.
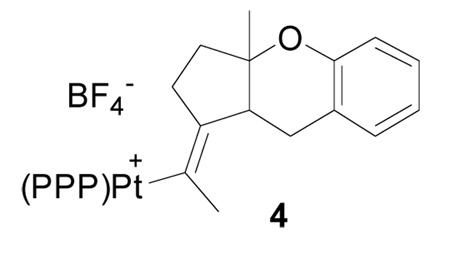
- 12.The two Pt-alkenyls where initially thought to arise from competing 6-endo (3) and 5-exo (4) cyclization modes. A reversible bicyclization and productive protonolysis of only 3 could have explained the convergence. The interconversion of these species in the absence of an acid mediator of retrocyclization argued against this possibility. See footnote 2b and Mullen CA, Campell AN, Gagné MR. Angew Chem., Int. Ed. 2008;47:6011–6014. doi: 10.1002/anie.200801423. for examples of reversible cyclizations.
- 13.Reaction with DBF4 cleanly generated 2-d labeled at the expected C2 position.
- 14.The two rotamers do not coalesce in the 31P NMR up to 120 °C.
- 15.DFT calculations on the products (B3LYP/6-31G*) indicated that 7 was more stable than 8 by 1.0 kcal/mol (MacSpartan 06).
- 16.It is worth noting that 8 does not convert to 7 on reacting with [Ph2NH2][BF4].
- 17.We cannot spectroscopically differentiate between the C1,2-d2 and the C1,3-d2 isomer of 10. For the purpose of providing a starting point for this preliminary discussion we have assumed that the Pt-center was ultimately replaced by D as shown.
- 18.D-NMR analysis indicated that D was located in both diastereotopic positions of C1.
- 19.The higher stability of the tertiary cation resulting from C2-protonolysis might explain why 3 proto-demetallates without isomerization.
- 20.Catalytic experiments with [(dppe)Pt-µ-Cl]2[BF4]2 gave a 1:1 mixture of 7 and 8.
- 21.For a review on enantioselective ene-yne cycloisomerization reactions, see: Fairlamb IJS. Angew. Chem. Int. Ed. 2004;43:1048–1052. doi: 10.1002/anie.200301699.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental procedures and crystallographic data are available. This information is free of charge via the Internet at http://pubs.acs.org.