

Abstract
A crucial step in human breast cancer progression is the acquisition of invasiveness. There is a distinct lack of human cell culture models to study the transition from preinvasive to invasive phenotype as it may occur “spontaneously” in vivo. To delineate molecular alterations important for this transition, we isolated human breast epithelial cell lines that showed partial loss of tissue polarity in three-dimensional reconstituted basement membrane cultures. These cells remained noninvasive; however, unlike their nonmalignant counterparts, they exhibited a high propensity to acquire invasiveness through basement membrane in culture. The genomic aberrations and gene expression profiles of the cells in this model showed a high degree of similarity to primary breast tumor profiles. The xenograft tumors formed by the cell lines in three different microenvironments in nude mice displayed metaplastic phenotypes, including squamous and basal characteristics, with invasive cells exhibiting features of higher-grade tumors. To find functionally significant changes in transition from preinvasive to invasive phenotype, we performed attribute profile clustering analysis on the list of genes differentially expressed between preinvasive and invasive cells. We found integral membrane proteins, transcription factors, kinases, transport molecules, and chemokines to be highly represented. In addition, expression of matrix metalloproteinases MMP9, MMP13, MMP15, and MMP17 was up-regulated in the invasive cells. Using small interfering RNA–based approaches, we found these MMPs to be required for the invasive phenotype. This model provides a new tool for dissection of mechanisms by which preinvasive breast cells could acquire invasiveness in a metaplastic context.
Introduction
Breast carcinomas are phenotypically and behaviorally heterogeneous (1). Subtypes of breast cancer with distinct behavioral characteristics have been recognized morphologically as well as by molecular analyses. For all breast cancers, conversion to the invasive phenotype is distinct from metastasis but is an obligate prerequisite for metastasis, thus representing a crucial step in cancer progression. Molecular profiling studies have begun to determine markers that are associated with progression in different breast cancer subtypes (2–17). Relevant models that recapitulate the morphologic and molecular aspects of progression within subtypes are needed to determine which of these molecular changes function in acquisition of the invasive phenotype.
Culture models of human breast cells could provide such an opportunity if relevance to cancer progression in vivo were established. Pathways associated with transition to malignancy have traditionally been studied using either transgenic mouse models or primary human cells engineered to express genes with oncogenic properties. To deal with the heterogeneity of human breast tumors, cell lines originating from a large number of malignant breast carcinomas have been used (18). It would also be useful to have models of “spontaneous” transition to malignancy, as opposed to transgenic, which could include intermediate noninvasive and preinvasive stages of carcinogenesis, as opposed to modeling only the invasive carcinoma stage. Such models could facilitate the discovery of steps in progression to the invasive phenotype in a context that recapitulates more the complexity of carcinogenesis.
The HMT-3522 human breast epithelial cell line series (S1, S2, and T4-2) were derived from a reduction mammoplasty specimen of a patient with fibrocystic breast disease more than 20 years ago. The epithelial component of the tissue was grown in defined medium to give rise to S1 cells that became immortalized spontaneously; these cells are epidermal growth factor (EGF) dependent for growth and are nontumorigenic (19). Continuous culturing in the absence of EGF gave rise to a cell population referred to as S2, a cell line that is heterogeneous and essentially still nonmalignant (20). Inoculation of S2 cells into mice produced a rare tumor that was cultured and passaged again through mice (20). The cell line derived from this tumor is referred to as T4-2 (20). Previously, we designed a three-dimensional laminin-rich basement membrane (3DlrBM) assay to model both the normal breast and breast cancers (ref. 21; Fig. 1A). In 3DlrBM, S1 cells produce growth-arrested acini that express differentiation markers mimicking in vivo polar patterns of localization of a number of markers (21): S2 colonies are heterogeneous (this study) and T4-2 cells form large and disorganized colonies (22).
Figure 1.
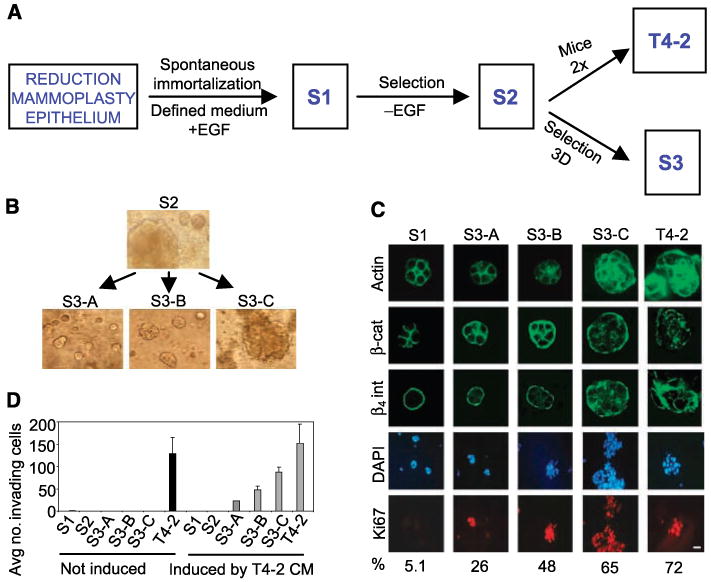
Partial loss of tissue polarity and preinvasive phenotypes of S3 cells. A, steps involved in the isolation of the HMT-3522 series of cell lines. B, 3DlrBM cultures of HMT-3522-S2 cells and small (S3-A), medium (S3-B), and large (S3-C) colonies isolated from the parent culture of S2 on day 10. C, confocal images of immunostained colonies in 3DlrBM, day 10. D, invasion assays: two experiments, triplicate samples. Black columns, untreated cells; gray columns, cells treated with T4-2 conditioned medium (CM).
Here, we isolated three essentially homogeneous cell populations from S2 cells, which we refer to as S3-A, S3-B, and S3-C, using colony size in 3DlrBM as a screening tool (Fig. 1A). These cells were noninvasive but had a higher potential for acquiring invasiveness than the parent S2. We found a substantial number of similarities between the model of S3 versus T4-2 and preinvasive to invasive transition in breast carcinogenesis based on progressive loss of tissue polarity, ability to form tumors with squamous and basal metaplastic histology, 76% similarity to recurrent comparative genomic hybridization (CGH) abnormalities found in breast tumors, and gene expression changes in classes of genes previously identified as being important for malignancy and invasion. These observations establish the S3 cell lines as the preinvasive counterpart of a model of breast cancer progression within a metaplastic context. Using matrix metalloproteinases (MMP), we show the utility of this model in identifying functionally significant changes in transition from the preinvasive to the invasive phenotype in metaplastic breast cancer. The usefulness of the model is further shown in another report (23), which describes the discovery of a novel pathway involved in invasion using this progression series.
Materials and Methods
Cell Culture
S1 and T4-2 cells were grown in tissue culture monolayers two-dimensionally using Falcon tissue culture plastic or three-dimensionally in laminin-rich basement membrane (Matrigel) in defined medium as previously described (21, 22); S2 and S3 cells were grown under the same conditions as T4-2.
Indirect Immunofluorescence Analysis and Image Acquisition in 3DlrBM
Cells in 3DlrBM were perfused in sucrose and frozen on dry ice in Tissue-Tek optimum cutting temperature (Miles Laboratories), sectioned, and immunostained as previously described (22).
Comparative Genomic Hybridization
Chromosomal CGH and BAC array CGH were done essentially as previously described (24). Hierarchical clustering analysis was done with GeneSpring software (Silicon Genetics), using standard correlation with a minimum distance metric of 0.001 to determine relatedness of cell lines. This was done using both the chromosomal and the BAC array data, with only significant amplification/deletion data points, as well as with the complete data sets, producing comparable results.
Analysis of Tumor Formation In vivo
We injected the cells into female BALB/c athymic nude mice (Simonsen Laboratories), measured tumors weekly with a caliper, and graphed the tumor frequency and volumes of tumors taken out after 9 to 10 weeks of tumor growth, at which time the animals were sacrificed. Statistical analysis of the mean tumor volumes was done by pairwise comparison using one-tailed homoscedastic t test analysis. We injected 10 million cells s.c. into the rear flanks, 5 million cells in 50% Matrigel s.c. into the front flanks, or 2 million cells into the left and right fourth inguinal mammary glands. Tumors from all injections and surrounding tissues were fixed in 4% formaldehyde, paraffin embedded, sectioned into 5-μm slices, and stained with H&E (HRL Laboratories). Three hundred fifty H&E sections were independently examined by two expert breast pathologists (A.D.B. and R.A.J.).
Global mRNA Expression Analysis by cDNA Microarrays
cDNA microarrays with ∼8,000 known gene spots on poly-l-lysine–coated chips (custom arrayed at LBNL using Research Genetics 8k human clones) were used. mRNA samples were directly compared with each other by cohybridization to the same slide using dendrimer technology to label with red-Cy5 or green-Cy3 (Genisphere). Total RNA (1 μg) isolated with Qiagen RNEasy reagents was used for each sample hybridized. Cells in 3DlrBM were extracted with 5 mmol/L EDTA in cold PBS to dissolve the Matrigel. For each comparison, three independent sets of cultures were processed, and cells were examined at day 10 of culture. For each comparison, four slides were hybridized. This corresponded to three sets of RNAs from independent cultures plus a dye-swap experiment in which the red and green labels were switched for the two samples in question. Data were analyzed as described in Supplementary File 8.
Reverse Transcription-PCR
Quantitative
The RNA prepared for microarrays was used to prepare cDNA after DNase I (Invitrogen) treatment, using Superscript First-Strand Synthesis System for reverse transcription-PCR (RT-PCR; Invitrogen). Quantitative real-time PCR was done using LightCycler (Roche) and FastStart SYBR Green (Roche). Primer sequences were as follows (5′-3′): MMP13 forward, aagatgcatccaggggtcct; MMP13 reverse, gtccaggtttcatcatcatca; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward, cccctggccaaggtcatccatgac; and GAPDH reverse, cataccaggaaatgagcttgacaaag. LightCycler reaction cycles were as recommended by Roche with the following modifications: for MMP13, 57°C for annealing, 27 seconds of extension, and signal acquisition at 84°C; for GAPDH, 56°C for annealing and 25 seconds for extension.
Semiquantitative
For MMP9, MMP13, MMP15, and MMP17, semiquantitative RT-PCR was done using the following primer sets (5′-3′): MMP9, atgcgtggagagtcgaaatc (forward) and tacacgcgagtgaaggtgag (reverse); MMP13, aagatgcatccaggggtcct (forward) and gtccaggtttcatcatcatca (reverse); MMP15, ccatatgtccaccatgcgtt (forward) and atgatggcattggggttgct (reverse); and MMP17, acgcaagaggagctgtctaag (forward) and acatggcttaacccaatggc (reverse). The conditions used (after having determined linear range) were, for MMP9, MMP15, and MMP17, 96°C 3 minutes, 34 × (96°C 30 seconds, 55°C 1 minutes, 72°C 1 minutes), 72°C 5 minutes; for MMP13, 96°C 3 minutes, 29 × (96°C 30 seconds, 55°C 30 seconds, 72°C 1 minutes), 72°C 5 minutes.
Expression Analysis of Cell-Surface Proteins by Fluorescence-Activated Cell Sorting
Live cells were immunostained in suspension before fixing with 2% paraformaldehyde. Primary antibodies were used at 1:10 dilution, and FITC-conjugated secondary antibodies were used at 1:100 dilution. Primary antibodies were MMP15, clone 162-22G5 (Oncogene); MMP17, rabbit polyclonal 475934 (Calbiochem). Fluorescence-activated cell sorting (FACS) analysis was done using EPICS XL-MCL data acquisition and display software on XL flow cytometry analyzers (University of California Berkeley, Flow Cytometry Facility). Gating of forward light scatter versus light scatter (90-degree scatter) allowed us to obtain data relevant to intact cells only; FITC fluorescence peak was evaluated for its median value and was corrected using samples that had not been treated with primary antibody.
Gelatin Zymography
Conditioned medium (for 48 hours) from 10-day cultures of S1, S2, S3, and T4-2 were used. Samples were analyzed by SDS-PAGE zymography (25) to determine the molecular weights and the relative abundance of the gelatinases present.
Invasion Assay
The ability to invade through laminin-rich basement membrane (Matrigel) was measured in Boyden chamber assays, essentially as described (26). The number of invading T4-2 or S3-C cells (of 1 × 105 seeded) was determined after 48 hours of incubation in regular growth medium, in medium containing T4-2 conditioned medium, in medium with DMSO with different concentrations of the MMP inhibitor GM6001 (AMS Scientific) or its inactive analogue (C1004), or in conditioned medium that had previously been treated with DMSO, GM6001, or C1004. For small interfering RNA (siRNA)–treated T4-2 cells, transfection of 30 to 150 nmol/L oligo with 4 μL of siPORT NeoFX (Ambion) per milliliter of media was done 1 day after plating cells at regular density. siRNAs were allowed to down-regulate protein levels for 2 days. Cells were then trypsinized and 1 × 105 cells were seeded for Boyden chamber assays. siRNAs against MMP9 (oligo 1, Ambion ID143941; oligo 2, Ambion ID 113182: oligo 3, Ambion ID 113183), MMP13 (oligo 1, Ambion ID 143556; oligo 2, Ambion ID 212725; oligo 3, Ambion ID 112915), MMP15 (oligo 1, Ambion ID 112917; oligo 2, Ambion ID 143557; oligo 3, Ambion ID 112916), MMP17 (oligo 1, Ambion ID 105396; oligo 2, Ambion ID 113514; oligo 3, Ambion ID 24016), or scrambled control siRNA (Ambion, Silencer-Cy3 labeled) were used.
Immunohistochemistry
Ki67
Formalin-fixed mouse xenograft tissue was paraffin embedded and sectioned into 5-μm-thick tissue sections (HRL Laboratories). The paraffin was removed by serial incubation in xylene, 100% ethanol, 95% ethanol, 70% ethanol, and water. Tissues were blocked with 3% hydrogen peroxide in PBS for 5 minutes. Antigen retrieval was done by incubating in 0.01% prewarmed trypsin in PBS for 15 minutes, followed by 10 minutes of microwaving in 10 mmol/L sodium citrate buffer. Tissues were blocked in 1.5% normal horse serum in PBS for 30 minutes and incubated with primary Ki67 antibody, which specifically recognizes human, but not mouse, Ki67 protein [Calbiochem, anti-Ki67 human (mouse), NA59] at 5 μg/mL overnight at 4°C. Slides were washed with PBS and incubated with biotinylated antirabbit antibody [1:200 dilution; Vector Laboratories, biotinylated antimouse IgG/antirabbit IgG (H + L), BA-1400] for 30 minutes at room temperature, followed by streptavidin-horseradish peroxidase (Vector Laboratories, Vectastain ABC kit, Elite PK-6100) for 30 minutes and complete 3,3′-diaminobenzidine tetrahydrochloride (Sigma) medium for 5 minutes. Slides were washed and counterstained with hematoxylin, followed by dehydration in 70% ethanol, 95% ethanol, 100% ethanol, and xylene; coverslips were mounted using Permount (Fisher Scientific, SP15). Strong nuclear staining was scored as positive and %Ki67 positive nuclei was determined for at least three tumor areas for each cell type injection, counting a minimum of 100 cells per area.
p63
Immunohistochemistry was done essentially as described for Ki67, except for omitting the trypsin step in antigen retrieval and increasing hydrogen peroxide blocking to 15 minutes. Primary antibody (Lab Vision, p63 Ab-1, clone 4A4) was used at 2 μg/mL.
CK5/6, estrogen receptor, and progesterone receptor
Immunohistochemistry was done as described in ref. 27, using CK5/6 primary antibodies from Zymed (1:100), estrogen receptor from DAKO (1:25), and progesterone receptor from DAKO (1:200); for HER2-neu, the Herceptest kit from DAKO was used.
Results
S3 cells display partial loss of tissue polarity in 3DlrBM, a preinvasive phenotype
Using colony size in 3DlrBM as an initial screen, we isolated multiple small-, medium-, and large-sized colonies; expanded and propagated these clones for at least six generations; and examined their morphology in the three-dimensional assay (Fig. 1B). We chose three isolates that essentially bred true for the size of colony formed. We refer to these as S3-A, S3-B, and S3-C cell lines where colony size is A < B < C. S3-A cultures contain ∼10% very large colonies (not shown); otherwise the colony size, especially for S3-Cs, is essentially homogeneous. S3 cells displayed intermediate phenotypes compared with S1 and T4-2 colonies in 3DlrBM, as determined by markers that have been extensively used to describe tissue (acinar) polarity (22, 28–31), such as basolateral expression of β-catenin, basal β4 integrin, cortical actin, and Ki67 labeling (Fig. 1C). In Boyden chamber assays, S1, S2, and S3 cell lines did not invade laminin-rich basement membrane, but T4-2 cells were invasive (Fig. 1D). When conditioned medium from T4-2 cells was added to the chambers, S1 and S2 cells did not invade, whereas S3-A, S3-B, and S3-C became invasive, with the S3-C cells displaying the highest propensity to invade: T4-2 > C > B > A.
S3 cells display an intermediate potential to form tumors
We determined tumorigenicity of the cells when injected into nude mice under three different microenvironments: in the flank with cells alone or cells plus Matrigel, and into mammary fat pads with cells alone (Fig. 2). In none of the injections did S1 cells or media control produce any measurable growth but T4-2 cells formed palpable tumors at high frequency, as shown also previously (refs. 20, 22, 32, 33; Fig. 2A–C, top) When injected s.c., S2 cells and the S3 series formed very small tumors at low frequencies. S.c. injection in the presence of Matrigel (Fig. 2A–C, middle) resulted in tumor formation in 50% of S2, 75% of S3-A, 73% of S3-B, 56% of S3-C (see following section on tumor histology), and 100% of T4-2 cell injection sites. Importantly, mammary fat pad injections (Fig. 2A–C, bottom) resulted in progressively increasing tumor frequency going from S2, S3-A, S3-B, S3-C, to T4-2, mirroring the rate of invasion observed in Boyden chambers (Fig. 1D). Comparing the P values for tumor volume (Fig. 2C) showed that the mammary fat pad most clearly distinguished the nontumorigenic S1, the preinvasive S3, and the malignant T4-2.
Figure 2.
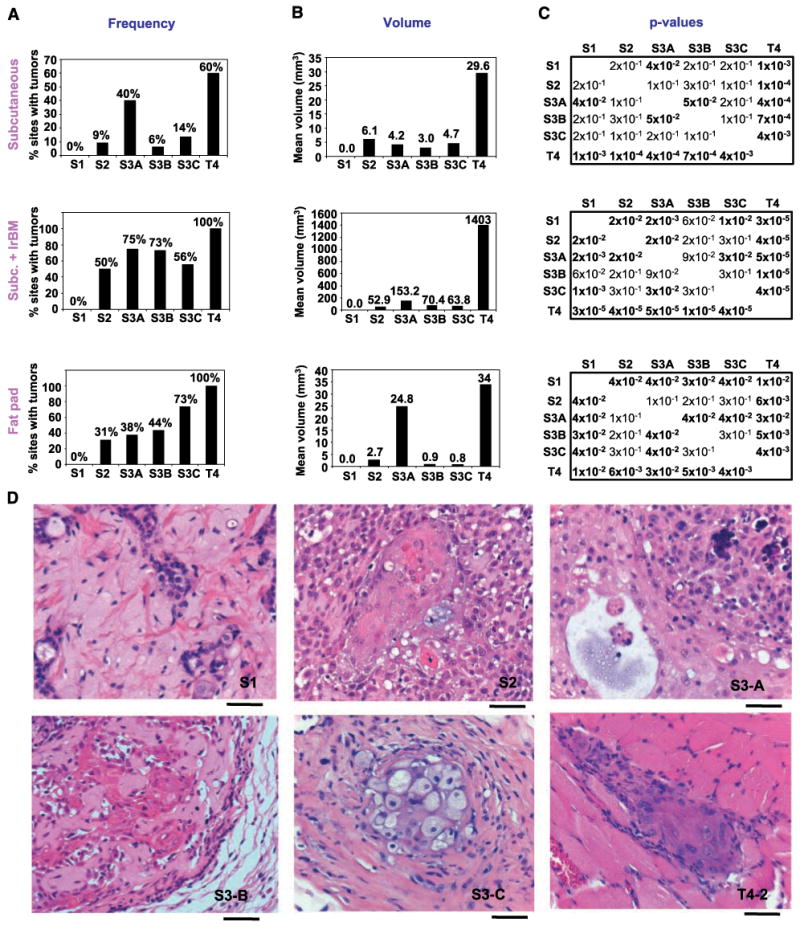
S3 cells display low tumorigenicity and squamous metaplastic phenotype in nude mice. A, percent injection sites that produced tumors sustained for the duration of the experiment. Top, s.c. injections (three experiments); 14 S1, 22 S2, 20 S3-A, 16 S3-B, 22 S3-C, 30 T4-2, and 3 media controls. Middle, s.c. + Matrigel (laminin-rich basement membrane, lrBM) injections (two experiments); 18 S1, 18 S2, 20 S3-A, 22 S3-B, 18 S3-C, and 18 T4-2 injections. Bottom, mammary fat pad injections; 15 S1, 16 S2, 16 S3-A, 16 S3-B, 15 S3-C, 24 T4-2, and 4 media controls. B, mean tumor volume (in mm3) excised from sacrificed animals. C, P values for the volume comparisons indicated; P < 0.05 in bold. Top, s.c. injections; middle, s.c. + Matrigel injections; bottom, mammary fat pad injections. D, H&E images of S1, S2, and S3-B s.c. + Matrigel tumors and of S3-A, S3-C, and T4-2 fat pad tumors. Bar, 50 μm. S1, low-grade adenosquamous carcinoma-like phenotype at the injection site; S2, squamous differentiation with whorls and bridges, transition from cuboidal cells to larger squamous cells in the center; S3-A, well-differentiated area with a small cyst, less well differentiated area with calcification; S3-B, cords of cells surrounding areas of extracellular matrix showing solid tumor areas with proliferation and squamous differentiation and displaying pleomorphic adenoma phenotype; S3-C, squamous differentiation, foamy squamous carcinoma cells, abundant stromal reaction; T4-2, squamous carcinoma invading the skeletal muscle.
Tumor histology reveals similarities to cancer progression within a metaplastic context
In s.c. injections (Supplementary File 1B), the T4-2 tumors had viable, dividing cells with squamous metaplastic morphology. The S2 and S3 tumors had nondividing cells, suggesting they did not maintain growth, but they contained keratin clusters and inflammatory cells, which are characteristics of well-differentiated squamous carcinomas of the breast. An average increase of 27-fold in tumor volume was observed when half the number of cells was injected in the presence of Matrigel. In the Matrigel injections (Fig. 2A–D, middle; Supplementary File 1A and C), 5 of 18 of the S1 injection sites showed a phenotype similar to low-grade adenosquamous carcinomas despite the lack of a palpable tumor at the time of sacrifice (Fig. 2J). Whereas 100% of the S2 and S3 tumors displayed the more benign mixed tumor histology, only 25% of the T4-2 tumors were mixed, with the remainder being pure squamous. In addition, T4-2 tumors had features of higher-grade squamous carcinoma than did the S2 and S3 tumors: 83% of the S2 and S3 tumors were well differentiated, compared with only 11% of the T4-2 tumors. Soft tissue involvement, indicating either invasion or proliferative expansion into neighboring tissues, was observed only in T4-2 tumors (Fig. 2J). Similarly, calcifications, which are an indication of necrosis usually observed in high-grade tumors, were found in almost all T4-2 tumors but only in a few of S2 or S3s. The fat pad tumor histology also showed similarities to squamous metaplasias of the breast (Fig. 2J; Supplementary File 1D), with T4-2 tumors being of higher “grade” in general based on the degree of differentiation, soft tissue involvement, and calcification phenotypes.
S3 cells display genomic aberrations that are distinct from those found in S1 and T4-2, but are relevant to some human breast cancers
We determined the genomically amplified or deleted regions by CGH (Supplementary File 2). The number of aberrations increased progressively when S1, S2-S3, and T4-2 cells were compared (Fig. 3A and B). Hierarchical clustering analysis (Fig. 3C) showed that the S2-S3 and T4-2 cell lines were more closely related to each other than they were to the S1 cells, and that the malignant T4-2 chromosomal aberration profiles were distinct from those of the S2-S3 cells. S2, S3, and T4-2 cell lines contained 76% (22 of 29) of the chromosomal gains or losses that are reported to be commonly found in primary breast tumors (Fig. 3D; Supplementary File 2). Of the aberrations recurrently found in carcinoma in situ (preinvasive), 6 of 10 were present in S3-A, S3-B, and S3-C cells, but all 10 were found in T4-2. The aberrations that were present in T4-2, but not in S3, were those found in high-grade in situ or invasive carcinomas.
Figure 3.

CGH profile of S3 cells compared with other HMT-3522 cells and with primary tumors (for details of CGH data, see Supplementary File 2). A, chromosomal amplifications (red) and deletions (green) for each cell type examined at the indicated passage number (e.g., S1_27: S1 cells at passage 27). B, chromosomal aberrations common to all cell lines shown on the left (e.g., “S1” means common to all S1 lineages shown in A). C, hierarchical clustering of 119 significantly amplified or deleted regions for the indicated cell lines. D, chromosomal aberrations commonly found in primary tumors, as described in each study cited; shown in bold if shared with HMT-3522 cell lines; complete list of aberrations shared between the cell lines and primary tumors listed at the bottom (TOTAL).
Transition from S3-C to T4-2 phenotype is associated with altered expression of gene classes previously implicated in human breast cancer progression
We determined differences in global gene expression between S3-C and T4-2 when cells were grown in either two-dimensional (monolayers) or 3DlrBM cultures (Supplementary File 3). To discover groups of genes with shared attributes in an unbiased manner, we analyzed these two sets of differentially expressed genes by model-based clustering according to their Gene Ontology (GO) terms and other annotations by developing and using a method called Attribute Profile Clustering (ref. 34; Supplementary File 8). The 141 genes altered between S3-C and T4-2 in two dimensions were divided into four groups summarized as integral membrane proteins, transcription factors, kinases, and transport molecules (Supplementary Files 4, 6, and 8). The groups for the 502 genes with different expression levels between S3-C and T4-2, when grown in three-dimensional cultures were integral membrane proteins, transcription factors, kinases, and chemokines (Supplementary Files 5, 7, and 8). The best-defined group in the comparison of S3-C and T4-2 in three dimensions, but not in two dimensions, revealed a class with 100% of the genes sharing the GO term “Chemokine Activity.” Chemokines have been shown previously to play a significant role in metastasis (35) and were shown to increase dramatically during human breast cancer progression (36).
MMP9, MMP13, MMP15, and MMP17 are functionally significant in the acquisition of invasiveness
In addition to the genes that were relevant to human breast cancer progression by attribute clustering analysis, the microarray data contained another class of genes, the MMPs, which showed differential expression when S3-C and T4-2 were compared only in 3DlrBM. Microarray analysis showed that MMP13 (collagenase-3), MMP15 (MT-2 MMP), and MMP17 (MT-4 MMP) had ∼30% higher expression in T4-2 versus S3-C in three-dimensional cultures (Supplementary File 3). For MMP13, we confirmed these results by quantitative RT-PCR (Fig. 4A) and Western blots of conditioned medium (not shown). For the membrane-bound MMP15 and MMP17, FACS analysis showed that cell-surface expression was higher in T4-2 than in S3-C cells (Fig. 4B and C). MMP15 has been shown to activate proMMP2 by cleavage (37). Therefore, we determined overall gelatinolytic activity, including MMP2 and MMP9 activity, present in S1, S2, S3, and T4-2 cell lines using zymograms. S1 cells expressed proMMP2 but not MMP9; S2, S3-A, and S3-B had no detectable expression of MMP2 or MMP9; S3-C had a small amount of proMMP9 expression; and T4-2 showed high levels of proMMP9 but no MMP2 expression (Fig. 4D).
Figure 4.
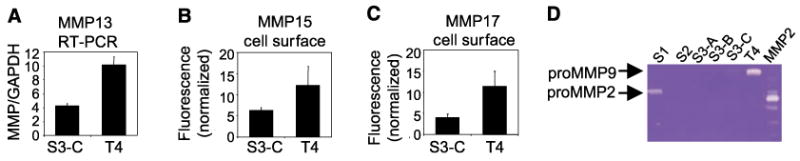
Candidate MMPs from microarrays are differentially expressed between S3-C and T4-2 cells. A, quantitative RT-PCR for MMP13, normalized to GAPDH internal control, for S3-C and T4-2 in 3DlrBM (P < 0.05). B and C, cell-surface expression of MMP15 and MMP17; four independent experiments (P < 0.05). D, zymogram detecting gelatinolytic activity in cell lines, compared with p-aminophenylmercuric acetate–activated recombinant MMP2 control.
In this model, we could directly assess the significance of MMP expression changes for the invasive phenotype. A broad-spectrum MMP inhibitor, GM6001 (38), abrogated T4-2 invasiveness whereas treatment of cells with C1004, an inactive analogue of GM6001, had no detectable effect (Fig. 5B). To confirm the inhibitor data specifically, we used siRNAs to transiently and individually knock down the levels of MMP9, MMP13, MMP15, and MMP17 (Fig. 5A) and found that siRNAs that successfully knocked down these MMPs decreased the amount of invasiveness of T4-2 cells compared with the scrambled control siRNA (Fig. 5C). MMPs were important for acquisition of invasiveness in S3-C cells as well: addition of GM6001, and not the control C1004, to either the S3-C or T4-2 media (the conditioned medium was taken from the latter) inhibited the ability of the T4-2 conditioned medium to induce invasiveness in S3-C (Fig. 5D).
Figure 5.
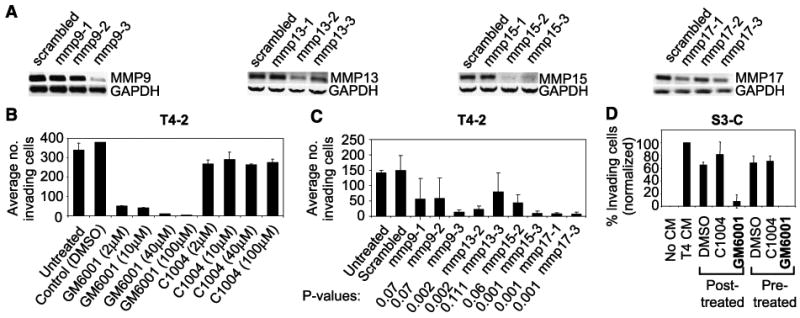
Candidate MMPs function in invasion through laminin-rich basement membrane. A, RT-PCR for MMP9, MMP13, MMP15, and MMP17 in cells transfected with siRNAs to the respective MMPs or a scrambled control. B, invasion assays for T4-2 cells treated with the indicated MMP inhibitor or control; two experiments, duplicate samples. C, invasion assays for T4-2 cells transfected with MMP9, MMP13, MMP15, or MMP17 siRNAs versus scrambled control; P values compared with scrambled control. D, invasion assays for S3-C cells; number of invading cells normalized to S3-C cells treated with T4-2 conditioned medium. Posttreated, conditioned medium was added to the S3-C medium after the indicated MMP inhibitor or control (40 μmol/L); Pretreated, conditioned medium was pretreated with the MMP inhibitor or control (40 μmol/L) for 48 h before being added to the S3-C medium in the invasion assays; three experiments.
Discussion
Modeling human breast cancer in culture traditionally has relied on carcinoma-derived cell lines or addition of potent oncogenic stimuli to nonmalignant cells. What we have lacked thus far are models with which we can recapitulate intermediate stages of cancer progression to simulate the complexity of different types of breast cancers (39). Here we describe one such human cell culture model. Neither the process of immortalization nor the process of selection of the preinvasive S3 or malignant T4-2 cells entailed introduction of oncogenic transgenes. The T4-2 population was generated by removal of EGF but provides a transgene-free opportunity to study the involvement of different and possibly unrelated pathways in malignant transition (Table 1; Supplementary File 8). The cell lines produced tumors with various metaplastic features, such as rare low-grade adenosquamous carcinoma, mixed benign tumors, and aggressive squamous metaplasia, suggesting progression within a metaplastic context (Supplementary File 1).
Table 1.
Attribute profile clustering of differentially expressed genes between S3-C and T4-2
| GenBank no. | Gene | Avg | P | GenBank no. | Gene | Avg | P | GenBank no. | Gene | Avg | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2D integral membrane (20/30) | 3D integral membrane (62/62) | ||||||||||
| NM_005050 | ABCD4 | 1.17 | 0.0295 | NM_000927 | ABCB1 | 0.82 | 0.0398 | AA450336 | NRXN3 | 0.74 | 0.0210 |
| NM_014567 | BCAR1 | 0.74 | 0.0222 | NM_012089 | ABCB10 | 0.61 | 0.0165 | AA598582 | OXA1L | 0.62 | 0.0054 |
| NM_004054 | C3AR1 | 1.20 | 0.0350 | NM_005689 | ABCB6 | 1.22 | 0.0029 | NM_002561 | P2RX5 | 0.74 | 0.0467 |
| NM_000722 | CACNA2D1 | 0.62 | 0.0450 | NM_019624 | ABCB9 | 1.33 | 0.0175 | NM_005446 | P2RXL1 | 0.68 | 0.0446 |
| NM_013230 | CD24 | 1.29 | 0.0093 | NM_000683 | ADRA2C | 1.24 | 0.0321 | NM_000919 | PAM | 1.24 | 0.0170 |
| NM_000760 | CSF3R | 1.17 | 0.0488 | NM_001671 | ASGR1 | 1.50 | 0.0384 | AA663440 | PLXNC1 | 0.83 | 0.0460 |
| NM_001381 | DOK1 | 0.80 | 0.0062 | NM_001681 | ATP2A2 | 1.43 | 0.0335 | NM_000952 | PTAFR | 1.22 | 0.0019 |
| NM_000118 | ENG | 0.64 | 0.0315 | NM_004335 | BST2 | 1.42 | 0.0106 | NM_002838 | PTPRC | 0.72 | 0.0020 |
| NM_014211 | GABRP | 0.68 | 0.0164 | NM_006137 | CD7 | 1.31 | 0.0421 | NM_002855 | PVRL1 | 1.09 | 0.0137 |
| NM_004482 | GALNT3 | 0.88 | 0.0219 | NM_001782 | CD72 | 1.24 | 0.0439 | NM_001037 | SCN1B | 0.69 | 0.0030 |
| H07878 | GPR19 | 1.23 | 0.0295 | NM_004356 | CD81 | 0.80 | 0.0182 | NM_005072 | SLC12A4 | 0.54 | 0.0230 |
| NM_000856 | GUCY1A3 | 1.16 | 0.0171 | NM_004233 | CD83 | 0.75 | 0.0092 | NM_015865 | SLC14A1 | 0.77 | 0.0417 |
| H11482 | IFNGR1 | 0.74 | 0.0029 | NM_005745 | DXS1357E | 0.84 | 0.0156 | NM_004207 | SLC16A3 | 0.79 | 0.0054 |
| NM_000612 | IGF2 | 1.13 | 0.0280 | NM_001400 | EDG1 | 1.69 | 0.0181 | NM_004694 | SLC16A6 | 1.23 | 0.0273 |
| NM_000598 | IGFBP3 | 0.62 | 0.0092 | NM_000604 | FGFR1 | 1.25 | 0.0311 | NM_005495 | SLC17A4 | 1.17 | 0.0471 |
| NM_002195 | INSL4 | 1.14 | 0.0360 | NM_006338 | GAC1 | 0.89 | 0.0454 | NM_007256 | SLC21A9 | 0.56 | 0.0133 |
| NM_012282 | KCNE1L | 1.15 | 0.0007 | NM_000160 | GCGR | 0.72 | 0.0317 | NM_000441 | SLC26A4 | 0.77 | 0.0389 |
| NM_002333 | LRP3 | 1.19 | 0.0085 | NM_000832 | GRIN1 | 1.50 | 0.0285 | NM_001860 | SLC31A2 | 0.83 | 0.0102 |
| AA496022 | MFAP4 | 0.60 | 0.0469 | NM_000840 | GRM3 | 0.60 | 0.0367 | NM_014437 | SLC39A1 | 1.31 | 0.0039 |
| AA598582 | OXA1L | 0.52 | 0.0009 | NM_005477 | HCN4 | 0.75 | 0.0172 | NM_002394 | SLC3A2 | 1.30 | 0.0077 |
| NM_002571 | PAEP | 1.15 | 0.0092 | AA464246 | HLA-C | 0.79 | 0.0248 | AA487543 | SORL1 | 0.83 | 0.0397 |
| NM_024411 | PDYN | 1.37 | 0.0492 | NM_033554 | HLA-DPA1 | 1.31 | 0.0498 | NM_000544 | TAP2 | 1.46 | 0.0217 |
| NM_004705 | PRKRIR | 0.80 | 0.0021 | H11482 | IFNGR1 | 0.59 | 0.0003 | NM_006335 | TIMM17A | 1.28 | 0.0042 |
| NM_000452 | SLC10A1 | 1.06 | 0.0296 | NM_000877 | IL1R1 | 0.80 | 0.0169 | NM_005834 | TIMM17B | 0.79 | 0.0361 |
| NM_006749 | SLC20A2 | 0.85 | 0.0041 | NM_004633 | IL1R2 | 0.63 | 0.0217 | NM_003265 | TLR3 | 0.71 | 0.0272 |
| NM_003127 | SPTAN1 | 1.17 | 0.0450 | NM_012276 | ILT7 | 1.28 | 0.0260 | NM_006827 | TMP21 | 1.17 | 0.0109 |
| NM_003235 | TG | 1.19 | 0.0297 | NM_002223 | ITPR2 | 0.75 | 0.0100 | NM_001192 | TNFRSF17 | 1.28 | 0.0190 |
| NM_003243 | TGFBR3 | 1.41 | 0.0218 | NM_002233 | KCNA4 | 0.73 | 0.0097 | NM_003810 | TNFSF10 | 1.15 | 0.0373 |
| NM_012471 | TRPC5 | 0.61 | 0.0346 | NM_002286 | LAG3 | 0.73 | 0.0064 | NM_006564 | TYMSTR | 0.45 | 0.0322 |
| NM_000371 | TTR | 0.87 | 0.0485 | NM_015364 | MD-2 | 0.68 | 0.0027 | NM_004738 | VAPB | 1.35 | 0.0212 |
| NM_000908 | NPR3 | 1.17 | 0.0156 | NM_021083 | XK | 1.23 | 0.0150 | ||||
| 2D transcription (23/26) | 3D transcription (51/51) | ||||||||||
| NM_004301 | BAF53A | 1.19 | 0.0280 | NM_005180 | BMI1 | 0.78 | 0.0355 | NM_006186 | NR4A2 | 1.18 | 0.0166 |
| NM_006317 | BASP1 | 0.92 | 0.0007 | NM_000092 | COL4A4 | 0.68 | 0.0387 | NM_003884 | PCAF | 0.62 | 0.0031 |
| NM_012116 | CBLC | 0.67 | 0.0249 | H54686 | DAB2 | 0.64 | 0.0163 | NM_021128 | POLR2L | 0.88 | 0.0464 |
| NM_003651 | CSDA | 0.86 | 0.0318 | NM_000399 | EGR2 | 1.37 | 0.0486 | NM_007244 | PROL4 | 1.43 | 0.0057 |
| NM_004111 | FEN1 | 0.82 | 0.0081 | NM_006874 | ELF2 | 0.74 | 0.0044 | NM_002892 | RBBP1 | 0.63 | 0.0012 |
| NM_001465 | FYB | 0.96 | 0.0002 | AA010400 | ETV4 | 1.36 | 0.0371 | NM_002919 | RFX3 | 1.22 | 0.0469 |
| NM_001515 | GTF2H2 | 0.86 | 0.0261 | NM_001987 | ETV6 | 1.28 | 0.0386 | NM_002938 | RNF4 | 1.22 | 0.0414 |
| NM_005324 | H3F3B | 1.10 | 0.0482 | AA181023 | EVI1 | 1.31 | 0.0024 | NM_002955 | RREB1 | 1.36 | 0.0482 |
| NM_005336 | HDLBP | 0.50 | 0.0200 | AI346582 | EVX1 | 0.69 | 0.0477 | NM_003071 | SMARCA3 | 1.30 | 0.0208 |
| NM_014213 | HOXD9 | 0.81 | 0.0059 | NM_003862 | FGF18 | 0.55 | 0.0374 | NM_007017 | SOX30 | 0.82 | 0.0371 |
| NM_016270 | KLF2 | 1.32 | 0.0361 | NM_000801 | FKBP1A | 0.87 | 0.0237 | NM_004509 | SP110 | 0.66 | 0.0173 |
| NM_002485 | NBS1 | 1.22 | 0.0159 | NM_005087 | FXR1 | 0.74 | 0.0038 | NM_012446 | SSBP2 | 1.35 | 0.0139 |
| NM_004555 | NFATC3 | 1.13 | 0.0302 | NM_002095 | GTF2E2 | 1.32 | 0.0018 | NM_005636 | SSX4 | 0.85 | 0.0450 |
| NM_002582 | PARN | 0.78 | 0.0034 | NM_003483 | HMGA2 | 1.05 | 0.4378 | NM_007375 | TARDBP | 1.36 | 0.0089 |
| NM_002779 | PSD | 0.59 | 0.0062 | NM_014213 | HOXD9 | 0.58 | 0.0021 | NM_007108 | TCEB2 | 1.19 | 0.0476 |
| NM_002938 | RNF4 | 1.17 | 0.0420 | NM_002200 | IRF5 | 1.22 | 0.0153 | NM_003201 | TFAM | 1.15 | 0.0263 |
| NM_007252 | RPF-1 | 0.60 | 0.0308 | NM_005853 | IRX5 | 0.59 | 0.0014 | NM_005077 | TLE1 | 1.18 | 0.0242 |
| NM_003079 | SMARCE1 | 1.14 | 0.0106 | NM_002228 | JUN | 1.30 | 0.0038 | NM_005120 | TNRC11 | 0.61 | 0.0064 |
| NM_003084 | SNAPC3 | 0.91 | 0.0081 | NM_014368 | LHX6 | 0.74 | 0.0210 | NM_001068 | TOP2B | 1.24 | 0.0498 |
| NM_005839 | SRRM1 | 1.05 | 0.0047 | NM_012321 | LSM4 | 0.78 | 0.0046 | NM_016936 | UBN1 | 1.27 | 0.0347 |
| NM_003152 | STAT5A | 1.14 | 0.0462 | NM_002397 | MEF2C | 0.84 | 0.0118 | AI925821 | USP22 | 0.76 | 0.0034 |
| NM_006284 | TAF10 | 1.17 | 0.0133 | NM_005933 | MLL | 0.58 | 0.0164 | NM_000378 | WT1 | 0.67 | 0.0150 |
| NM_005644 | TAF12 | 1.24 | 0.0035 | NM_004641 | MLLT10 | 1.24 | 0.0292 | NM_003443 | ZNF151 | 0.62 | 0.0241 |
| NM_005481 | TRAP95 | 0.59 | 0.0271 | NM_002479 | MYOG | 0.73 | 0.0299 | NM_016423 | ZNF219 | 1.27 | 0.0198 |
| NM_004236 | TRIP15 | 0.65 | 0.0394 | NM_016170 | NCX | 1.36 | 0.0116 | NM_005741 | ZNF263 | 1.49 | 0.0055 |
| NM_005082 | ZNF147 | 0.63 | 0.0243 | AA703115 | NFAT5 | 1.22 | 0.0183 | ||||
| 2D kinases (4/4) | 3D kinases (23/23) | ||||||||||
| NM_001105 | ACVR1 | 1.24 | 0.0206 | NM_000020 | ACVRL1 | 0.64 | 0.0383 | NM_014002 | IKKE | 0.81 | 0.0493 |
| NM_001259 | CDK6 | 0.79 | 0.0258 | NM_004327 | BCR | 1.26 | 0.0102 | NM_003010 | MAP2K4 | 1.23 | 0.0109 |
| NM_004783 | TAO1 | 1.24 | 0.0053 | NM_004333 | BRAF | 1.28 | 0.0071 | NM_005923 | MAP3K5 | 0.79 | 0.0114 |
| NM_003565 | ULK1 | 0.80 | 0.0293 | NM_016508 | CDKL3 | 1.17 | 0.0413 | NM_002378 | MATK | 0.73 | 0.0410 |
| NM_001274 | CHEK1 | 0.74 | 0.0073 | AA496964 | NEK1 | 0.76 | 0.0475 | ||||
| 2D intracellular transport (4/4) | NM_001278 | CHUK | 0.71 | 0.0341 | NM_005030 | PLK | 0.81 | 0.0046 | |||
| NM_001328 | CTBP1 | 1.34 | 0.0251 | NM_005406 | ROCK1 | 0.80 | 0.0333 | ||||
| NM_016451 | COPB | 0.67 | 0.0461 | NM_004734 | DCAMKL1 | 1.32 | 0.0095 | NM_003161 | RPS6KB1 | 1.36 | 0.0272 |
| NM_007357 | COG2 | 0.82 | 0.0162 | NM_001982 | ERBB3 | 1.21 | 0.0281 | NM_005627 | SGK | 0.77 | 0.0403 |
| NM_006323 | SEC24B | 0.91 | 0.0056 | NM_001465 | FYB | 0.63 | 0.0039 | NM_004783 | TAO1 | 1.31 | 0.0137 |
| NM_004766 | COPB2 | 1.31 | 0.0074 | NM_005734 | HIPK3 | 1.31 | 0.0080 | NM_003331 | TYK2 | 0.77 | 0.0382 |
| NM_000875 | IGF1R | 1.30 | 0.0233 | ||||||||
| 3D chemokines (4/4) | |||||||||||
| NM_006419 | SCYB13 | 1.38 | 0.0039 | NM_000609 | SDF1 | 1.37 | 0.0246 | ||||
| NM_001735 | C5 | 1.23 | 0.0203 | NM_000584 | IL8 | 1.45 | 0.0157 | ||||
NOTE: The titles (e.g., transport, kinase, etc.) describe the most common gene function in each class; the ratio of genes in the class that fit this functional definition is shown in parentheses. For each gene, the row of information contains the GenBank number (e.g., NM_005050), gene name (e.g., ABCD4), the mean value of the ratio of T4-2/S3-C expression over four microarray experiments (e.g., 0.15), and t-test penalized P value (P < 0.05).
Abbreviations: 2D, two-dimensional; 3D, three-dimensional; Avg, average.
Pure squamous carcinomas of the breast are rare, although mixed adenocarcinomas are diagnosed at a higher frequency (40). Some studies describe squamous metaplastic breast cancers as being extremely aggressive and they have been characterized to be at least as aggressive as grade 3 hormone receptor–negative adenocarcinomas (40–43). Five-year survival for patients with pure squamous carcinomas of the breast seems to be only marginally worse than what is reported for all breast cancers, although these comparisons suffer from lack of a sufficient number of squamous carcinomas with follow up data (41). Squamous metaplasias of the breast share similarities with the aggressive basal-like subtype of carcinomas in clinical behavior as well as in the expression of a panel of markers (40, 44). HMT-3522 cells resemble the basal-like subtype of breast cancers at the molecular level by gene expression profiling (45). In addition, the HMT-3522 xenograft tumors are p63 and CK5/6 positive as well as estrogen receptor, progesterone receptor, and HER2 negative (Supplementary File 1F and G), similar to squamous metaplastic carcinomas (46). In addition to the rare pure squamous carcinomas of the breast, 39% of breast cancers in premenopausal African Americans and the majority of tumors in BRCA1 mutation carriers are estrogen receptor and progesterone receptor negative and have basal characteristics (47, 48). Understanding whether such similarities in marker expression translate to overlap in the mechanism of transition to invasiveness would require developing models that recapitulate these subtypes, as described for at least one subtype developed here.
The HMT-3522 cells display xenograft tumor histology consistent with the breast cancer type represented by their molecular makeup. This is a rare situation. Most human breast cancer cell line xenografts display an undifferentiated phenotype (observations of A.D.B.). To our knowledge, in addition to the cell line model described here, the MCF10A series of Ha-Ras transformed cell lines compose the only other model that recapitulates the histologic characteristics of human carcinomas (ductal carcinoma in situ and infiltrating ductal carcinoma) as xenograft tumors (49). To capture a more complete picture of the malignant progression in vivo, more complex microenvironments need to be created in three-dimensional cultures, and a number of laboratories including ours are engaged in this endeavor.
Comparison of the phenotypes of S3 cells in culture to their ability to form tumors in three different microenvironments in vivo show that these cells are particularly sensitive to their host microenvironment, producing complex phenotypes in vivo. For example, the S3-A cell line throws off ∼10% large colonies in the 3DlrBM, but these do not invade through laminin-rich basement membrane in Boyden chambers, possibly because the duration of the assay is too short (2 days). However, S3-A tumor frequency is higher than the other S3s in the skin injections, but interestingly not in the skin + Matrigel or fat pad injections, and the S3-A tumor volumes in the skin + Matrigel and fat pad injections are higher than the other S3s (Fig. 2). The other S3 cell lines also seem to be very sensitive to their particular host microenvironment, resulting in the breakdown of a one-to-one correlation between the progressive phenotypes observed in culture for S3-A, S3-B, S3-C, and the more complex phenotypes found in the xenografts. It is conceivable that, given the length of the 9- to 10-week time period, the host microenvironment within which the injected cells are in dynamic and reciprocal interactions with the stromal and humoral factors could allow for selection of the aggressive subpopulations, resulting in gain or loss of necessary functions to become malignant. In addition, results confirm that the cells poised to become malignant have not yet inactivated all pathways or gained all the new functions needed for a stable malignant phenotype.
What was found to be consistent across all assays done (invasion, three-dimensional polarity, CGH profile, tumorigenicity) is that all of the S3 cells displayed a phenotype distinct from both S1 and T4-2 cells in vivo as well as in the 3DlrBM assay. For tumorigenicity, this was supported by statistical analysis of tumor volume, as well as by histologic analysis showing that S3 tumors manifested more benign characteristics than the T4-2 tumors. In addition, S3 cells displayed more homogeneous phenotypes and a higher propensity to become invasive than the parent S2 cells when induced with T4-2 conditioned medium. Therefore, by these criteria, S3 cells have indeed progressed further than their parent S2 cells toward malignancy and are intermediate between the nontumorigenic S1 and malignant T4-2s.
Here we have described this model as one of transition from preinvasive to invasive phenotype to mimic the multistep carcinogenesis hypothesis of breast cancer (50, 51). However, the emerging cancer stem/progenitor cell hypothesis also needs to be considered. Sontag and Axelrod (1) have proposed four separate models to describe how atypical hyperplasia, ductal carcinoma in situ, invasive ductal carcinoma, and metastasis may relate to each other. These are the linear, nonlinear, branched, and parallel pathways. Unlike the other three hypotheses, the parallel pathway does not assume that in situ carcinoma develops into invasive carcinoma, but rather that in situ and invasive carcinomas can arise from the same progenitor. In our cell lines, S2 gave rise to both the preinvasive S3-C and the invasive T4-2 cells via different steps of manipulation in culture and in mice, respectively (Fig. 1A). Therefore, it is tempting to speculate that the model described here could be placed as supporting the parallel progression hypothesis. Regardless of which mathematical model we consider, what we have developed are human breast epithelial cells with preinvasive and invasive characteristics, which recapitulate some aspects of preinvasive and invasive carcinomas with basal and squamous histologic and molecular phenotypes.
Analysis of this malignant transition in three-dimensional cultures allowed us to identify candidate genes as potential therapeutic targets. For example, of the MMPs we identified as having a function in invasion, MMP9, MMP13, and, recently, MMP17 have been shown to be associated with, and increased in, breast cancer progression (52–54). Although broad-spectrum inhibitors of MMPs have failed in clinical trials, targeting specific genes for certain subtypes of cancers may eliminate nonspecific effects, thereby producing more desirable clinical outcomes (55). In addition to the list of genes differentially expressed between S3-C and T4-2 (Table 1; Supplementary File 8), this model system is conducive to functional siRNA screens probing either gain of invasion function in S3-C or loss of invasion of T4-2 to find new genes involved in invasion. Using the same model, we recently discovered a new pathway of invasion regulated by polo-like kinase I (23). In general, the results described here provide a proof-of-principle for the development of spontaneous transition models for additional subtypes of breast cancers, with potential utility in discovering other mechanisms of invasion and other potential targets for therapy.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: U.S. Department of Energy, Office of Biological and Environmental Research grant DEAC0376SF00098 (M.J. Bissell); NIH grant CA64786 (M.J. Bissell and O.W. Petersen); Department of Defense (DOD) Breast Cancer Research Program (BCRP) Innovator Award DAMD170210438 (M.J. Bissell); California Breast Cancer Research Program (CBCRP) Postdoctoral Fellowship Award 8FB0184 (A. Rizki); DOD BCRP Innovative, Developmental, and Exploratory Award DAMD170110368, NIH grant CA78731, and American Cancer Society New Investigator Internal Award IRG7800223 (V.M. Weaver); DOD BCRP Predoctoral Fellowship Award DAMD170310421 (G.I. Rozenberg); NIH grant CA88858 (J.D. Mott); NIH grants CA80067 and CA098131 (R.A. Jensen); CBCRP grant 8PB0171 and NIH grants P01 AG017242 and U01 ES011044 (I.S. Mian); DOD BCRP Predoctoral Fellowship Award BC021231 (J.L. Bascom); and NIH grant CA58207 (K. Chin and J.W. Gray). NIH grant CA50519 and DOD BCRP grant DAMD170210439 (D.J. Chen). M.J. Bissell is a recipient of a distinguished fellowship from the U.S. Department of Energy, Office of Biological and Environmental Research.
We thank Eva Lee, Hui Zhang, Genee Lee, and Shraddha Ravani (Lawrence Berkeley National Laboratory) and Travis Mullan (Virginia Commonwealth University) for technical assistance and/or advice.
References
- 1.Sontag L, Axelrod DE. Evaluation of pathways for progression of heterogeneous breast tumors. J Theor Biol. 2005;232:179–89. doi: 10.1016/j.jtbi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Buerger H, Otterbach F, Simon R, et al. Comparative genomic hybridization of ductal carcinoma in situ of the breast-evidence of multiple genetic pathways. J Pathol. 1999;187:396–402. doi: 10.1002/(SICI)1096-9896(199903)187:4<396::AID-PATH286>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 3.Etzell JE, Devries S, Chew K, et al. Loss of chromosome 16q in lobular carcinoma in situ. Hum Pathol. 2001;32:292–6. doi: 10.1053/hupa.2001.22759. [DOI] [PubMed] [Google Scholar]
- 4.Gong G, DeVries S, Chew KL, Cha I, Ljung BM, Waldman FM. Genetic changes in paired atypical and usual ductal hyperplasia of the breast by comparative genomic hybridization. Clin Cancer Res. 2001;7:2410–4. [PubMed] [Google Scholar]
- 5.Isola J, Chu L, DeVries S, et al. Genetic alterations in ERBB2-amplified breast carcinomas. Clin Cancer Res. 1999;5:4140–5. [PubMed] [Google Scholar]
- 6.Isola JJ, Kallioniemi OP, Chu LW, et al. Genetic aberrations detected by comparative genomic hybridization predict outcome in node-negative breast cancer. Am J Pathol. 1995;147:905–11. [PMC free article] [PubMed] [Google Scholar]
- 7.Loveday RL, Greenman J, Simcox DL, et al. Genetic changes in breast cancer detected by comparative genomic hybridisation. Int J Cancer. 2000;86:494–500. doi: 10.1002/(sici)1097-0215(20000515)86:4<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 8.Ma XJ, Salunga R, Tuggle JT, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:5974–9. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishizaki T, DeVries S, Chew K, et al. Genetic alterations in primary breast cancers and their metastases: direct comparison using modified comparative genomic hybridization. Genes Chromosomes Cancer. 1997;19:267–72. doi: 10.1002/(sici)1098-2264(199708)19:4<267::aid-gcc9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 10.Nishizaki T, Chew K, Chu L, et al. Genetic alterations in lobular breast cancer by comparative genomic hybridization. Int J Cancer. 1997;74:513–7. doi: 10.1002/(sici)1097-0215(19971021)74:5<513::aid-ijc6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Polyak K. Molecular alterations in ductal carcinoma in situ of the breast. Curr Opin Oncol. 2002;14:92–6. doi: 10.1097/00001622-200201000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Porter DA, Krop IE, Nasser S, et al. A SAGE (serial analysis of gene expression) view of breast tumor progression. Cancer Res. 2001;61:5697–702. [PubMed] [Google Scholar]
- 13.Sgroi DC, Teng S, Robinson G, LeVangie R, Hudson JR, Jr, Elkahloun AG. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;59:5656–61. [PubMed] [Google Scholar]
- 14.Thor AD, Eng C, Devries S, et al. Invasive micro-papillary carcinoma of the breast is associated with chromosome 8 abnormalities detected by comparative genomic hybridization. Hum Pathol. 2002;33:628–31. doi: 10.1053/hupa.2002.124034. [DOI] [PubMed] [Google Scholar]
- 15.Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi OP. Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer. 1998;21:177–84. [PubMed] [Google Scholar]
- 16.Waldman FM, DeVries S, Chew KL, Moore DH, II, Kerlikowske K, Ljung BM. Chromosomal alterations in ductal carcinomas in situ and their in situ recurrences. J Natl Cancer Inst. 2000;92:313–20. doi: 10.1093/jnci/92.4.313. [DOI] [PubMed] [Google Scholar]
- 17.Waldman FM, Hwang ES, Etzell J, et al. Genomic alterations in tubular breast carcinomas. Hum Pathol. 2001;32:222–6. doi: 10.1053/hupa.2001.21564. [DOI] [PubMed] [Google Scholar]
- 18.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23:181–8. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 20.Briand P, Nielsen KV, Madsen MW, Petersen OW. Trisomy 7p and malignant transformation of human breast epithelial cells following epidermal growth factor withdrawal. Cancer Res. 1996;56:2039–44. [PubMed] [Google Scholar]
- 21.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A. 1992;89:9064–8. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weaver VM, Petersen OW, Wang F, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizki A, Mott JD, Bissell MJ. Polo-like kinase I is involved in invasion through extracellular matrix. Cancer Res. 2007;67:11106–10. doi: 10.1158/0008-5472.CAN-07-2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinkel D, Segraves R, Sudar D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–11. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 25.Herron GS, Werb Z, Dwyer K, Banda MJ. Secretion of metalloproteinases by stimulated capillary endothelial cells. I. Production of procollagenase and prostromelysin exceeds expression of proteolytic activity. J Biol Chem. 1986;261:2810–3. [PubMed] [Google Scholar]
- 26.Lochter A, Srebrow A, Sympson CJ, Terracio N, Werb Z, Bissell MJ. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J Biol Chem. 1997;272:5007–15. doi: 10.1074/jbc.272.8.5007. [DOI] [PubMed] [Google Scholar]
- 27.Taylor MA, Cote RJ. Immunomicroscopy: a diagnostic tool for the surgical pathologist. Philadelphia: W.B. Saunders; 1994. pp. 300–78. [Google Scholar]
- 28.Liu H, Radisky DC, Wang F, Bissell MJ. Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J Cell Biol. 2004;164:603–12. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weaver VM, Lelievre S, Lakins JN, et al. β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–16. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lelievre SA, Weaver VM, Nickerson JA, et al. Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc Natl Acad Sci U S A. 1998;95:14711–6. doi: 10.1073/pnas.95.25.14711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muschler J, Levy D, Boudreau R, Henry M, Campbell K, Bissell MJ. A role for dystroglycan in epithelial polarization: loss of function in breast tumor cells. Cancer Res. 2002;62:7102–9. [PubMed] [Google Scholar]
- 33.Wang F, Weaver VM, Petersen OW, et al. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci U S A. 1998;95:14821–6. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semeiks JR, Rizki A, Bissell MJ, Mian IS. Ensemble attribute profile clustering: discovering and characterizing groups of genes with similar patterns of biological features. BMC Bioinformatics. 2006;7:147. doi: 10.1186/1471-2105-7-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 36.Allinen M, Beroukhim R, Cai L, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 37.d'Ortho MP, Will H, Atkinson S, et al. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur J Biochem. 1997;250:751–7. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- 38.Grobelny D, Poncz L, Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31:7152–4. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 39.Leaf C. Why we're losing the war on cancer (and how to win it) Fortune. 2004;149:76–82. 4–6, 8. passim. [PubMed] [Google Scholar]
- 40.Hennessy BT, Krishnamurthy S, Giordano S, et al. Squamous cell carcinoma of the breast. J Clin Oncol. 2005;23:7827–35. doi: 10.1200/JCO.2004.00.9589. [DOI] [PubMed] [Google Scholar]
- 41.Aparicio I, Martinez A, Hernandez G, Hardisson D, De Santiago J. Squamous cell carcinoma of the breast. Eur J Obstet Gynecol Reprod Biol. 2007 doi: 10.1016/j.ejogrb.2007.03.021. online ahead of print. dx.doi.org/10.1016/j.ejogrb.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 42.Tse GM, Tan PH, Putti TC, Lui PC, Chaiwun B, Law BK. Metaplastic carcinoma of the breast: a clinicopathological review. J Clin Pathol. 2006;59:1079–83. doi: 10.1136/jcp.2005.030536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Behranwala KA, Nasiri N, Abdullah N, Trott PA, Gui GP. Squamous cell carcinoma of the breast: clinico-pathologic implications and outcome. Eur J Surg Oncol. 2003;29:386–9. doi: 10.1053/ejso.2002.1422. [DOI] [PubMed] [Google Scholar]
- 44.Reis-Filho JS, Milanezi F, Steele D, et al. Metaplastic breast carcinomas are basal-like tumours. Histopathology. 2006;49:10–21. doi: 10.1111/j.1365-2559.2006.02467.x. [DOI] [PubMed] [Google Scholar]
- 45.Kenny PA, Lee GY, Myers CA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tse GM, Tan PH, Chaiwun B, et al. p63 is useful in the diagnosis of mammary metaplastic carcinomas. Pathology. 2006;38:16–20. doi: 10.1080/00313020500444625. [DOI] [PubMed] [Google Scholar]
- 47.Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 48.Honrado E, Benitez J, Palacios J. Histopathology of BRCA1- and BRCA2-associated breast cancer. Crit Rev Oncol Hematol. 2006;59:27–39. doi: 10.1016/j.critrevonc.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 49.Santner SJ, Dawson PJ, Tait L, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–10. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- 50.Allred DC, Mohsin SK. Biological features of premalignant disease in the human breast. J Mammary Gland Biol Neoplasia. 2000;5:351–64. doi: 10.1023/a:1009573710675. [DOI] [PubMed] [Google Scholar]
- 51.Burstein HJ, Polyak K, Wong JS, Lester SC, Kaelin CM. Ductal carcinoma in situ of the breast. N Engl J Med. 2004;350:1430–41. doi: 10.1056/NEJMra031301. [DOI] [PubMed] [Google Scholar]
- 52.Nielsen BS, Rank F, Lopez JM, et al. Collagenase-3 expression in breast myofibroblasts as a molecular marker of transition of ductal carcinoma in situ lesions to invasive ductal carcinomas. Cancer Res. 2001;61:7091–100. [PubMed] [Google Scholar]
- 53.Rha SY, Yang WI, Kim JH, et al. Different expression patterns of MMP-2 and MMP-9 in breast cancer. Oncol Rep. 1998;5:875–9. doi: 10.3892/or.5.4.875. [DOI] [PubMed] [Google Scholar]
- 54.Chabottaux V, Sounni NE, Pennington CJ, et al. Membrane-type 4 matrix metalloproteinase promotes breast cancer growth and metastases. Cancer Res. 2006;66:5165–72. doi: 10.1158/0008-5472.CAN-05-3012. [DOI] [PubMed] [Google Scholar]
- 55.Fingleton B. Matrix metalloproteinases: roles in cancer and metastasis. Front Biosci. 2006;11:479–91. doi: 10.2741/1811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).