

Abstract
Aims/hypothesis
There are strong associations between measures of inflammation and type 2 diabetes, but the causal directions of these associations are not known. We tested the hypothesis that common gene variants known to alter circulating levels of inflammatory proteins, or known to alter autoimmune-related disease risk, influence type 2 diabetes risk.
Methods
We selected 46 variants: (1) eight variants known to alter circulating levels of inflammatory proteins, including those in the IL18, IL1RN, IL6R, MIF, PAI1 (also known as SERPINE1) and CRP genes; and (2) 38 variants known to predispose to autoimmune diseases, including type 1 diabetes. We tested the associations of these variants with type 2 diabetes using a meta-analysis of 4,107 cases and 5,187 controls from the Wellcome Trust Case Control Consortium, the Diabetes Genetics Initiative, and the Finland-United States Investigation of NIDDM studies. We followed up associated variants (p<0.01) in a further set of 3,125 cases and 3,596 controls from the UK.
Results
We found no evidence that inflammatory or autoimmune disease variants are associated with type 2 diabetes (at p≤0.01). The OR observed between the variant altering IL-18 levels, rs2250417, and type 2 diabetes (OR 1.00 [95% CI 0.99-1.03]), is much lower than that expected given (1) the effect of the variant on IL-18 levels (0.28 SDs per allele); and (2) estimates, based on other studies, of the correlation between IL-18 levels and type 2 diabetes risk (approximate OR 1.15 [95% CI 1.09-1.21] per 0.28 SD increase in IL-18 levels).
Conclusions/interpretation
Our study provided no evidence that variants known to alter measures of inflammation, autoimmune or inflammatory disease risk, including type 1 diabetes, alter type 2 diabetes risk.
Keywords: Autoimmune disease, Genes, Genetic epidemiology, Inflammation, Mendelian randomisation, SNP, Type 2 diabetes
Introduction
Inflammatory and immunological factors are associated with type 2 diabetes, but the causal direction of this association is not known. Evidence for a possible causal role of inflammatory and immunological processes in type 2 diabetes comes from several sources. These include prospective studies that show raised levels of pro-inflammatory, and lower levels of anti-inflammatory, cytokines many years before the onset of type 2 diabetes [1]. Specific examples include the presence of raised circulating IL-18 [1] and macrophage migration inhibitory factor (MIF) [2] levels in the KORA study, where average follow-up time to diagnosis of type 2 diabetes is 10 years, and similar findings observed with raised C-reactive protein (CRP) or IL-6 levels in several other prospective studies [3-5]. Second, mouse knockout models of certain cytokine genes, including IL18, IL1RN, IL1A and IL1B, result in adverse effects on obesity and insulin resistance in mouse models [6-8]. For example, the CD11B (also known as MAC-1 or ITGAM) knockout mouse is susceptible to diet-induced obesity compared with wild-type litter mates [9]. A common variant in the CD11B gene has recently been established as a risk factor for systemic lupus erythematosus (SLE) [10], suggesting a possible link between autoimmune diseases and metabolic traits. Common variants in the CDKAL1 gene are associated with type 2 diabetes, psoriasis and Crohn's disease [11], although the associated polymorphisms are different and not correlated (in linkage disequilibrium) with each other. Finally, recent data indicating a critical role for IL-1β in beta cell function [12] have led to a small randomised controlled trial of recombinant human IL-1 receptor antagonist (IL-1RA) (anakinra) treatment in type 2 diabetes that showed improved beta cell function and reduced levels of markers of systemic inflammation in 35 anakinra-administered patients with type 2 diabetes [13].
Further evidence is needed to establish whether inflammation has a causal role in type 2 diabetes in humans. Observed changes in inflammatory protein levels/concentrations with disease progression may be secondary to early disease processes and knockout animal studies suggest that if inflammatory processes have a role it is more likely to be through obesity-induced changes. Genetics studies can add to the evidence for or against the causal role of a trait in disease. This is because genetic variants cannot be influenced by disease processes and are much less likely to be confounded than non-genetic factors. This principle of ‘Mendelian randomisation’ has been used before to indicate that raised CRP levels are likely to be a consequence rather than cause of continuous metabolic traits [14].
Detailed studies of genetic variation across genes encoding inflammatory proteins have identified an increasing number of common variants that alter circulating levels of these proteins. These include variants in or near the IL18 [15-17], IL1RN [17, 18], IL6R, PAI1 (also known as SERPINE1)[17, 19], MIF [20] and CRP genes [17, 21, 22], all associated with robust statistical confidence with levels of their respective protein products. In addition the variant in IL6R is associated with IL-6 levels as well as levels of its soluble receptor (sIL-6R) [23, 24].
Recent genome-wide association (GWA) studies have resulted in a greatly increased knowledge of common gene variants that predispose to systemic or organ-specific auto-immune and inflammatory diseases. These include convincing genetic associations between variants at 30 loci, outside the HLA region, and Crohn's disease[25, 26], type 1 diabetes [27], rheumatoid arthritis [28, 29], coeliac disease [30, 31], ankylosing spondylitis [32], SLE [10, 33, 34] and multiple sclerosis [32, 35]. Five of these loci include variants that alter the risk of more than one inflammatory disease.
It has been suggested that type 1 and type 2 diabetes share a partially overlapping aetiology [36] but there is mixed evidence to support this. Recent studies show that known type 2 diabetes variants in the TCF7L2 and FTO loci do not predispose to type 1 diabetes [37, 38] In contrast, a recent Scandinavian study indicates that individuals with adult latent autoimmune diabetes have an increased frequency of both type 1 and type 2 diabetes risk alleles [39].
In this study we aimed to provide further insight into the possible role of inflammatory and autoimmune processes in type 2 diabetes. To do this we tested the hypothesis that common gene variants known to alter either circulating levels of inflammatory proteins or autoimmune and inflammatory disease risk also alter the risk of type 2 diabetes.
Methods
Study participants
Initial type 2 diabetes GWA meta-analysis
We used the p values and ORs from a meta-analysis of three type 2 diabetes GWA scans (http://www.well.ox.ac.uk/DIAGRAM/) that had recently been carried out using 2.2 million singlenucleotide polymorphisms (SNPs) (directly typed and imputed) with 4,107 type 2 diabetes cases and 5,187 controls, to identify associations between the selected polymorphisms and type 2 diabetes. The meta-analysis included samples used in GWA scans from the Wellcome Trust Case Control Consortium (WTCCC) (1,924 type 2 diabetes cases and 2,938 population controls, all from the UK), the Diabetes Genetics Initiative (DGI) (1,022 type 2 diabetes cases and 1,075 matched controls and 326 sibships discordant for diabetes) and the Finland-United States Investigation of NIDDM Genetics (FUSION) (1,161 type 2 diabetes cases and 1,174 normal glucose-tolerant controls, from Finland) studies [40]. Each SNP passed quality control criteria in each individual type 2 diabetes GWA study, as recently described [40]. Each study analysed the data under a model that is additive on the log-odds scale. ORs from each sample, excluding the DGI sibships, were combined using a fixed-effects model [41]. Patients in the WTCCC and DGI studies positive for anti-GAD antibodies were not included [40, 42]. The FUSION GWA case sample is composed of 789 cases from FUSION and 372 cases from FINRISK 2002. FUSION cases were excluded if they were positive for anti-GAD antibodies, had a short time to onset of insulin treatment following diagnosis, or had known or probable type 1 diabetes first-degree relatives [43]. Brief details of study participants are given in Electronic supplementary material (ESM) Table 1.
Replication study participants
For replication we genotyped SNPs nominally associated with type 2 diabetes using the UK Type 2 Diabetes Genetics Consortium Collection of 3,125 cases and 3,596 controls. All study participants were recruited from the Tayside region in Dundee, Scotland and were of European origin. Cases with monogenic forms of diabetes or with a history of treatment with regular insulin therapy within 1 year of diagnosis were excluded [44]. Controls were restricted to individuals <80 years of age and participants with evidence of hyperglycaemia were excluded from the study [44]. Details are given in ESM Table 1.
SNP selection
We selected three sets of SNPs from previous studies that have either been directly genotyped [28, 30-32, 35, 40, 42, 43] or imputed in the three-study diabetes meta-analysis [40]. These included: (1) Eight SNPs previously associated with circulating IL-1RA [17, 18], sIL-6R [24, 45], IL-18 [16, 17], CRP [17], plasminogen activator inhibitor (PAI)-1 [19] or MIF levels [20]. All SNPs were ‘cis’ effects in that they were present in or near the gene that codes for the protein they are associated with. This together with the statistical confidence (p≤10-5) of the associations between the SNP and protein levels means the associations are very unlikely to be false-positive results. For IL1RN and IL18 genes [16-18] there is evidence for two independent association signals (r2 between variants <0.5) and we therefore included both the SNPs associated with respective protein levels in each case. (2) Seven SNPs from five loci: IL23R, PTPN2, PTPN22, SH2B3 and IL2RA, which are associated with more than one inflammatory/autoimmune disease [25-27, 29, 31, 32, 35]. (3) Thirty-one independently associated variants from 30 loci proven to be autoimmune disease risk variants after robust replications (overall p≤ 10-8) and excluding the SNPs in the MHC locus. These SNPs are associated with seven diseases (Crohn's disease, type 1 diabetes, rheumatoid arthritis, coeliac disease, anky-losing spondylitis, SLE and multiple sclerosis).
All the 46 SNPs tested for an association passed QC control criteria in each individual study, as recently described [40]. Briefly all SNPs below a minor allele frequency (MAF) of 1% were excluded from analysis and all genotyped SNPs were in Hardy-Weinberg equilibrium in each individual study (p>10-4 for WTCCC, p≥10-6 for DGI and p≥10-6 for FUSION). Where genotyping data were unavailable, imputed SNPs were used for analysis as has previously been described [40]. In total 54% (25), 50% (23) and 52% (24) of the tested 46 SNPs lacked direct genotype information in the FUSION, WTCCC and DGI studies, respectively, and were therefore imputed. All imputed SNPs, except in five situations, had quality scores (r2hat in DGI and FUSION, imputation information scores in WTCCC) >80% and all, except in 17 situations, had quality scores >90%.
Individual genotyping in the replication study
We used a p value of <0.01 in the three-way meta-analysis to take SNPs forward into the replication samples. The exception to this was SNPs in the IL1RN and IL18 genes, where we used the less stringent p<0.1. SNPs in these two genes influence circulating levels and have a stronger prior case for their products' involvement in type 2 diabetes given previous trial and prospective studies. Genotyping was performed by KBiosciences (Hoddesdon, UK) using their own system of fluorescence-based competitive allele-specific PCR (KASPar).
Statistics
Meta-analysis of p values using an additive effects model in WTCCC, DGI and FUSION samples has been previously described [40]. The ORs were calculated excluding the related component of the DGI study. To combine the previously published meta-analysis data with replication data we performed a meta-analysis using a fixed-effects inverse variance method using the summary ORs and 95% CIs from the three-way analysis as one study and the ORs and 95% CIs from the replication samples as a second study. This meta-analysis was performed using the ‘Metan’ module in STATA version 9.1. In the three-study meta-analysis we had 80% power to detect an OR of 1.20 for a SNP with a MAF of 0.09 (the lowest MAF of all 46 SNPs) at p=0.05. For a more common SNP (MAF=0.30) we had 80% power to detect an OR of 1.10 at p=0.05. Power calculations were performed using STATA version 9.1.
Sensitivity analysis using directly genotyped SNPs
For 34 of the 46 SNPs the direct genotypes, or direct genotypes at an r2>0.8 proxy, were available in the WTCCC and DGI studies (total 2,946 cases and 4,013 controls typed on the Affymetrix 500 K chip). For eight of these 34 SNPs the r2>0.8 proxy was also available in FUSION. To confirm that the use of imputed data was not substantially influencing our results, we therefore repeated the analyses using only directly genotyped variants for these 34 SNPs.
Estimating expected effect sizes of inflammatory protein variants on type 2 diabetes risk
We calculated the approximate effect size we would expect a SNP that alters inflammatory protein levels to have on the risk of type 2 diabetes, if the inflammatory protein is causal to type 2 diabetes. We used the triangulation approach outlined in Fig. 1 to (1) estimate the expected per allele effect of a SNP on type 2 diabetes risk (A in Fig. 1), given (2) the per allele effect of the SNP on inflammatory protein levels (B in Fig. 1), and (3) the correlation between inflammatory protein levels and type 2 diabetes (C in Fig. 1). We did not have measures of inflammatory-related proteins in our cases and controls and so we used data from previous studies to obtain approximate effect sizes. For example, each allele of rs2250417, in the IL18 gene, raises IL-18 levels by 0.28 SDs in the InCHIANTI study [17]. In the same study, a 0.28 SD (68 pg/ml) rise in IL-18 levels is associated with an OR of 1.15 (95% CI 1.09-1.21) for type 2 diabetes risk (L. Ferrucci, unpublished results). The prospective KORA study of type 2 diabetes showed that individuals in the top quartile of IL-18 levels, had IL-18 levels 242 pg/ml higher than those in the bottom quartile [46], and a risk ratio for type 2 diabetes of 1.96 (95% CI 1.49-2.58). Using these figures we calculated that an allele effect of 68 pg/ml will equate to an approximate expected effect of a risk ratio of 1.21 (95% CI 1.12-1.30) for type 2 diabetes. Similar calculations using the previously reported effect of rs8192284 in the IL6R gene on IL-6 levels [24] and risk estimates from prospective studies of IL-6 with type 2 diabetes [47], suggest we would expect an approximate per allele effect of rs8192284 of 1.06 (95% CI 1.04-1.08) for type 2 diabetes risk. Using the previously reported effect of rs12093699 in the CRP gene on CRP levels [17] and risk estimates from prospective studies of CRP with type 2 diabetes [5] we would expect an approximate per allele effect of rs12093699 of a risk ratio of 1.34 (95% CI 1.23-1.39) for type 2 diabetes risk (ESM Table 2). These estimates are very approximate because they use circulating measures from different studies. In the case of prospective studies, estimates are based on risk ratios rather than ORs but the incidence of type 2 diabetes (5-10%) means that risk ratios and ORs will be approximately equivalent.
Fig. 1.
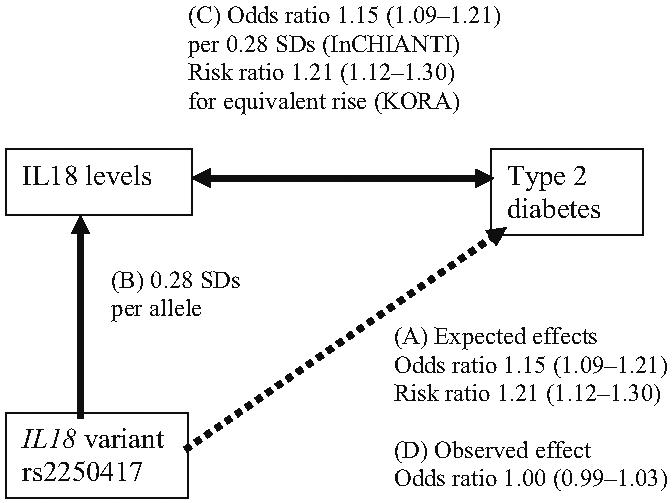
Associations between the SNP rs2250417 and IL-18 levels, IL-18 levels and type 2 diabetes and the expected and observed effects of rs2250417 on type 2 diabetes. ORs and risk ratios for the IL-18 levels-type 2 diabetes association are those estimated for a 0.28 SD increase in IL-18 levels, the effect of rs2250417 on IL-18 levels, from the InCHIANTI [17] and KORA studies [46]. Odds ratios and risk ratios are shown with 95% CIs
Results
SNPs known to alter circulating levels of inflammatory proteins
We found no strong evidence that eight SNPs, robustly associated with circulating levels of inflammatory proteins, alter the risk of type 2 diabetes. Associations with type 2 diabetes risk for two SNPs, one in each of the IL18 and IL1RN genes, reached p<0.1 in the three-study meta-analysis. The protein products of these two genes have both been strongly implicated in type 2 diabetes in trial and prospective studies and so we assigned a less-stringent statistical threshold for follow-up. At the time of study the SNP in each of these genes that represented the strongest association with circulating levels was uncertain. We therefore decided to increase the confidence of our genetic association findings (negative or positive) by increasing the sample size for all four SNPs in IL1RN and IL18. Meta-analysis using a total GWA and replication sample of 7,232 cases and 8,783 controls showed no strong evidence of association (Table 1). Subsequent analysis [17] indicated that rs2250417 had the strongest association with circulating IL-18 levels. The OR observed between the IL-18-altering variant, rs2250417, and type 2 diabetes is 1.00 (95% CI 0.99-1.03) (Fig. 1d). This is much lower than that expected given (1) the effect of the variant on IL-18 levels (0.28 SDs per allele [17]), and (2) estimates, based on other studies, of the correlation between IL-18 levels and type 2 diabetes risk, which are 1.15 (95% CI 1.09-1.21) per 0.28 SD increase in IL-18 levels in the InCHIANTI study (L. Ferrucci, unpublished results), and a risk ratio of 1.21 (95% CI 1.12-1.30) based on the KORA prospective study [46] (Fig. 1).
Table 1.
Cytokine cis-effect SNPs and associations with type 2 diabetes: three-way meta-analysis results across WTCCC, DGI and FUSION studies
| Disease SNP | Gene | Total cases | Total controls | MAF | Imputed |
OR for developing type 2 diabetes given pro-inflammatory allele (95% CI) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WTCCC | DGI | FUSION | Stage 1 | p value | Stage 2 (including replication samples) | p value | |||||
| For IL1RN and IL18 genes, data from the replication study are included in the meta-analysis | |||||||||||
| rs4251961 | IL1RN | 7,493 | 8,949 | 0.40 | No | No | Yes | 1.06 (0.99-1.12) | 0.07 | 1.01 (0.99-1.03) | 0.28 |
| rs6761276 | IL1RN | 7,888 | 8,484 | 0.44 | Yes | Yes | No | 0.98 (0.92-1.05) | 0.60 | 1.00 (0.99-1.03) | 0.39 |
| rs2250417 | IL18 | 7,674 | 9,175 | 0.47 | Yes | Yes | No | 1.00 (0.95-1.07) | 0.87 | 1.00 (0.99-1.03) | 0.39 |
| rs5744256 | IL18 | 7,479 | 8,509 | 0.21 | Yes | Yes | Yes | 1.08 (1.01-1.15) | 0.03 | 1.03 (1.00-1.05) | 0.03 |
| rs8192284 | IL6R | 4,549 | 5,579 | 0.35 | Yes | Yes | Yes | 0.96 (0.90-1.02) | 0.18 | ||
| rs12093699 | CRP | 4,549 | 5,579 | 0.32 | Yes | Yes | No | 1.02 (0.96-1.09) | 0.52 | ||
| rs2227631 | PAI1 | 4,549 | 5,579 | 0.40 | Yes | Yes | No | 0.97 (0.91-1.0) | 0.45 | ||
| rs1007888 | MIF | 4,549 | 5,579 | 0.42 | Yes | Yes | Yes | 0.96 (0.90-1.02) | 0.19 | ||
All ORs and p values from the meta-analysis are given in Table 1. The upper 95% CIs for risk alleles did not exceed 1.10. There was no evidence of association when limiting the analysis to the directly genotyped proxies, which captured six of the eight SNPs through directly genotyped proxies at r2>0.8, in the WTCCC and DGI studies (ESM Table 3).
SNPs known to alter the risk of autoimmune diseases
We found no strong evidence that SNPs robustly associated with multiple autoimmune diseases alter the risk of type 2 diabetes. ORs and p values from the three-study meta-analysis are given in Table 2. The upper 95% CIs for risk alleles did not exceed 1.12. We found no strong evidence that SNPs robustly associated with individual autoimmune diseases, including type 1 diabetes, alter the risk of type 2 diabetes. ORs and p values from the three-study meta-analysis are given in Table 3. The upper 95% CIs for risk alleles did not exceed 1.19. There was also no evidence of association when limiting the analysis to directly genotyped proxies, which captured 28 of the 38 SNPs at r2>0.8 in the WTCCC and DGI studies (ESM Table 3).
Table 2.
Pleiotropic autoimmune disease SNPs and their associations with type 2 diabetes: three-way meta-analysis results across WTCCC, DGI and FUSION studies
| Disease SNP | Disease | Gene | Imputed |
Disease risk allele frequency HapMap-CEPH | Meta-analysis OR for developing type 2 diabetes given disease risk allele (95% CI) | OR p value | ||
|---|---|---|---|---|---|---|---|---|
| WTCCC | DGI | FUSION | ||||||
| Hapmap CEPH refers to individuals of European ancestry typed in the International Hapmap project | ||||||||
| rs11209032 | Ankylosing spondylitis | IL23R | Yes | Yes | No | 0.31 | 0.96 (0.89-1.02) | 0.16 |
| rs118053303 | Crohn's disease | IL23R | No | No | Yes | 0.29 | 0.93 (0.87-0.99) | 0.04 |
| rs2542151 | Crohn's disease and type 1 diabetes | PTPN2 | No | No | Yes | 0.19 | 0.94 (0.87-1.02) | 0.17 |
| rs6679677 | Rheumatoid arthritis, Crohn's disease and type 1 diabetes | PTPN22 | No | No | Yes | 0.14 | 1.02 (0.93-1.12) | 0.69 |
| rs2104286 | Type 1 diabetes and multiple sclerosis | IL2RA | No | No | Yes | 0.23 | 1.01 (0.94-1.08) | 0.79 |
| rs17696736 | Type 1 diabetes and coeliac disease | SH2B3 | No | No | Yes | 0.35 | 1.03 (0.97-1.09) | 0.66 |
| rs653178 | Type 1 diabetes and coeliac disease | SH2B3 | Yes | Yes | Yes | 0.41 | 1.00 (0.94-1.06) | 0.99 |
Table 3.
Autoimmune disease SNPs and their associations with type 2 diabetes: three-way meta-analysis results across WTCCC, DGI and FUSION studies
| Disease SNP | Disease group | Gene | Imputed |
Disease risk allele frequency HapMap-CEPH | Meta-analysis OR for developing type 2 diabetes given disease risk allele (95% CI) | p value for OR | ||
|---|---|---|---|---|---|---|---|---|
| WTCCC | DGI | FUSION | ||||||
| Hapmap CEPH refers to individuals of European ancestry typed in the International Hapmap project | ||||||||
| Rs30187 | Ankylosing spondylitis | ARTS1 | Yes | Yes | No | 0.30 | 0.99 (0.93-1.05) | 0.81 |
| rs6822844 | Coeliac disease | IL21, IL2, TENR (also known as ADAD1), KIAA1109 | Yes | Yes | No | 0.79 | 1.02 (0.94-1.11) | 0.64 |
| rs2816316 | Coeliac disease | RGS1 | Yes | Yes | No | 0.78 | 1.02 (0.94-1.10) | 0.68 |
| rs13015714 | Coeliac disease | IL1RL1, IL18R1, IL18RAP, SLC9A4 | Yes | Yes | No | 0.23 | 1.05 (0.97-1.12) | 0.22 |
| rs17810546 | Coeliac disease | IL12A | Yes | Yes | No | 0.10 | 1.03 (0.94-1.13) | 0.50 |
| rs1464510 | Coeliac disease | LPP | Yes | Yes | No | 0.57 | 1.02 (0.96-1.08) | 0.51 |
| rs1738074 | Coeliac disease | TAGAP | Yes | Yes | No | 0.49 | 1.00 (0.94-1.07) | 0.88 |
| rs10883365 | Crohn's disease | NKX2-3 | No | No | Yes | 0.50 | 0.98 (0.92-1.03) | 0.41 |
| rs2241880 | Crohn's disease | ATG16L1 | Yes | Yes | No | 0.54 | 0.98 (0.93-1.04) | 0.60 |
| rs10077785 | Crohn's disease | IBD5 | No | No | Yes | 0.75 | 0.96 (0.89-1.02) | 0.20 |
| rs10801047 | Crohn's disease | Chr 1q31 | No | No | Yes | 0.09 | 1.06 (0.95-1.19) | 0.27 |
| rs6596075 | Crohn's disease | Chr 5q31 | No | No | Yes | 0.83 | 1.03 (0.95-1.17) | 0.50 |
| rs12035082 | Crohn's disease | Chr 1q24 | No | No | Yes | 0.37 | 0.99 (0.94-1.06) | 0.88 |
| rs2836754 | Crohn's disease | Chr 21q22 | No | No | Yes | 0.65 | 0.97 (0.92-1.04) | 0.42 |
| rs17221417 | Crohn's disease | CARD 15 (also known as NOD2 | No | No | Yes | 0.36 | 1.07 (0.99-1.15) | 0.05 |
| rs4958847 | Crohn's disease | IRGM | No | No | No | 0.09 | 1.06 (0.96-1.15) | 0.24 |
| rs9858542 | Crohn's disease | Chr 3p21 | No | No | Yes | 0.24 | 0.99 (0.93-1.05) | 0.66 |
| rs17234657 | Crohn's disease | Chr 5p13 | No | No | Yes | 0.17 | 1.06 (0.97-1.15) | 0.18 |
| rs10761659 | Crohn's disease | Chr 10q21 | No | No | Yes | 0.45 | 0.96 (0.91-1.02) | 0.22 |
| rs6920220 | Rheumatoid arthritis | Chr 6q23 | No | No | Yes | 0.17 | 1.05 (0.98-1.13) | 0.17 |
| rs10499194 | Rheumatoid arthritis | Chr 6q23 | Yes | Yes | Yes | 0.82 | 1.03 (0.96-1.10) | 0.41 |
| rs12708716 | Type 1 diabetes | KIAA0350 (also known as CLEC16A) Chr 16p13 | No | Yes | Yes | 0.29 | 1.03 (0.97-1.10) | 0.31 |
| rs763361 | Type 1 diabetes | CD226 | Yes | Yes | No | 0.47 | 0.94 (0.88-0.99) | 0.04 |
| rs3788964 | Type 1 diabetes | IFIH1 | No | No | No | 0.86 | 0.95 (0.87-1.02) | 0.17 |
| rs2292239 | Type 1 diabetes | ERBB3 | No | No | Yes | 0.30 | 0.97 (0.91-1.04) | 0.45 |
| rs3087243 | Type 1 diabetes | CTLA4 | No | No | Yes | 0.54 | 0.98 (0.92-1.04) | 0.46 |
| rs6897932 | Multiple sclerosis | IL7RA | No | No | No | 0.76 | 1.03 (0.96-1.10) | 0.35 |
| rs6445975 | SLE | PXK | Yes | Yes | No | 0.21 | 1.01 (0.95-1.08) | 0.73 |
| rs12537284 | SLE | IRF5, TNPO3 | Yes | Yes | No | 0.16 | 1.07 (0.98-1.16) | 0.11 |
| rs4963128 | SLE | KIAA1542 (also known as PHRF1) | Yes | Yes | No | 0.65 | 1.00 (0.94-1.08) | 0.89 |
| rs9888739 | SLE | ITGAM | Yes | Yes | No | 0.11 | 1.03 (0.94-1.14) | 0.51 |
Discussion
We have found no evidence that common gene variants known to alter circulating levels of inflammatory proteins, or those robustly associated with autoimmune or inflammatory-based diseases, influence risk of type 2 diabetes. We have not corrected for multiple testing because, given that our results are consistent with no association, this would be anti-conservative, rather than conservative. We included variants that influence inflammatory protein levels that are altered many years before the onset of diabetes. Included in our study were variants known to alter IL-1RA levels. This inflammatory protein is of particular interest because administration of recombinant human IL-1RA (anakinra) in a recent parallel group trial improved beta cell function in type 2 diabetic patients. However, we found no evidence that lifetime exposure to slightly lower IL-1RA levels, as determined by IL1RN genotypes, predisposes to type 2 diabetes.
Our results are consistent with chronic inflammation being secondary to type 2 diabetes disease processes rather than being causal. This does not rule out a role for other inflammatory or autoimmune processes, for which there is no known genetic variant. There are a number of limitations to our study. The main limitation is that we have not measured inflammatory proteins in our cases and controls. This means that we cannot perform a formal Mendelian randomisation analysis, where it is preferable to have the SNP, inflammatory protein levels and disease status measured in the same study. However, using data from other studies we estimated that variants that alter IL-18, IL-6 and CRP levels should predispose to type 2 diabetes with ORs of approximately 1.15 to >1.21, 1.06 and 1.34, respectively (ESM Table 2), if these pro-inflammatory proteins are causally related to type 2 diabetes. The upper 95% CI of the effect sizes we observed (1.05, 1.02 and 1.09 for IL18, IL6R and CRP variants, respectively) are lower than the expected effect size. This is consistent with raised IL-18, IL-6 and CRP levels being secondary to, rather than causally predisposing to, type 2 diabetes. Further studies are needed with measures of inflammatory proteins in large numbers of cases and controls. It is also possible that these variants are not altering inflammatory pathways even if they are altering circulating levels.
A second limitation is that variants known to alter the risk of autoimmune and inflammatory diseases may not have an inflammatory effect in the general population. However, the identification of variants associated with inflammatory and autoimmune-related diseases indicates that the variant may be altering the expression, processing or function of a nearby gene that is critical to an inflammatory or autoimmune process prior to disease onset. Most of the autoimmune disease-associated variants have only been discovered very recently and so the disease mechanisms are not completely understood. However, early studies indicate that the variant in the IL2RA region predisposing to type 1 diabetes and multiple sclerosis alters circulating levels of IL2RA protein [48], the variant predisposing to coeliac disease in the IL18RAP locus alters mRNA levels of IL-18 receptor accessory protein in lymphocytes [31] and the variant in the IL7RA (also known as IL7R) gene predisposing to multiple sclerosis alters levels of soluble IL-7 receptor antagonist because of differential splicing of a key exon [49]. It is also suggested that the variant predisposing to ankylosing spondylitis in the ARTS1 (also known as ERAP-1) gene may have a general pro-inflammatory effect because it cleaves cell surface receptors for IL-1, IL-6 and TNF-α [32].
A final possible limitation is that we have used imputed SNP genotypes for association analysis for the majority of the 46 studied variants. However, this is unlikely to have resulted in appreciable differences in OR estimates, as has previously been shown [40, 50] and there were no positive associations when we used the strongest available directly genotyped proxies (r2>0.8) instead of the imputed SNPs (ESM Table 3).
A further implication of our study is that it does not support the hypothesis of a genetic overlap between type 1 and type 2 diabetes. Our results are consistent with the recent studies which show that the known type 2 diabetes-and BMI-associated variants in the TCF7L2 [37] and FTO [38] genes do not predispose to type 1 diabetes.
In conclusion, in a large case-control study of type 2 diabetes, we tested 46 SNPs associated with inflammatory diseases or inflammatory protein levels, and identified no associations with type 2 diabetes. Our study is consistent with inflammatory processes being secondary, rather than causal, to type 2 diabetes.
Supplementary Material
Acknowledgements
M. N. Weedon is a Vandervell Foundation research fellow. The FUSION study was funded by an intramural grant (NIH DK 062370) from the US National Institutes of Health (NIH). E. Zeggini is a Wellcome Trust Career Development Fellow. D. Melzer's work is supported in part by NIH/National Institute on Aging (NIH/NIA) Grant R01 AG24233. Details of the DIAGRAM (Diabetes Genetics Replication And Meta-analysis) Consortium can be found at http://www.well.ox.ac.uk/DIAGRAM/index.html.
Abbreviations
- CRP
C-reactive protein
- DGI
Diabetes Genetics Initiative
- FUSION
Finland-United States Investigation of NIDDM
- GWA
genome-wide association
- IL-1RA
IL-1 receptor antagonist
- MAF
minor allele frequency
- MIF
macrophage migration inhibitory factor
- PAI
plasminogen activator inhibitor
- sIL-6R
soluble IL-6 receptor
- SLE
systemic lupus erythematosus
- SNP
single-nucleotide polymorphism
- WTCCC
Wellcome Trust Case Control Consortium
Footnotes
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-008-1160-3) contains supplementary material, which is available to authorised users.
References
- 1.Kolb H, Mandrup-Poulsen T. An immune origin of type 2 diabetes. Diabetologia. 2005;48:1038–1050. doi: 10.1007/s00125-005-1764-9. [DOI] [PubMed] [Google Scholar]
- 2.Herder C, Kolb H, Koenig W, et al. Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: results from the Cooperative Health Research in the Region of Augsburg, Survey 4 (KORA S4) Diabetes Care. 2006;29:368–371. doi: 10.2337/diacare.29.02.06.dc05-1474. [DOI] [PubMed] [Google Scholar]
- 3.Duncan BB, Schmidt MI, Pankow JS, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- 4.Barzilay JI, Abraham L, Heckbert SR, et al. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes. 2001;50:2384–2389. doi: 10.2337/diabetes.50.10.2384. [DOI] [PubMed] [Google Scholar]
- 5.Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 6.Netea MG, Joosten LA, Lewis E, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12:650–656. doi: 10.1038/nm1415. [DOI] [PubMed] [Google Scholar]
- 7.Matsuki T, Horai R, Sudo K, Iwakura Y. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J Exp Med. 2003;198:877–888. doi: 10.1084/jem.20030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chida D, Osaka T, Hashimoto O, Iwakura Y. Combined interleukin-6 and interleukin-1 deficiency causes obesity in young mice. Diabetes. 2006;55:971–977. doi: 10.2337/diabetes.55.04.06.db05-1250. [DOI] [PubMed] [Google Scholar]
- 9.Dong ZM, Gutierrez-Ramos JC, Coxon A, Mayadas TN, Wagner DD. A new class of obesity genes encodes leukocyte adhesion receptors. Proc Natl Acad Sci U S A. 1997;94:7526–7530. doi: 10.1073/pnas.94.14.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nath SK, Han S, Kim-Howard X, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 11.Wolf N, Quaranta M, Prescott NJ, et al. Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J Med Genet. 2008;45:114–116. doi: 10.1136/jmg.2007.053595. [DOI] [PubMed] [Google Scholar]
- 12.Donath MY, Storling J, Maedler K, Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med. 2003;81:455–470. doi: 10.1007/s00109-003-0450-y. [DOI] [PubMed] [Google Scholar]
- 13.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 14.Timpson NJ, Lawlor DA, Harbord RM, et al. C-reactive protein and its role in metabolic syndrome: Mendelian Randomisation Study. Lancet. 2005;366:1954–1959. doi: 10.1016/S0140-6736(05)67786-0. [DOI] [PubMed] [Google Scholar]
- 15.Tiret L, Godefroy T, Lubos E, et al. Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation. 2005;112:643–650. doi: 10.1161/CIRCULATIONAHA.104.519702. [DOI] [PubMed] [Google Scholar]
- 16.Frayling TM, Rafiq S, Murray A, et al. An interleukin-18 polymorphism is associated with reduced serum concentrations and better physical functioning in older people. J Gerontol A Biol Sci Med Sci. 2007;62:73–78. doi: 10.1093/gerona/62.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melzer D, Perry JR, Hernandez D, et al. A genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rafiq S, Stevens K, Hurst AJ, et al. Common genetic variation in the gene encoding interleukin-1-receptor antagonist (IL-1RA) is associated with altered circulating IL-1RA levels. Genes Immun. 2007;8:344–351. doi: 10.1038/sj.gene.6364393. [DOI] [PubMed] [Google Scholar]
- 19.Kathiresan S, Gabriel SB, Yang Q, et al. Comprehensive survey of common genetic variation at the plasminogen activator inhibitor-1 locus and relations to circulating plasminogen activator inhibitor-1 levels. Circulation. 2005;112:1728–1735. doi: 10.1161/CIRCULATIONAHA.105.547836. [DOI] [PubMed] [Google Scholar]
- 20.Herder C, Klopp N, Baumert J, et al. Effect of macrophage migration inhibitory factor (MIF) gene variants and MIF serum concentrations on the risk of type 2 diabetes: results from the MONICA/KORA Augsburg Case-Cohort Study, 1984-2002. Diabetologia. 2008;51:276–284. doi: 10.1007/s00125-007-0800-3. [DOI] [PubMed] [Google Scholar]
- 21.Russell AI, Cunninghame Graham DS, Shepherd C, et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet. 2004;13:137–147. doi: 10.1093/hmg/ddh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zee RY, Ridker PM. Polymorphism in the human C-reactive protein (CRP) gene, plasma concentrations of CRP, and the risk of future arterial thrombosis. Atherosclerosis. 2002;162:217–219. doi: 10.1016/s0021-9150(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 23.Reich D, Patterson N, Ramesh V, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiq S, Frayling TM, Murray A, et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8:552–559. doi: 10.1038/sj.gene.6364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomson W, Barton A, Ke X, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL-2 and IL-21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunt KA, Zhernakova A, Turner G, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton PR, Clayton DG, Cardon LR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harley JB, Alarcon-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 35.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 36.Wilkin TJ. The accelerator hypothesis: a unifying explanation for type-1 and type-2 diabetes. Nestle Nutr Workshop Ser Clin Perform Programme. 2006;11:139–150. doi: 10.1159/000094447. discussion 150-133. [DOI] [PubMed] [Google Scholar]
- 37.Field SF, Howson JM, Smyth DJ, Walker NM, Dunger DB, Todd JA. Analysis of the type 2 diabetes gene, TCF7L2, in 13,795 type 1 diabetes cases and control subjects. Diabetologia. 2007;50:212–213. doi: 10.1007/s00125-006-0506-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Field SF, Howson JM, Walker NM, Dunger DB, Todd JA. Analysis of the obesity gene FTO in 14,803 type 1 diabetes cases and controls. Diabetologia. 2007;50:2218–2220. doi: 10.1007/s00125-007-0767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cervin C, Lyssenko V, Bakhtadze E, et al. Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes. 2008;57:1433–1437. doi: 10.2337/db07-0299. [DOI] [PubMed] [Google Scholar]
- 40.Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kavvoura FK, Ioannidis JP. Methods for meta-analysis in genetic association studies: a review of their potential and pitfalls. Hum Genet. 2008;123:1–14. doi: 10.1007/s00439-007-0445-9. [DOI] [PubMed] [Google Scholar]
- 42.Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 43.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galicia JC, Tai H, Komatsu Y, Shimada Y, Akazawa K, Yoshie H. Polymorphisms in the IL-6 receptor (IL-6R) gene: strong evidence that serum levels of soluble IL-6R are genetically influenced. Genes Immun. 2004;5:513–516. doi: 10.1038/sj.gene.6364120. [DOI] [PubMed] [Google Scholar]
- 46.Thorand B, Kolb H, Baumert J, et al. Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA Augsburg Study, 1984-2002. Diabetes. 2005;54:2932–2938. doi: 10.2337/diabetes.54.10.2932. [DOI] [PubMed] [Google Scholar]
- 47.Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes. 2004;53:693–700. doi: 10.2337/diabetes.53.3.693. [DOI] [PubMed] [Google Scholar]
- 48.Lowe CE, Cooper JD, Brusko T, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 49.Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 50.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.