

Abstract
Current Good Manufacturing Practices (cGMP) for botanicals stipulates the use of appropriate methods for identification of raw materials. Due to natural variability, chemical analysis of plant material is a great challenge and requires special approaches. This paper presents a comprehensive proposal to the process of validating qualitative high-performance thin-layer chromatographic (HPTLC) methods, proving that such methods are suitable for the purpose. The steps of the validation process are discussed and illustrated with examples taken from a project aiming at validation of methods for identification of green tea leaf, ginseng root, eleuthero root, echinacea root, black cohosh rhizome, licorice root, kava root, milk thistle aerial parts, feverfew aerial parts, and ginger root. The appendix of the paper, which includes complete documentation and method write-up for those plants, is available on the J. AOAC Int. Website (http://www.atypon-link.com/AOAC/loi/jaoi).
Current Good Manufacturing Practices (cGMPs) for dietary supplements (1) have just recently become effective. After a long period of discussion and anticipation, the dietary supplement industry is now expected to implement and utilize a rigorous quality management system to ensure safety, quality, and consistency of products. Because quality can not be tested into a product, it is important that quality is already an attribute of the raw material of any given production process. For “botanicals,” which can be fresh, or processed plants, extracts, or other preparations, the identity is one of the central elements of quality. cGMP regulation therefore requires identity testing for each raw material that enters a process. Appropriate and scientifically sound methods should be applied. From the frame work of quality assurance, it is understood that such methods must be properly documented, validated, and reviewed. Data generated with these methods must be secure, transparent, and traceable. The only problem seems to be where to get all the required methods. That the U.S. Food and Drug Administration (FDA) does not limit “appropriate” methods to official or compendial ones does not change the situation, because it becomes now the responsibility of the analyst to find elsewhere or develop methods that meet this criterion.
It is the purpose of this paper to propose a concept for validation of the “appropriateness” of high-performance thin-layer chromatographic (HPTLC) methods for identification of botanical raw materials. We have developed further the ideas published by Koll et al. (2) and elaborated a general protocol. This protocol was successfully applied to the validation of methods for identification of green tea leaf, ginseng root, eleuthero root, echinacea root, black cohosh rhizome, licorice root, kava root, milk thistle aerial parts, feverfew aerial parts, and ginger root. Throughout this paper, we will use examples from those projects. The validated methods and corresponding validation data are published as a separate appendix in this journal.
General Considerations
Analysis of botanicals is a great challenge because they are highly complex mixtures of compounds covering a broad range of substance classes and exhibit natural variability. Tools and acceptance criteria developed in an attempt to comply with cGMP for synthetic pharmaceuticals have only limited applicability to these challenges. It seems necessary to explore new approaches, which are fit for the purpose.
In the context of this paper, we aim to understand quality in terms of agreement between an expected or declared property with the results of a suitable measurement of that property. High and low quality expressed as good or poor agreement is of course arbitrary unless we clearly define acceptance criteria, which can then be used in quality control (QC) to pass or fail a material. Analytically, any material has qualitative and quantitative aspects, dealing with the questions what it is and how much of it there is. For example, let us look at aspirin, a common drug taken against pain: A 300 mg aspirin tablet contains 300 mg of the chemically defined substance acetylsalicylic acid and some (defined) inert excipients, coating, etc. During analysis, we must first determine that there is acetylsalicylic acid present and not, for example, acetaminophen. This can be done by comparing chromatographic retention and spectral data of the sample against that of a standard. Then we determine whether we find the declared amount of the active substance. With more tests, we could also find out whether there are known or unknown impurities in the tablet (3). In essence, quality assurance is not a problem, as long as the bulk acetylsalicylic acid that is pressed into the tablet is of “good” quality, e.g., at least 99.5% pure, and the manufacturing process is under strict control. We can safely assume, regardless of the brand name, that acetylsalicylic acid is always acetylsalicylic acid.
Now, how about a capsule containing 50 mg of a black cohosh extract standardized to 2.5% triterpeneglycosides calculated as 27-deoxyactein? This very common dietary supplement may support health of women experiencing menopausal symptoms. The information on the label suggests an equally simple approach to quality assurance, but this time we are dealing with a bulk extract as the starting material of which we only know, it was standardized to contain a certain total amount of substances—how many different ones and how much of each is not declared. What makes up the other 97.5% of the extract? What exactly happens to the original black cohosh plant material when it is extracted? Which of the components of the extract are expected and which must be regarded as impurities? For the majority of dietary supplements, such questions have no definitive answers. Therefore, we will not look further into the associated analytical problems but focus on the identity of the plant material as primary criterion of quality. But, speaking analytically, what exactly is black cohosh?
Black cohosh is a plant with the scientific name Actaea racemosa (syn. Cimicifuga racemosa) that grows in North America, where also its relatives Actaea pachypoda, A. podocarpa, and A. rubra (white, yellow, and red cohosh) are native. Actaea foetida, A. dahurica, and A. heracleifolia grow in China and are commonly used as medicinal plants. Based on macroscopic and microscopic examination, experienced botanists can identify to which species a given plant belongs. For each species, an HPTLC fingerprint can be generated, which is able to comprehensively characterize the chemical constitution. HPTLC fingerprints of different species are expected to be significantly different, but different specimens of the same species may be quite different as well, depending on growing region, nutrition, developmental stage, harvesting practice, etc.
That means Actaea racemosa as such is, in contrast to acetylsalicylic acid, analytically not clearly defined. To arrive at a quality standard against which we can evaluate a sample, we must include further criteria to the definition, such as organoleptic characters and any known active or nonactive constituents. This quality standard is called a botanical reference material (BRM). It is botanically defined, usually with a voucher specimen deposited in an herbarium, and meets all other quality criteria. BRMs are available from different sources and treated similar to chemical reference substances (CRS).
During analysis, a sample of correct identity will produce a fingerprint similar to that of the BRM. This similarity has a certain fuzziness to it and is fundamentally different from the identification of acetylsalicylic acid, in which sample and standard are either identical or not. The problem is the natural variability of the plant raw material. Details and approaches to a solution are discussed in ref. 4.
To properly identify the black cohosh extract of our example, we need not only a BRM of Actaea racemosa but also BRMs of related species to help determine what can be regarded as similar and what as different. Then, as part of cGMP, a link between the fingerprint of the starting material and the fingerprint of the product can be established, which now in turn serves as the point of reference.
What Validation Can Accomplish
This paper looks at validation as an iterative process, closely connected with method development and optimization. Following AOAC’s approach, validation must establish the performance characteristics while demonstrating the fitness of a method for the intended purpose (5).
At the level of the plant, a method of identification must be suitable to establish with certainty whether or not a sample represents the same plant species as a BRM. Therefore, the method must be specific to distinguish the presence of wrong species instead of the expected one. Specificity is commonly regarded as the only validation point for an identification method (6). We include the aspect of natural variability of all involved species.
For large composite samples (e.g., multiple samples of large batches of unhomogenized raw material) or samples of plant derived products (e.g., extracts), selectivity must also address the possibility of accidental or willful adulteration. This however is also a quantitative issue, concerning detection limits of unwanted components. In the following, we focus only on the discrimination of species because this is the area where HPTLC methods are most versatile.
HPTLC is an off-line method that offers enormous flexibility in the selection of parameters for the individual steps of the process (7). Aside from the stationary and mobile phase, these parameters include the development conditions (chamber saturation, developing distance), derivatization (reagent concentration, time, heat), and waiting times between the subsequent steps. The method to be validated should define all parameters rigorously. We have adopted the concept of precision (repeatability, intermediate precision, reproducibility), which is commonly used only for quantitative methods, as a separate validation point (8). Precision helps to evaluate how predictably a fingerprint can be generated from a given botanical reference material.
As an evaluation of the effects of some environmental and user-dependent methodological parameters on the method’s performance, we propose the inclusion of robustness tests in the validation. Applied in routine, a method validated according to this concept will be able to distinguish whether a deviation of a fingerprint from that of a BRM is within the method’s uncertainty, due to natural variability or caused by the presence of an adulterant. More importantly, the identity of the investigated sample is established as correct with certainty when the fingerprint corresponds to that of the BRM.
This has a practically important consequence: If the validated method is performed once with a BRM or reference extract, the resulting fingerprint, stored as an electronic image, could be used as reference (atlas, official method collection, QC document, etc.). Thus fingerprints obtained subsequently during routine analyses can be evaluated against the reference image over a long period of time without the need to include a BRM in the test. BRM would only be required for revalidation after method transfer and after implementing changes to the method (e.g., change of plate manufacturer, use of different equipment, etc.).
The Validation Process and Its Elements
Validation should not be seen separately from the development of a method. The entire process can be visualized with the scheme in Figure 1. It starts from a clearly defined analytical goal, covers method selection/optimization or method development, respectively, and so-called prevalidation considerations before arriving at the elaboration of a validation protocol, which, seen formally, is the starting point of the actual validation. After performing all experiments described in the validation protocol, obtained data are evaluated and compared with the acceptance criteria. If all criteria are met, the method can be regarded as valid. In a less formal approach, some validation data may be incorporated from experiments, which were conducted previously as part of the method development/optimization.
Figure 1.
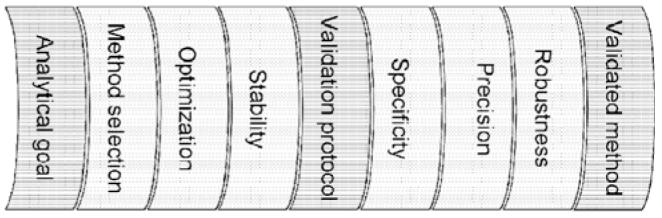
Validation process.
Method Selection
In a cGMP-regulated environment, 3 different situations may exist when it comes to establishing QC with respect to the identity of the starting material: (1) no method is available; (2) a compendial method is available; (3) no compendial methods are available, but one or more methods published in the literature for the same or similar analytical tasks.
The first case requires method development and is not discussed here. Information can be found in ref. 9. The second case would only require a simple method transfer for which most companies have established procedures. Although generally accepted as suitable and, in most cases, treated like validated, compendial methods may neither be state-of-the-art nor optimized. With our focus on the performance characteristics of the method, some optimization should be performed before the method is (re-)validated. In the third case, we can consider any method for evaluation. This is done in a paper review. As an example, Table 1 lists some of the methods that were evaluated in 2003 for identification and discrimination of ginseng species.
Table 1.
Methods included in paper review (2003) as part of selection of methods for identification of ginseng
| Source | Reference materials | Mobile phase on TLC silica gel | Derivatization |
|---|---|---|---|
| U.S. Pharmacopeia/National Formulary (USP/NF) 20, 2nd supplement |
Escin and arbutin | Ethyl acetate—water—1-butanol (25 + 50 + 100), upper phase |
Anisaldehyde |
| Pharmacopoeial Forum 30 (2) | Reference extracts of American and Asian ginseng: 2D |
1: chloroform—methanol—water (13 + 7 + 2), lower phase 2: water—butanol—ethyl acetate (5 + 4 + 1), upper phase |
Anisaldehyde |
| Pharmeuropa 15 (3) | Escin and arbutin | As USP/NF 20 | Anisaldehyde |
| Chinese Pharmacopoeia | Rb1, Re, Rg1 | Chloroform—ethyl acetate—methanol—water (15 + 40 + 22 + 10), store at 10°C, use lower layer |
Anisaldehyde |
| Plant Drug Analysis (Wagner, Bladt) | Rb1, Rb2, Rc, Rd, Re, Rg1, Rg2 |
Chloroform—methanol—water (70 + 30 + 4) | Vanillin |
|
Journal of Planar Chromatography (2002) 15 (2) |
Rb1, Re, Rg1 | Chloroform—methanol—water (65 + 50 + 10) | Anisaldehyde |
| Xie and Yan, Journal of High Resolution Chromatography & Chromatography Communications (1987) 10 |
Ginsenoside mixture |
Chloroform—ethyl acetate—methanol—water (15 + 40 + 22 + 10), use lower layer; plate drying over P2O5 |
Sulfuric acid |
Selected methods are experimentally evaluated using BRM and/or CRS, qualified equipment, and reagents of defined quality. Initially, we follow the instructions of the method as far as the experimental details are given. If such details are missing or not specific enough, a standardized HPTLC methodology [standard operating procedure (SOP); appendix to this paper] is applied. Criteria for selecting a method are determined by the analytical goal but also include simplicity, time consumption, and safety factors.
Optimization of Method
During optimization, all TLC methods are converted into HPTLC methods (10, 11) and a standardized methodology is applied. Figure 2 shows an example.
Figure 2.
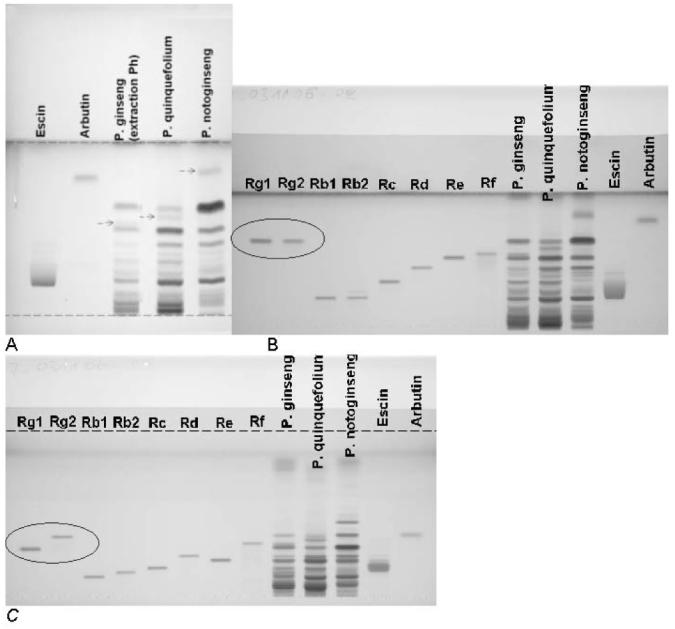
Method optimization (A) TLC of USP/NF20/Pharmeuropa (15.3) method for identification of ginseng species (details in Table 1); (B) same method on HPTLC plate; (C) optimized HPTLC. Color image is available on the J. AOAC Int. Website (http://www. atypon-link.com/AOAC/loi/jaoi).
Also evaluated is whether sample preparation can be simplified. We found that in most cases a 5 min sonication with methanol, followed by centrifugation and using the supernatant as test solution, yields satisfactory results. Furthermore, derivatization is optimized with the goal of convenience, safety, and reproducibility. At this point, we include BRM of known adulterants to ensure sufficient specificity of the method. Small modifications of the mobile phase composition may be applied to fine-tune separation. Each step of the optimization process is documented for complete traceability.
Stability
Before the validation of the optimized method can begin, the stability of the analyte on the plate, in solution, and during chromatography, as well as the stability of the visualized chromatogram, have to be investigated.
As a rule of thumb, we recommend testing the stability of the analyte in solution and on the plate over 3 h. If the workflow in the laboratory or special sample preparation requires delay between sample preparation and application and/or application and development, longer time periods should be investigated. One portion of the BRM is prepared according to the selected method. An aliquot of this solution is applied onto a plate. The sample and the plate with the applied sample (wrapped in aluminum foil) are set aside. After 3 h, another portion of the same BRM is prepared. Two aliquots of this solution are applied next to the first sample on the plate that was set aside, followed by an aliquot of the sample that was set aside. The 4 samples on the plate (Figure 3) represent the following: Track 1 = sample on the plate for 3 h prior to chromatography; Tracks 2, 4 = fresh sample applied immediately prior to chromatography; Track 3 = sample prepared 3 h prior to chromatography and stored in solution.
Figure 3.
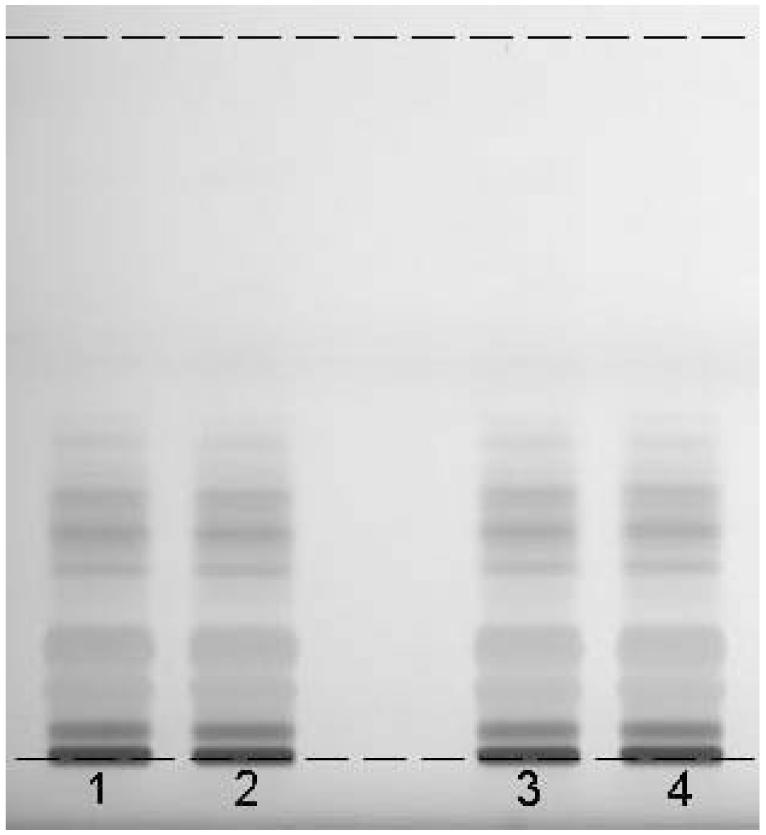
Stability of black cohosh on the plate and in solution. For details, see text. Color image is available on the J. AOAC Int. Website (http://www.atypon-link.com/AOAC/loi/jaoi).
Following chromatography, there should not be any difference in the fingerprints if the sample is stable on the plate and in solution.
Stability of the sample during chromatography is investigated by 2-dimensional (2D) development. One portion of the BRM is prepared according to the selected method and applied as spot at the lower right corner of a 10 × 10 cm plate (10 mm from each edge). The plate is developed and dried according to the selected method and then turned 90° to the right. The plate is developed a second time according to the method with a fresh portion of developing solvent. If the sample is stable during chromatography, all components can be detected on the diagonal line connecting the application position and the intersection of the 2 solvent fronts (Figure 4). Spots located off this line indicate the formation of artifacts. Methods that produce artifacts must be treated with caution.
Figure 4.

Investigation of stability during chromatography for (A) eleuthero, stable; (B) angelica, not stable. For details, see text. Color image is available on the J. AOAC Int. Website (http://www.atypon-link. com/AOAC/loi/jaoi).
If visualization of the fingerprint requires a derivatization step, the stability of result must be evaluated. In this experiment, we have taken one image immediately after completion of derivatization and subsequent images after 5, 10, 20, 30 min, and 1 h. The images are compared visually and with the help of video-densitometry (Figure 5).
Figure 5.

Evaluation of stability of the chromatographic result for black cohosh (colors of frames correspond to colors of curves). Color image is available on the J. AOAC Int. Website (http://www.atypon-link.com/AOAC/loi/jaoi).
Validation Protocol
The validation protocol is a key instrument for structuring, regulating, and documenting the validation process. Depending on the quality management system, it will follow the general document format of the company. The following elements must be included:
Goal of the method to be validated
General acceptance criteria
Personnel (study director, analysts of primary and secondary laboratory)
Detailed description of the method
- Validation
- Material (chemicals, solvents, references, instruments, plates, etc.)
- Stability (may be imported from previous experiments)
- Specificity (natural variability, adulterants, excipients in products, etc.)
- Precision
- Repeatability
- Intermediate precision
- Reproducibility (optional)
- Robustness
Results, releases, signatures
For 5.2–5.5, experimental details are described and acceptance criteria are given.
Specificity
Validation of specificity of a method for identification requires some convention about what we regard as “the same,” “similar,” and “different.” A strictly theoretical point of view leads to the assumption that we could actually measure the similarity of 2 samples and define numerical values as basis for decision. The practical problem is natural variability and our inability to predict the absolute chemical composition of the plant material based on its botanical identity. The degree of sophistication of an analytical approach is proportional to the number of differences found for 2 samples and the difficulty to decide which features are to be regarded as essential, common, and distinguishing (12).
We have adopted a simple pragmatic approach: visual comparison of fingerprints as electronic images. The sequence (number, color, intensity, and position) of the zones in a fingerprint is evaluated. Even for very complex fingerprints, this is not difficult as illustrated in Figure 6. Several samples, including preferably at least 3 authenticated samples of each species, are included in the investigation. They come ideally from different origin and help illustrate natural variability. It is good practice to use composite samples, i.e., homogenized samples that include more than one specimen.
Figure 6.
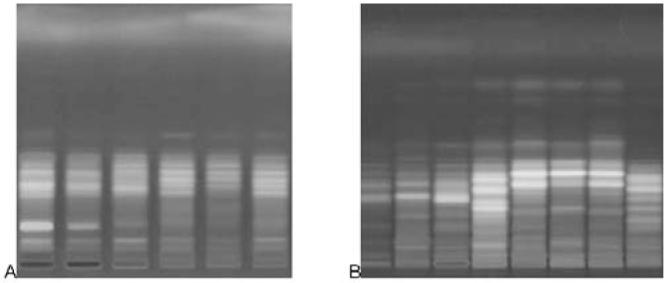
Similarity (left) and differences (right) of fingerprints: (A) samples of Actaea racemosa and (B) other Actaea species. Color image is available on the J. AOAC Int. Website (http://www.atypon-link. com/AOAC/loi/jaoi).
Acceptance criteria
A method is specific if during validation a sample representing the target species gives a fingerprint similar to that of the BRM and samples representing other species give different fingerprints. Any non-BRM sample investigated in the test may pass or fail the similarity test but authenticated samples of the target species must pass. Otherwise their authenticity must be questioned.
Precision
The precision of a qualitative analysis, in this case the sequence of zones of the HPTLC fingerprint, can be expressed as precision of the positions of separated zones (Rf-values). We propose to look at 3 levels.
For repeatability, 3 portions of the BRM are individually prepared according to the selected method. Onto 3 plates, 3 aliquots of each sample are applied. The plates are chromatographed subsequently using the same chamber but fresh portions of the mobile phase. The fingerprints of all tracks on a plate must be identical. This validates the absence of any inhomogeneity in chromatography (waves, “banana fronts,” etc.). The variability of Rf-values for 3 markers across each plate, and the variability of the average Rf (n =9)ofthose markers on 3 plates are evaluated. For determination of intermediate precision, the same experiment is repeated with 1 sample on 3 days. Determination of reproducibility requires the participation of at least 1 substantiating laboratory and could be extended to a full collaborative study. Due to practical considerations and in the attempt to simulate a method transfer requiring the use of identical methodology and identical instrumentation, we have chosen to include only the primary laboratory and 1 substantiating laboratory (1 in Switzerland, 1 in North Carolina). The substantiating laboratory analyzed the same BRM on 3 plates.
Acceptance criteria
All fingerprints must be identical. The variability of the Rf-values (Rf) of 3 markers should not exceed 0.01 across each plate, 0.02 for repeatability, 0.05 for intermediate precision, and 0.07 for reproducibility.
Robustness
Generally, robustness is not considered a required validation point. Effects of many experimental parameters can be evaluated already during method development. They include plates from different manufacturers, different chamber types and chamber configurations, mobile phase composition, temperature during chromatography, heating temperature and time during derivatization, etc. For practical reasons, we have limited our tests to the use of 1 different chamber type (with the same configuration, e.g., “saturated”) and a separation distance increased by 5 mm because these 2 can be regarded as “likely” deviations from our standard methodology under routine conditions.
The HPTLC result is generally affected by the relative humidity to which the plate was exposed prior to chromatography (13). Because only very few laboratories have controlled (fixed) humidity, we have included an evaluation of humidity effects to establish the range in which the method performs as expected (Figure 7).
Figure 7.
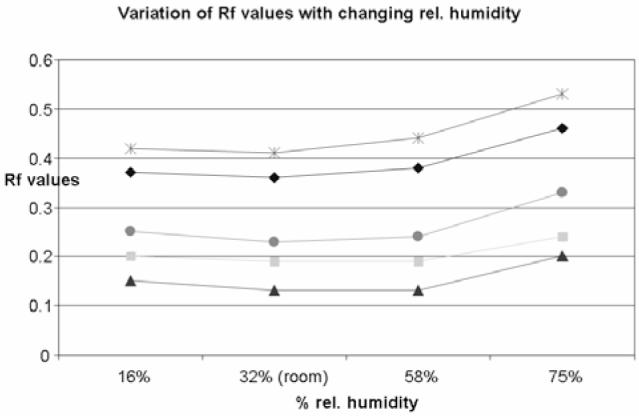
Humidity effects on the fingerprint of 5 components of licorice. Color image is available on the J. AOAC Int. Website (http://www.atypon-link.com/ AOAC/loi/jaoi).
Acceptance criteria
—There are 2 criteria for evaluation of robustness tests. (1) The Rf-values of all markers must lie within the acceptance criteria of the intermediate precision (Rf ≤ 0.05). (2) The system suitability test must pass [Note: We have introduced the concept of system suitability test (SST), even though it has already a certain (different) meaning in quantitative analysis, particularly in column chromatography. Each HPTLC plate must be considered as a new chromatographic system, which requires its own SST. Because the analysis of all samples on the plate takes place in parallel, the SST is performed at the same time. Typically specific features of the fingerprint of the sample (e.g., critical separation of 2 zones) are used to define SST. The only requirement is that the SST is meaningful. It must ensure that the analysis was performed properly and none of the parameters was out of range. If the SST fails, the plate can not be evaluated.]
Validated Method
The presented approach to validation of qualitative HPTLC methods for identification does not anticipate validation to fail, but rather to establish, what is to be expected from a method during routine use. It is possible that the write-up of the validated method includes some changes to the initial write-up in the validation protocol. Such changes may include restrictions or warnings with respect to relative humidity, waiting times, precision, etc. All changes are explained in the result section of the validation protocol.
Supplementary Material
Acknowledgments
We acknowledge the work of Kathrin Koll (14) who has, in the context of QC of herbal medicinal products, developed some principal ideas that went into the project.
The selection of plants followed the prioritization by the AOAC Presidential Task Force on Dietary Supplements, the American Herbal Products Association (AHPA), the American Herbal Pharmacopoeia (AHP), and the United States Pharmacopeia (USP). Some of the methods have already been included and published in AHP monographs.
The validation part of the project was financially supported in part by the Office of Dietary Supplements.
We thank ChromaDex (Santa Ana, CA), Phytolab (Vestenbergsgreuth, Germany), and USP (Rockville, MD) for providing chemical reference materials. Most plant samples were provided by AHP (Soquel, CA). Additional samples were donated from Botanical Liaison (Boulder, CO), Frutarom (Wädenswil, Switzerland), the EDQM (Strasbourg, France), NSF International (Ann Arbor, MI), USP, Nikyang Enterprise (Zhuhai, China), and VitaPlant (Witterswil, Switzerland).
We thank Kerri LeVanseler (NSF International) and Al Pohland (AOAC INTERNATIONAL) for their constructive input during discussions of the validation concept.
We thank Valeria Widmer (CAMAG Laboratory, Muttenz, Switzerland) for editorial help with the manuscript.
Appendix
Associated sample documents available electronically on the J. AOAC Int. Website (http://www.atypon-link.com/ AOAC/loi/jaoi):
0a-SOP-HPTLC—1 Supp–4 Supp
1b-Green Tea Validation Protocol—5 Supp–31 Supp
1c-Green Tea Validated Method—32 Supp–38 Supp
2a-Ginseng Method Evaluation—39 Supp–47 Supp
2b-Ginseng Validation Protocol—48 Supp–70 Supp
2c-Ginseng Validated Method—71 Supp–75 Supp
3a-Eleuthero Method Evaluation—76 Supp–86 Supp
3b-Eleuthero Validation Protocol—87 Supp–113 Supp
3c-Eleuthero Validated Method—114 Supp–118 Supp
4a-Echinacea Method Evaluation—119 Supp
4b-Echinacea Validation Protocol—120 Supp–149 Supp
4c-Echinacea Validated Method—150 Supp–157 Supp
5a-Black Cohosh Method Evaluation—158 Supp–163 Supp
5b-Black Cohosh Validation Protocol—164 Supp–188 Supp
5c-Black Cohosh Validated Method—189 Supp–195 Supp
6a-Licorice Method Evaluation—196 Supp–210 Supp
6b-Licorice Validation Protocol—211 Supp–233 Supp
6c-Licorice Validated Method—234 Supp–238 Supp
7a-Kava Method Evaluation—239 Supp–241 Supp
7b-Kava Validation Protocol—242 Supp–250 Supp
7c-Kava Validated Method—251 Supp–254 Supp
7d-Kava Method Development—255 Supp–262 Supp
8a-Milk Thistle Method Evaluation—263 Supp–271 Supp
8b-Milk Thistle Validation Protocol—272 Supp–280 Supp
8c-Milk Thistle Validated Method—281 Supp–285 Supp
9a-Feverfew Method Evaluation—286 Supp–294 Supp
9b-Feverfew Validation Protocol—295 Supp–303 Supp
9c-Feverfew Validated Method—304 Supp–307 Supp
10a-Ginger Method Evaluation—308 Supp–318 Supp
10b-Ginger Validation Protocol—319 Supp–327 Supp
10c-Ginger Validated Method—328 Supp–332 Supp
Footnotes
This paper provides a summary of a project that aimed at (1) development and validation of 10 HPTLC methods for identification of botanical raw materials; (2) elaboration of a general concept for validation of such methods; (3) proving that their validation is possible and necessary with respect to cGMP; and (4) providing to the botanical industry guidance in establishing practical and meaningful methods for quality assurance.
The paper is intended to serve as basis for a broad discussion of the subject among scientists and technicians from academia, industry, and regulatory agencies. The validation protocols and methods of the appendix may be considered as a possible template for future validation of methods, allowing convenient evaluation and transparency.
Supplemental information is available on the J. AOAC Int. Website (http://www.atypon-link.com/AOAC/loi/jaoi). Supplemental information is for information only. It has not been reviewed through the AOAC peer-review process.
References
- (1).Current Good Manufacturing Practice in Manufacturing, Packing, Labelling, or Holding Operations for Dietary Supplements (cGMPs) 2007U.S. Food and Drug Administration; Rockville, MD: http://www.cfsan.fda. gov/~lrd/fr07625a.html (accessed August 21, 2007) [PubMed] [Google Scholar]
- (2).Koll K, Reich E, Blatter A, Veit M. J. AOAC Int. 2003;86:909–915. [PubMed] [Google Scholar]
- (3).Ferenczi-Fodor K, Végh Z, Renger B. TRAC-Trend Anal. Chem. 2006;25:778–789. [Google Scholar]
- (4).Reich E, Schibli A. High-Performance Thin-Layer Chromatography for the Analysis of Medicinal Plants. Thieme Medical Publishers Inc.; New York, NY: 2007. pp. 132–156. [Google Scholar]
- (5).AOAC Guidelines for Single-Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals 2002AOAC INTERNATIONAL; Gaithersburg, MD: http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/ slv_guidelines.pdf (accessed August 21, 2007) [Google Scholar]
- (6).Validation of Analytical Procedures: Text and Methodology Q2(R1) ICH Harmonised Tripartite Guideline 2005International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use; http://www.ich.org/LOB/media/MEDIA417.pdf (accessed August 21, 2007) [Google Scholar]
- (7).Reich E, Schibli A. J. Planar Chromatogr. 2004;17:438–443. [Google Scholar]
- (8).Ferenczi-Fodor K, Végh Z, Nagy-Turák A, Renger B, Zeller M. J. AOAC Int. 2001;84:1265–1276. [PubMed] [Google Scholar]
- (9).Reich E, Schibli A. High-Performance Thin-Layer Chromatography for the Analysis of Medicinal Plants. Thieme Medical Publishers Inc.; New York, NY: 2007. pp. 175–192. [Google Scholar]
- (10).Reich E, Blatter A, Meier B. Pharmeuropa. 2003;15:424–430. [Google Scholar]
- (11).European Pharmacopoeia. 5th Council of Europe, European Directorate for the Quality of Medicines (EDQM); Strasbourg, France: 2005. Chapter 2.2.46. [Google Scholar]
- (12).Liang Y, Xie P, Chan K. J. Chromatogr. B. 2004;812:53–70. doi: 10.1016/j.jchromb.2004.08.041. [DOI] [PubMed] [Google Scholar]
- (13).Geiss F. Fundamentals of Thin-Layer Chromatography. Dr. Alfred Hüthig Verlag; Heidelberg, Germany: 1987. [Google Scholar]
- (14).Koll K. Dissertation. University of Bonn; Germany: 2003. Optimierung und Validierung dünnschichtchromatograhischer Verfahren in der Qualitätsanalytik von Phytopharmaka (German) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.