

Abstract
Many available methods aimed at incorporating the receptor flexibility in ligand docking are computationally expensive, require a high level of user intervention, and were tested only on benchmarks of limited size and diversity. Here we describe the Four-dimensional (4D) docking approach that allows seamless incorporation of receptor conformational ensembles in a single docking simulation and reduces the sampling time while preserving the accuracy of traditional ensemble docking. The approach was tested on a benchmark of 99 therapeutically relevant proteins and 300 diverse ligands (half of them experimental or marketed drugs). The conformational variability of the binding pockets was represented by the available crystallographic data, with the total of 1113 receptor structures. The 4D docking method reproduced the correct ligand binding geometry in 77.3% of the benchmark cases, matching the success rate of the traditional approach, but employed on average only one fourth of the time during the ligand sampling phase.
Keywords: 4D Docking, Cross-docking, Ensemble Docking, ICM, Internal Coordinate Mechanics, Induced Fit, Receptor Flexibility, Drug Binding, Structure Based Drug Design, Clean Benchmark
Introduction
Introducing receptor flexibility in a standard ligand docking protocol is the only way to account for conformational changes induced by ligand binding.1 While almost all the docking protocols employed today achieve a satisfactory performance in self-docking experiments, calculations based on a single rigid receptor fail to reproduce the native complex geometry more than 50% of the cases in cross-docking runs.2-4 Even very small changes in the conformation of the binding pocket can dramatically affect the final outcome. Numerous attempts have been made over the past several years to develop a method that can accurately simulate receptor flexibility, with varying degrees of success (reviewed by Teodoro and Kavraki5 and more recently by Totrov and Abagyan6). The most practical approach to receptor flexibility in docking is so-called ensemble docking: in a set of independent simulations, the docking procedure is systematically applied to a collection of receptor conformations. The collected results may be assembled together, further refined, and possibly rescored.
The conformational ensemble can consist of experimental structures, computationally generated models, or both.7 In principle, the computer assisted generation of receptor variants can produce unprecedented rearrangements of the binding pocket and, therefore, enhance the possibility of discovering truly novel ligands. On the other hand, when only high quality experimental structures are considered, the range of possibilities can be comparatively narrow. However, the receptor conformations do not need any further validation, and tend to represent the specific regions of the conformational space that best suit a binding event. Moreover, a collection of experimental structures allows a clear-cut representation of the protein backbone and loop transitions, movements quite difficult to represent efficiently in computationally generated conformations.8, 9
Several examples of Multiple Receptor Conformers (MRC) docking protocols tested on ensembles of X-ray structures have been reported. In 1997, Knegtel and coworkers10 employed a modified version of DOCK 3.511 energy grids to account for the conformational variability of the binding site. Two different protocols, based on geometrically and energetically averaged grid complements, were tested on four ensembles of five crystal structures each. The idea of average grids was further developed by introducing weights in the calculation, in order to reduce the bias associated to strongly disallowed or highly favorable regions in the single members of the ensemble.4 The weighted average method was applied to a customized version of the AutoDock12 docking scheme and tested on twenty-one HIV-1 protease crystal structures. Another interesting attempt to simultaneously consider multiple targets in a single run is the In Situ cross-docking approach: grids representing different binding sites are joined together, one next to the other, in a single three-dimensional object. This procedure was separately tested on six different protein ligand co-crystals in a sort of virtual receptor screening setup,13 on different conformations of the same protein in order to explore the protein flexibility, and on mixed ensembles of similar proteases in ligand specificity studies.14 FlexE15 implements the receptor flexibility in the FlexX16 docking paradigm through a united protein description; namely, different protein structures are superimposed and the regions that display structural variations are combinatorially merged to generate new conformations which, in turn, are later employed along the original ones. Huang and Zou17 proposed a MRC protocol based on DOCK 4.0.18 The method's main feature consists of an application of the Simplex local minimization to pinpoint the structure in the ensemble best suitable for accommodating the ligand; in the optimization procedure, the protein conformational state is added to the six roto-translational ligand degrees of freedom as an additional dimension. A milestone study on the advantages and risks of employing multiple receptor conformations was reported by Barril and Morley.19 In their elegant analysis, they investigated the full ensemble docking potential using two large sets of X-ray structures available for Cyclin dependent Kinase 2 and for Heat Shock Protein 90. The calculations were independently carried out on each receptor conformation by means of an in-house purpose-written code. Recently, the FITTED genetic algorithm has been proposed.20 The genetic operators can implement the receptor flexibility in the procedure by (i) simply switching among the different protein conformations from the set (semi flexible run), or (ii) rearranging side chains and backbone variables independently. Another feature of the FITTED algorithm is the ability to predict efficiently the number and the position of potential water molecules that bridge the ligand to the receptor. Applications of the MRC protocol to screening have also been reported.21, 22
All the above MRC strategies were tested on sets of crystal structures of comparatively small size, representative of a limited number of proteins, of a limited number of conformations for each protein, or both. Moreover, the issue of the increasing calculation time when multiple variants of the binding pocket are considered was not always satisfactorily discussed.
In the present study, we report a novel ensemble docking strategy called Four-dimensional Docking (4D Docking). Eisenstein, commenting on the work of Grünberg and colleagues,23 was the first to adopt the term fourth dimension to describe the plasticity of protein structures in flexible protein protein docking protocols.24 Correspondingly, our ligand docking method is based on the idea that receptor flexibility can be represented as the fourth discrete dimension of the small molecule conformational space, with multiple recomputed 3D grids from optimally superimposed conformers merged into a single 4D object. In order to benchmark the overall method performance, a diverse, clean, yet challenging test set of known ligand-receptor complexes was derived. The 4D method's results in terms of speed and accuracy are reported and systematically compared to those of a standard MRC docking protocol.
Materials and Methods
Dataset of Protein-Ligand Complex Structures and Ensembles
Protein sequences with publicly available 3D information25 were retrieved from SwissProt26, 27 (release February of 2008). The sequences were searched against a non redundant subset of PDB sequences with common protein expression tags (e.g. HHHHH) removed. 3D domains were annotated based on PDB sequence boundaries, and their structures were clustered to 95% sequence identity. Each protein domain with more than one X-ray structure was considered a potential ensemble docking test case.
Next, a comprehensive collection of ∼3000 non trivial drug-like molecules from the PDB was built by (i) excluding ubiquitous substrates and (ii) applying relaxed Lipinski rules filter to the entire PDB Chemical Component Dictionary. That collection was merged with the above protein domain ensemble set to obtain multiple conformation ensembles for 864 proteins co-crystallized with at least one relevant compound.
All protein structures in the ensemble were superimposed using only the backbone atoms in the immediate vicinity of the ligands. The superimposition algorithm28 adopted here was based on an iterative procedure that, through an unbiased weight assignment to different atomic subsets, gradually found the better alignable core between the template and the other structures. The procedure starts from two equivalent atom arrays and proceeds precedes as follow: (i) the atomic equivalences are established and the weights Wi for each atom pair i are all set to 0, (ii) the weighted superimposition is performed and the RMSD is evaluated, (iii) the deviations Di are calculated for each atom pair i and sorted, (iv) the 50-percentile D50 is selected, and v) new weights are calculated according to the formula
While well superimposed atoms small deviations will are be assigned weights close to 1, the weights associated towith strongly deviating atom pairs larger deviations will get progressively smaller. Steps from (ii) through (v) are iterated until the RMSD value stops improving or the maximum number of iterations (set equal to 10 in this case) is reached. In this way, the presence of a minority of deviating atoms between otherwise similar structures cannot compromise the overall quality of the superimposition. The obtained superimposed complexes were automatically annotated in terms of the receptor binding site composition: homo- and hetero-multimeric receptors, catalytic metal ions, cofactors and their analogs were automatically identified based on the consistency of each of these features throughout the ensemble. Compositional and conformational differences between the individual ensemble structures were recorded. The ligands were analyzed for correctness of their covalent geometry and checked against the electron density data from the Uppsala Electron Density Server.29 In order to evaluate the ligand fit into its real space crystallographic density, an in-house algorithm was developed. For ideally fitting ligands, the procedure returns a density fit value of 1. Molecules that are ambiguously or incorrectly placed in the density, have high temperature factors, or contain unrealistic atom positions are characterized by values ranging from −1 to 0.7 (Kufareva et al., manuscript in preparation).
The benchmark was further filtered to fairly test the 4D docking accuracy. A conformational ensemble for one protein had to (i) represent at least three different crystal structures, and (ii) include at least one co-crystallized ligand structure. Receptors and ligands from covalently bound co-crystals and duplicated copies of a co-crystal structure were eliminated (if bound to structures belonging to different ensembles, multiple instances of the same ligand were allowed). Structures where any druggable binding site30 could not be automatically identified (indicative of the ligand binding at a crystallographic interface) were excluded as well. In the binding region, members of the same ensemble had to display exactly the same composition, both in terms of amino acids and cofactors. If more than one compositional variant satisfying the minimum requirements could be identified, the original ensemble was split and the resulting groups assigned consecutive numbers. In order to be included in the set, ligand structures had to consist of a single fragment small organic molecule with: (i) more than 20 non-hydrogen atoms, (ii) less than 12 rotatable bonds, (iii) no ring with 9 or more members, (iv) a druglikeness28 ≥ -0.3, and (v) a density fit value ≥ 0.8. Lastly, ligands that could not be accurately re-docked into their own cognate receptor binding site were excluded since our previous studies established that the majority of those failures are indicative of crystallographic, protonation or tautomerization errors in either ligand or receptor. The sequence of filtering criteria applied to select the validation set is summarized in Table 1. Note that the number of ensembles is slightly larger than the number of proteins because some proteins need to be represented by two or three different ensembles.
Table 1.
Sequential filtering criteria applied in the selection of the 4D validation set.
| Selection Step | Selection Criteria and Filters | Proteins | Ensembles | Receptors Structures | Ligands |
|---|---|---|---|---|---|
| Receptor Conformations Availability | 3 or more X-ray high quality structures of the same protein, at least one of them co-crystallized with a ligand, are retrieved. | 387 | 387 | 5779 | 4304 |
| Set Cleaning up | Redundant structures and non drug-like ligands are filtered out. | 211 | 211 | 2152 | 623 |
| Compositional Identity | Only structures with exactly the same amino acidic composition of the binding pocket can be part of the same ensemble. Several proteins provide more than one ensemble. | 126 | 134 | 1328 | 453 |
| Self Docking | Complexes whose bound conformation cannot be reproduced in a self docking simulation are excluded. | 99 | 107 | 1113 | 300 |
Preparation of Receptor Structures
According to the rule of intra-ensemble compositional identity, chains, heteroatoms, and prosthetic groups not involved in the binding site definition as well as water molecules were deleted. The inclusion in the binding site definition of crystallographic water molecules that through specific interactions bridge the receptor and the ligand was recently reported to improve the quality of the docking predictions for some specific complexes.20, 31 However, the introduction of explicit water molecules would have compromised in many cases the compositional identity of the binding site on which the 4D docking approach presently depends on. The assumption made here is that the role of water molecules in the binding site can be approximated after rescoring by cavities of a high distance-dependent dielectric constant.32 Afterwards, the correct atom types were assigned, and hydrogen atoms and missing heavy atoms were added. Zero occupancy side chains and polar hydrogen atoms were optimized and assigned the lowest energy conformation. Tautomeric states of histidines and the positions of asparagine and glutamine side chain amidic groups were optimized to improve the hydrogen bonding patterns. The cognate ligands were deleted from the complexes only after hydrogen optimization.
Preparation of Ligand Structures
Ligand atomic coordinates were extracted from the native structures. Bond orders, tautomeric forms, stereochemistry, hydrogen atoms and protonation states were assigned automatically by the ICM25 converting procedure. Each ligand was assigned the MMFF26 force field atom types and charges, and was then subjected to Cartesian minimization.
Binding Pocket Definition
In the self docking step associated to the filtering process, the boundaries of the binding box were assumed to be known and directly derived from the ligand bound position. Based on the ligand position, the Cartesian axes system was re-oriented to allow an optimal box fit. A mesh representing the ligand molecular surface33 at the binding site was generated. All the residues with at least one side chain non-hydrogen atom in the range of 3.5 Å from the molecular surface were considered parts of the binding pocket.
In the cross-docking experiments, the binding pocket definition was based on the largest envelope predicted in each receptor by the Pocketome Gaussian Convolution algorithm.30 The tolerance value was set equal to 5.0. The binding pocket was generated selecting all the residues with at least one side chain non-hydrogen atom in the range of 3.5 Å from the mesh.
In the ensemble docking experiments, the largest envelopes predicted in each structure of the group were merged together in a single mesh so that all the structures in the ensemble shared a common definition of the binding pocket.
Single Receptor Conformer Docking Procedure
The single rigid receptor conformer docking was employed during the validation set selection in the self docking filtering step, in the single rigid conformer cross-docking exercise, and as a part of the MRC docking approach. In both cases, it was carried out by means of the Biased Probability Monte Carlo (BPMC) stochastic optimizer as implemented in ICM.34-36 The ligand binding site at the receptor was represented by pre-calculated 0.5 Å spacing potential grid maps, representing van der Waals potentials for hydrogens and heavy probes, electrostatics, hydrophobicity, and hydrogen bonding. The van der Waals interactions were described by the 6-12 Lennard-Jones potential. However, the 6-12 standard implementation is extremely sensitive to even small deviations in atomic coordinates and can generate a large amount of noise in the intermolecular energy calculations. A smoother form of the potential with the repulsive contribution capped at a cutoff value Emax was here adopted.
Because of the soft van der Waals potential, the electrostatic contribution is buffered artificially increasing the distance between two charged atoms. The buffering prevents two oppositely charged atoms to collapse when the electrostatic attractive energy prevails on the softened van der Waals repulsion. Emax was set equal to 4 Kcal/mol and 1 Kcal/mol in re-docking and in MRC docking, respectively.
The molecular conformation was described by means of internal coordinate variables. The adopted force field was a modified version of the ECEPP/3 force field37 with a distance-dependent dielectric constant. Given the number of rotatable bonds in the ligand, the basic number of BPMC steps to be carried out was calculated by an adaptive algorithm (thoroughness 1.0). Before sampling, the ligand torsional variables were randomized. The binding energy Ebind was assessed by means of the standard ICM empirical scoring function36, 38, 39
Multiple Receptor Conformers Docking Procedure
In MRC docking, a single rigid receptor docking run was independently carried out for each receptor conformation in the stack. The solutions retrieved from each run were assembled together and geometrically clustered to eliminate redundant poses. The distance between the poses was calculated as the static RMSD among the ligand non-hydrogen atoms (chemical equivalences were taken into account). The vicinity parameter was set equal to 1Å. After compression, the poses within a range of 1 kcal/mol from the lowest energy conformation achieved in Monte Carlo underwent the all atom rescoring procedure.
4D Docking Procedure
In the 4D docking approach, all the structures in the ensemble were simultaneously considered in a single docking run as a result of 4D grids complement. In order to generate the 4D grids, the structures in the ensemble had to be converted into a conformational stack; in other words, separate entities had to be expressed as conformational variants of the same object. The structure that displayed the most complete sequence according to the UniProt26 database along with the best resolution was selected as the template and all the structures in the ensemble were superimposed to it. The superimposition was performed by means of the iterative weighted process described in the Dataset of Protein-Ligand Complex Structures and Ensembles.28 Since in the selection procedure the compositional identity was only established in the binding region, the residual non conserved residues outside the binding region were deleted. The template was then employed to build the conformational stack by means of a tethering function. Regular 3D grids representing the receptor interaction potentials were generated sequentially for all receptor conformations. The integer index of a receptor conformation is added as a discrete variable to the global optimization. During Monte Carlo sampling in the docking protocol, the conformer index changed alongside the regular conformational changes of the ligand, Cartesian translations and rotations. In the current implementation, the entire ligand would move from one 4D ‘plane’ to another, thus switching between different receptor conformations. The sampling length was scaled according to number of conformations considered (thoroughness equal to 0.5 multiplied by the number of conformations; the value was capped at 5.0). All poses within a range of 1 kcal/mol from the lowest energy conformation achieved in Monte Carlo sampling underwent the all atom scoring procedure.
Software and Hardware
The receptor and ligand preparations, the docking simulations, and the energy evaluations were carried out with ICM 3.5 (Molsoft L.L.C., La Jolla, CA).
The hardware facilities employed in the present study were an Intel CoreTM 2 Duo 2.40 GHz CPUs and 2 GBytes of memory workstation and a 3020 64-bit Intel XEON-EMT CPUs Linux Compute Cluster at The Scripps Research Institute (La Jolla, CA).
Results
The 4D Docking Procedure: Using Conformers as 4th Dimension
The aim of the 4D docking procedure is to include in ligand docking the conformational variability of the receptor in a simple and computationally efficient way. The outline of the 4D procedure and its comparison with the MRC procedure are shown on Figure 1A. In a standard MRC run, the ligand is independently docked to each receptor conformation and the results from each run are combined together in a post processing step (see Materials and Methods for details). In the 4D docking method, since all the receptor conformations are represented by a single set of 4D grids, no post-process step is needed. The present section contains two detailed reports on 4D docking results to the Estrogen Related Receptor  and Abl kinase, followed by a large scale evaluation of the 4D docking procedure and its comparison with the MRC docking using a benchmark of ∼100 proteins, more than 1000 conformers and 300 ligands.
and Abl kinase, followed by a large scale evaluation of the 4D docking procedure and its comparison with the MRC docking using a benchmark of ∼100 proteins, more than 1000 conformers and 300 ligands.
Figure 1.
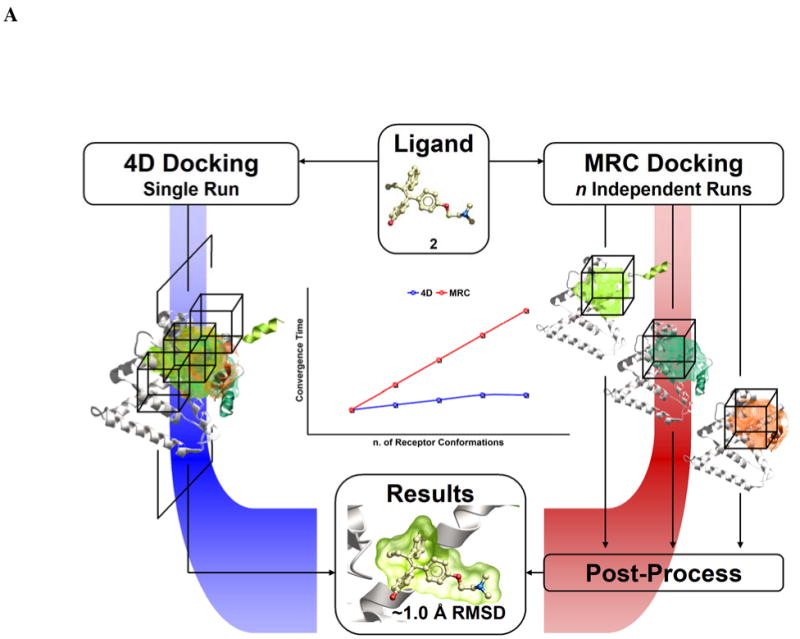
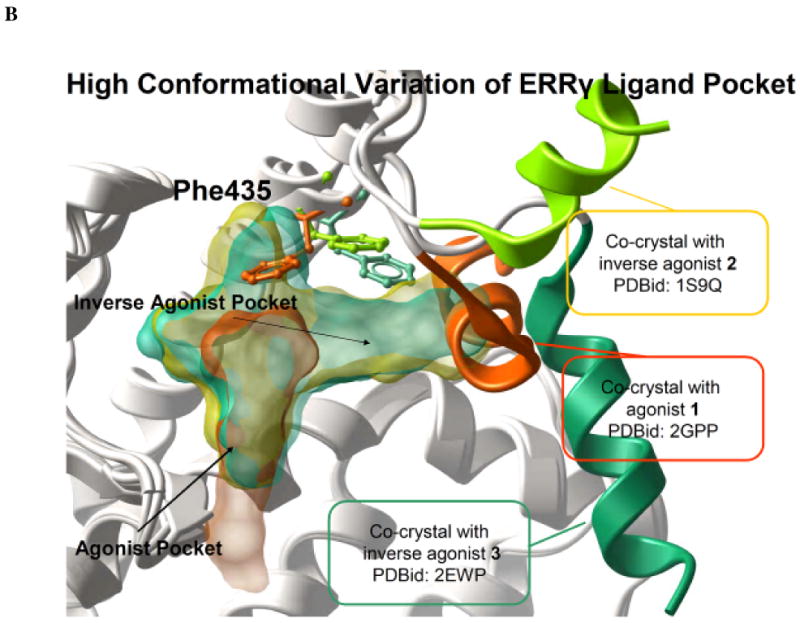
The extent of induced fit for the ERR and its treatment. (A) Schematic representation of the two different ensemble docking approaches compared in the present study. (B) Three different crystal structures of the EER Ligand Binding Domain are represented after superimposition without ligands. The variations of Phe435, AF-2 helix, and the binding pocket shape are highlighted.
Evaluating 4D Docking Accuracy on a Diverse Set of Conformers of Estrogen Related Receptor
The Estrogen Related Receptor (ERR) is an orphan receptor that acts as a constitutive activator of transcription.40 Although no endogenous ligand is known, ERR has been reported to interact with several SERMs.41 The ERR Ligand Binding Domain (LBD) exists in two distinct conformations: in the agonist bound structure the helix AF-2 (also known as H12 or activation helix) is tightly connected to the core of the LBD in a pose that promotes the basal activity; conversely, in the inverse agonist bound structure, the Phe435 side chain adopts a conformation that displaces AF-12 away from the LBD core, thus interfering with the basal activity. This example represents a very challenging case for docking because of the large scale conformational changes around the binding site.
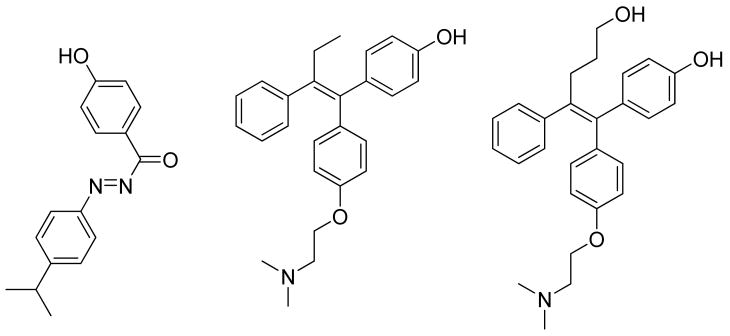
In the present study, three different co-crystals of ERR were employed: one agonist bound complex (red in Figure 1B, PDBid: 2GPP) where the receptor was co-crystallized with 1 (GSK4716)42, one inverse agonist complex (yellow in Figure 1B, PDBid: 1S9Q) with the non specific ligand 2 (4-OH-Tamoxifen)43, and one inverse agonist complex (green in Figure 1B, PDBid: 2EWP) with the specific ligand 3 (GSK5182).44 It is interesting to note that the AF-2 helix is displaced in rather different positions in the two inverse agonist bound conformations. The detailed docking results for the case study are shown in Table 2.
Table 2.
Comparison of MRC docking vs. 4D docking in reproducing the binding mode of three ERR modulators (positive values of the binding score are indicative of incorrect binding modes). Both methods achieved a highly accurate docking pose as the lowest score.
 |
||||||
|---|---|---|---|---|---|---|
| 1 (agonist) |
2 (inverse agonist) |
3 (inverse agonist) |
||||
| RMSD (Å) | Binding Score | RMSD (Å) | Binding Score | RMSD(Å) | Binding Score | |
| MRC – 1S9Q | 8.1 | 62.1 | 1.0 | -40.0 | 1.8 | -33.5 |
| MRC – 2EWP | 10.3 | 141.1 | 1.1 | -33.5 | 0.6 | -42.7 |
| MRC – 2GPP | 0.4 | -29.8 | 9.7 | -25.9 | 10.1 | -21.5 |
| MRC – Overall | 0.4 | -29.8 | 1.0 | -40.0 | 0.6 | -42.7 |
| 4D Docking | 0.4 | -30.0 | 0.9 | -32.0 | 0.6 | -42.2 |
In the MRC docking, the native binding mode of each ligand could be successfully reproduced within a RMSD of 1 Å by the best scoring pose after that the results of each single run were combined together. In every case, the best RMSD value and the best docking score were provided by the cognate receptor structure. Despite the different position of the AF-12 helix, the two inverse agonists displayed a high level of cross-docking efficiency, although the predictions were not as accurate as in self docking. The agonist and inverse agonist conformations were completely incompatible, and a simple rigid receptor cross-docking of agonist to inverse agonist pockets (and vice verse) always failed. However, a single run of 4D docking found the correct solutions for all three ligands in terms of geometry and scoring.
4D Docking Speed on DFG-in and -out conformers of ABL kinase
The previous example had 3 sufficiently different conformers and their impact on the speed of the procedure was difficult to assess. The Abl kinase set of conformers was more challenging; they included both type I and type II inhibitor bound structures and conformers with more subtle variations within each group.
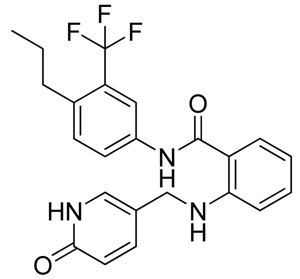
The ABL1 tyrosine kinase domain (or simply Abl kinase) is an important pharmaceutical target in the treatment of Chronic Myelogenous Leukemia as well as other types of cancer.45 Abl kinase was co-crystallized with: (i) traditional type I kinase inhibitors that bind the ATP binding pocket of the kinase in its active, DFG-in state, and (ii) type II inhibitors that induce a distinct, DFG-out conformation of the kinase and occupy an additional hydrophobic pocket created by this rearrangement. Both classes of inhibitors typically establish one to three hydrogen bonds with the main-chain atoms of the kinase hinge region. However, the ability to bind the extended pocket grants the type II inhibitors additional specificity. The 4D and MRC docking runs on a diverse Abl kinase ensemble were compared for their ability to predict the binding pose of a type II inhibitor, 4 (NVP-AEG082).46 The ensembles were built from three type I bound and two type II bound Abl kinase conformers from 5 co-crystal structures. When a single rigid receptor conformer docking run was performed, only the cognate structure (PDBid: 2HZ0) was suitable to reproduce 4 native binding mode (RMSD 0.3 Å). The convergent minimum energy pose could be sampled on average in 15.7 seconds. The MRC docking and the 4D docking could both efficiently reproduce the native binding mode in the top ranking pose (see Table 3); the only difference between the two was the convergence time.
Table 3.
Comparison of MRC docking vs. 4D docking in reproducing the binding mode of the Abl kinase Type II inhibitor 4. Both methods achieved a highly accurate docking pose as the lowest score despite the mixture of three DFG-in and two DFG-out conformers in the 4D maps.
 |
|||
|---|---|---|---|
| 4 (type II inhibitor) |
|||
| RMSD (Å) | Binding Score | ||
| MRC – 2HZ0 | DFG-in | 0.3 | -54.9 |
| MRC – 2HIW | DFG-in | 2.3 | -34.0 |
| MRC – 1M52 | DFG-out | 9.9 | -20.3 |
| MRC – 2F4J | DFG-out | 9.2 | -24.4 |
| MRC – 2G1T | DFG-out | 9.3 | -18.3 |
| MRC – Overall | 0.3 | -54.9 | |
| 4D Docking | 0.3 | -51.5 | |
In order to better understand the dependency of the 4D docking convergence time on the number of conformers in the ensemble and the structural differences among them, four diverse scenarios were explored. In the simplest ensemble docking situation, the input ensemble consists only of identical copies of the cognate receptor. In this case, the MRC convergence time scaled linearly with the number of conformations employed; it could therefore be expressed by multiples of 15.7 s (white curve in Figure 2A). Conversely, the 4D docking convergence time appeared to be unaffected by the number of identical conformations employed (cyan blue curve in Figure 2A). In a different setup, the conformational ensemble consisted of a single copy of the cognate receptor and multiple copies of an alternative receptor conformation. In the first case, the pocket variant was provided by an Abl kinase type I inhibitor co-crystal structure (PDBid: 1M52); the convergence time was only slightly affected by the number of conformations and, when five or more were considered, the variation became comparable to the statistical fluctuations (pink curve in Figure 2B). In the second case, the pocket variant was provided by an Abl kinase co-crystal with a type II inhibitor. Despite the overall similarity between the native receptor and this variant, the sampling time did not appear to be linearly related to the number of identical pocket variants (yellow curve in Figure 2B). 4D docking could handle equally well either the structural variants close to the native structure, escaping possible local minima, as well as conformers not compatible with the crystallographic ligand binding mode, avoiding the waste of sampling time in unfavorable regions of the conformational space. Finally, we investigated how the 4D docking behaved when all the conformations in the ensemble were genuine crystallographic variants rather than artificially introduced copies. The results are plotted in the orange curve in Figure 2C. The curve displayed a flat profile and the convergence time stabilized around the value of 30 seconds.
Figure 2.
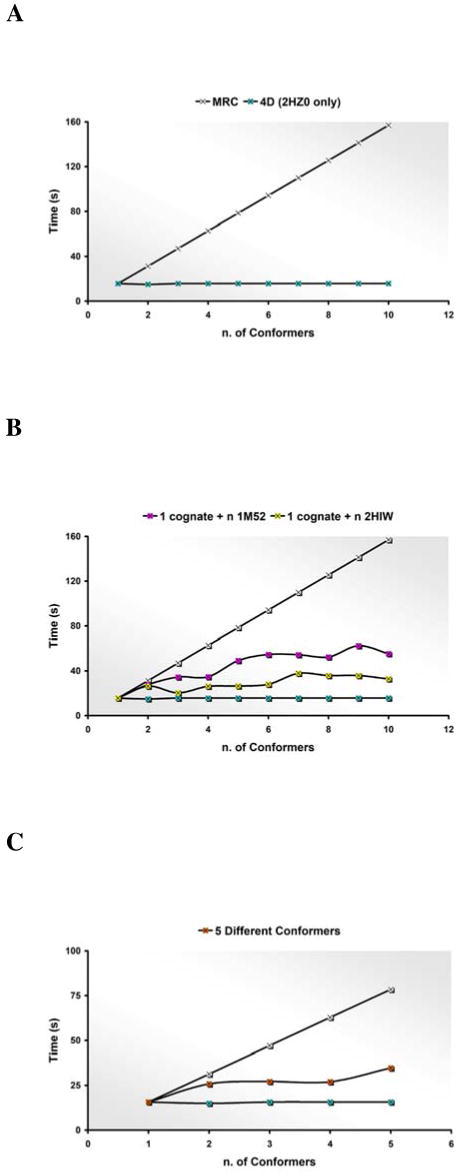
Evaluating the time required for 4D docking convergence as a function of the number of conformers in a 4D ensemble and their conformational variation. (A) The 4D docking time in case of identical copies of the cognate receptor (cyan blue curve). (B) The 4D docking time with two types of conformers: a cognate conformer and multiple copies of an extra conformer. The pocket variants are provided by a Type I inhibitor bound structure (pink curve) and by a Type II inhibitor bound structure (yellow curve). (C) The 4D docking time to all available different pocket conformers (orange curve). Each point represents the median value of all the possible combinations of n<=5 conformers. In each panel, the MRC docking ideal case (white curve) is reported for comparison. Each reported value is averaged over 100 runs.
Theoretically, the length of the 4D grid simulation required for convergence may vary depending on the diversity of the receptor conformations. Indeed, if Lsingle is the simulation length required for convergence for a single receptor conformation docking run and Nconf is the number of receptor conformations, 4D grid simulation may require from Lsingle to Nconf Lsingle. These two limits correspond to two extreme cases: if all receptor conformations are essentially identical, convergence length would be Lsingle, while if they differ so much that for each receptor conformation a completely different set of low energy ligand poses has to be explored, convergence length would be Nconf Lsingle. In reality, multiple receptor conformations, while different in certain parts, typically share a lot of common features. Therefore, regions of receptor/ligand conformational space corresponding to different receptor conformations need not be explored completely independently, resulting in convergence somewhere in between Lsingle to Nconf Lsingle and saving simulation time as compared to MRC docking.
In order to prove that the initial superimposition of the binding sites has a very strong bearing on the procedure, the 4D grid calculation was repeated adopting an unweighted scheme where highly flexible regions and the structurally conserved ones contributed equally to the spatial alignment. In the crystallographic complex,46 compound 4 forms four specific hydrogen bonds with the protein: two hydrogen bonds with the backbone of Glu316 and Met318 in the hinge region, one with the backbone NH of Ala380 right before the DFG motif, and one with Glu286 in the C-helix. The displacement of the hinge region and that of the Ala380 NH are rather small; the C-helix is only displaced in one of the DFG-out structures (PDBid: 2G1T) where it bends away from the binding site. When the weighted superimposition scheme was adopted, the atoms involved in these key interactions were automatically considered part of a high similar alignable core and assigned heavier weights. Conversely, the DFG motif, the glycine-rich loop, and the activation loop, which accounted for the structural diversity among the crystals, were assigned progressively smaller weights and their contribution to the final superimposition outcome was limited. The average RMSD from the crystallographic structure calculated on the non-hydrogen atoms of the hinge region after weighted superimposition was 0.7 Å, the average RMSD of the amide nitrogen of Ala380 0.3 Å. When the weights were not considered, the average RMSD from the crystal of the non-hydrogen atoms of the hinge region and of the amide atom of Ala380 increased to 1.4 Å and 1.6 Å respectively. Moreover, the predicted envelopes describing the binding sites were shifted and the volume of the bounding box built around the merged mesh increased of almost 30%. While the accuracy of the 4D docking predictions were not affected, the convergence time, despite being still much faster then in a standard MRC run, showed a closer dependence on the number of receptor conformers (42 seconds when five different conformers were considered).
Compiling 4D docking validation benchmark of 100 multi-conformer proteins
The above studies illustrated the ability of the 4D docking procedure to dock ligands in a fast and accurate fashion. However both ERR and Abl kinase were rather special examples. To prove the applicability of the procedure to a diverse set of conformationally variable pockets, we carefully compiled an unbiased benchmark (see Materials and Methods). It consisted of 1113 X-ray protein structures representing 99 different proteins and 300 ligands (267 unique structures). Due to the nature of the filtering sequence, the cognate receptor of each ligand was always included among the selected protein structures. The 99 diverse proteins were further subdivided into 107 structural ensembles (7 proteins were represented by two ensembles and one protein by three). The number of conformers in each ensemble varied from 3 to 29 with a median value between 9 and 10. The median intra-ensemble RMSD value, calculated on the position of the non-hydrogen atoms of the binding site side chains, was 1.92 Å. The median number of ligands per ensemble was three.
The set selected in the present study appears to be therapeutically relevant. According to the DrugBank47 database, 32 of the selected proteins are known to be targeted by at least one marketed small molecule drug and 62 are targets (or very close homologs of targets) of small molecule experimental drugs in different development stages. Among the 267 non-redundant ligand structures, there are 28 marketed drugs and 121 experimental drugs. Interestingly, there is a substantial overlap between the 4D docking validation set and the Astex diverse set,31 a clean set of 85 protein – ligand co-crystals, recently reported by Hartshorn and coworkers and assembled to test self docking protocols on targets of pharmaceutical relevance. The two sets have 32 proteins and 22 ligand structures in common (27 and 13 from exactly the same X-ray structures, respectively).
The complete list of structures included in the 4D docking validation set is reported in the Supplementary Information and is available upon request.
Comparing 4D Docking with MRC and Single Cross-docking
It was critical to illustrate that the 4D docking procedure is significantly superior to single cross-docking; since there is always a danger that presenting a ligand with a larger set of conformers actually deteriorates the performance due to increased “noise”. We also sought to establish whether a faster 4D docking procedure in which the 4th dimension is randomly sampled concurrently with the ligand optimization is as accurate as a systematic, slow, and more cumbersome MRC procedure. As a measure of success, we used the fraction of cases at which a near-native ligand binding pose (within 2 Å rmsd from the co-crystal structure after receptor superimposition) was scored first. Despite the implementation of more lenient thresholds (2.5 Å and top 5 ranks) that have been reported for several ensemble docking exercises15, 17, we chose to adopt the conventional 2 Å RMSD limit and a single top pose as more strict but practical success criterion.48
To establish a baseline for docking accuracy, every ligand in the set was cross docked at each receptor in the same ensemble by means of a single rigid conformer docking protocol. That exercise was successful in only 46.6% of the 107 ensembles. This finding is in good agreement with previously reported data,2, 4, 49 and confirms the limitation of the single rigid conformer docking approach.
A traditional MRC protocol was then applied to the validation set. Each ligand was independently docked at the binding site of every structure in the ensemble and the results were combined together. The poses within a window of 1 Kcal from the best grid potentials-based energy in the combined ranking were re-scored in an explicit receptor environment and re-ranked accordingly. The mean number of poses in the considered top energy window was comprised between 1 and 2, never exceeding 5. The MRC docking was able to reproduce the native binding mode for as many as 239 ligands out of 300 (79.6 % success rate). For 53 out of the 61 failed cases, the correct poses were found within top 10 ranking solutions. For the rest 8 cases (2.7%) no near native solution was identified.
The inability of the MRC approach to achieve a 100% success rate, even on a set of ligands that can be successfully docked into their cognate receptors, is primarily explained by the increased rate of false positives due to the use of multiple structures. A ligand that docks well into its own receptor, can nevertheless score better in an incorrect pose with a different receptor conformation.19 For our benchmark, that was the case in 17.7% of the ligands (e.g., 53 out of 300). Careful examination of the 8 cases where the correct position was not identified showed a shift of the bounding box as the major reason for the failure. The shift, in turn, was due to our use of a predicted bounding box to avoid any bias from the known ligand pose. In agreement with our previous findings,2 the unbiased definition of the binding site and of the bounding box compromised to some extent the prediction quality but made the numbers more realistic.
On the present benchmark, the average sampling time required by the MRC method for each ligand was 312 seconds (∼32.8 seconds per receptor conformer). Naturally, the time depends linearly on the number of conformers.
In order to prove that the 4D docking could achieve accuracies equal or close to those obtained by the MRC docking with calculation times much closer to a single run, the proposed method was tested on the same validation set. The native binding mode was correctly reproduced for as many as 232 ligands out of 300 (77.3 % success rate) and the 4D strategy was only slightly less accurate then the MRC procedure. The small performance drop could be mainly ascribed to insufficient sampling of the 4th dimension or the dependence of the 4D procedure on the conformer's 3D superposition (see Materials and Methods for further details). In this light, it was not surprising that the fraction of binding modes that could not be properly reproduced because of an incorrect sampling rose from 2.7% to 12.4 %. The average sampling time for the 4D docking was 75 seconds (∼7.9 seconds per receptor conformer), roughly four times faster than the MRC method. Longer sampling times did not improve the quality of the 4D results. The accuracy and the average simulation length for single rigid conformer cross-docking, MRC docking, and 4D docking are summarized in Figure 3.
Figure 3.
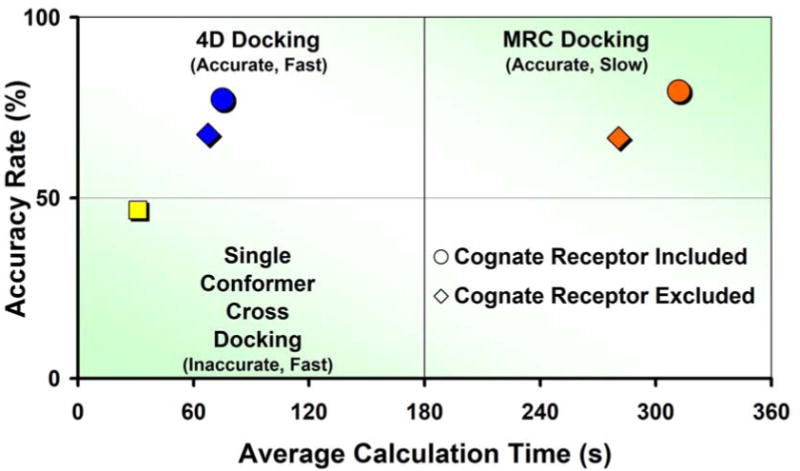
The 4D docking protocol (blue markers, upper left quadrant) combines the accuracy of the MRC docking (orange markers, upper right quadrant) with the speed of single conformer cross-docking (yellow marker, lower left quadrant).
We have established that an average 4D docking is better than an average cross-docking to one conformer. However, too many conformers may also lead to performance deterioration. How does the number of receptor conformers affect the 4D docking performance? To analyze the influence of the number of structures in the ensemble on the docking accuracy we divided the whole benchmark into three subsets roughly equivalent in size: (i) low number of receptor conformations, found in 47 ensembles that consist of less than nine structures and 104 ligands, (ii) medium number of receptor conformations, found in 36 ensembles that consist of nine to fourteen protein structures and 112 ligands, and (iii) high number of receptor conformations, found in 24 ensembles that consist of more than fifteen protein structures and 84 ligands. In Figure 4A, the results of each subset considered separately are reported.
Figure 4.
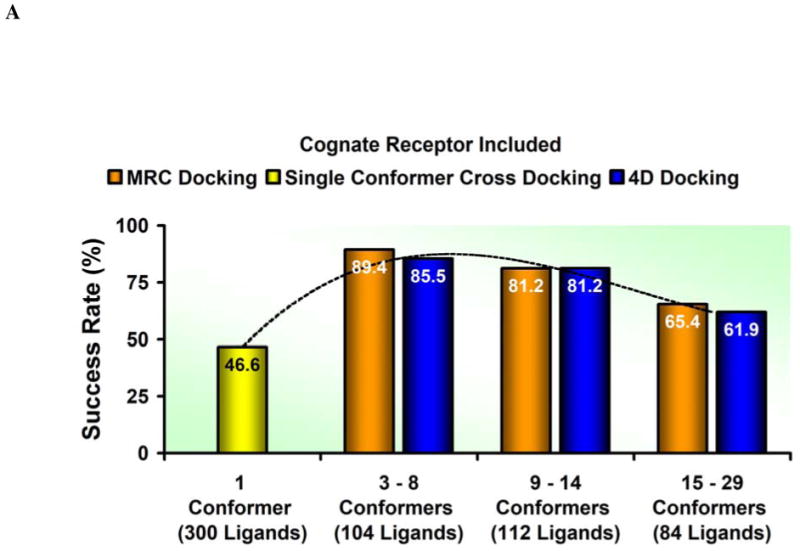
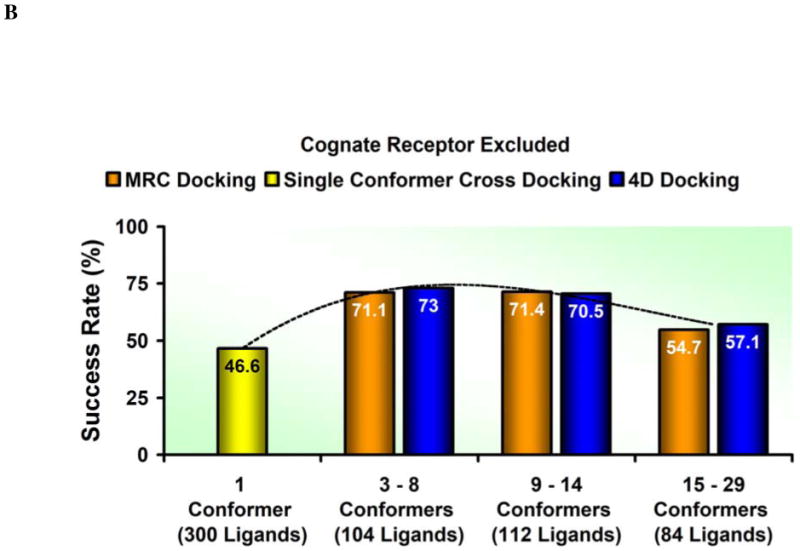
A diagram of the accuracy versus the ensemble size. The histograms compare the MRC (orange) and 4D (blue) results considering, instead of the whole validation set, three subsets divided according to the number of structures in each ensemble (between 3 and 8, between 9 and 14, and between 15 and 29, respectively) to the performance of the single rigid conformer docking (yellow histogram). A dashed line represents the accuracy trend. (A) The cognate receptor structure is included in the ensemble docking calculations. (B) The cognate receptor structure is not included in the ensemble docking calculations.
Both methods displayed a very similar profile and responded in the same way when structural ensembles of different size were considered. Such a consistent pattern ruled out the possibility that the accuracy loss associated with large ensembles reflected an intrinsic limitation of the 4D approach. It suggested instead that the amount of noise generated when the number of structures grew beyond a certain threshold compromised the efficacy of the scoring scheme that each method adopted.19 In a straightforward attempt to reduce the size of the conformational ensembles containing more than fourteen structures, a hierarchical clustering analysis was carried out. The distance criterion adopted was the RMSD calculated on the non-hydrogen atoms of the binding site side chains. Two different threshold values (0.5 Å and 1.0 Å, respectively) were tested to determine two possible functional partitions but neither of them appeared to increase the quality of the results. The full outcome of the clustering exercise is reported in the Supplementary Information.
Finally, we tested the docking accuracy in the absence of the cognate receptor structure. While the above results could be considered a best case scenario, the goal of this exercise was to establish the accuracy limit for the 4D docking protocol in a more realistic case. We purposely and systematically deleted from the ensemble the receptor conformation which would have likely provided a correct pose during sampling to assess if, in these circumstances, an ensemble docking approach could still outperform the single rigid conformer cross-docking. The results are reported in Figure 3 and Figure 4B. The MRC docking correctly reproduced the native binding mode in as many as 200 ligands out of 300 and 4D docking provided very similar results with 203 accurate predictions. As expected, when compared to the success rate of the runs including the cognate receptor, the accuracy of both methods decreased (the MRC rate dropped from 79.6% to 66.6%, the 4D docking rate from 77.3% to 67.6%) but remained superior to the single rigid conformer cross-docking scheme. When the cognate receptor was excluded, the use of several structures improved the chance that at least one of them, even if not perfectly adapted to the ligand, could still allow a near native pose to be sampled and properly scored. Because the validation set was selected promoting ligand diversity among the complexes, these results could not be simply explained by trivial redundancy in the set. Even when present, the cognate receptor was not necessarily the structure that provided the best prediction either in terms of geometry and/or score; this behavior has already been reported in the literature19 and, at least in part, is due to variations of quality of individual structures contributing a particular ensemble.
Discussion
In this study, we present an objective comparison of three cross-docking approaches: single receptor cross-docking, ensemble docking, and 4D docking. To the best of our knowledge, this is the first effort to assess these protocols on a very large automatically compiled benchmark (∼100 proteins, >1000 structures). Under the stringent definition of success (2 Å ligand heavy atom RMSD), we obtained the realistic estimates of 46.6%, 79.6%, and 77.3% for the expected success rate of the single receptor cross-docking, ensemble docking, and 4D docking. While the accuracy of ensemble docking and 4D docking appeared to be almost identical, the time requirements of the latter approach are significantly lower. This, therefore, validates the 4D docking as a fast and reliable method for incorporating receptor flexibility in ligand docking.
As a practical alternative to simultaneous explicit sampling of protein and ligand, ensemble docking recently attracted the attention of many research groups. However, previous studies focused on significantly smaller hand-picked datasets in a somewhat idealized conditions, avoiding the issues of automatic structure pre-processing, protonation and tautomerization states assignment, and binding box composition and boundaries definition. For example, the FlexE15 united protein description was tested on ten protein ensembles, encompassing 105 structures and 60 ligands. Only 21 ligand poses (35%) could be correctly predicted in the top ranking position within a threshold of 2 Å. The FlexE module ran 1.75 times faster than the reiterated application of FlexX on each structure in the ensemble. However, since FlexE achieved a performance almost equivalent in terms of accuracy, the role of the new conformations generated by the combination of the experimental structures' non overlapping regions was not completely elucidated. Huang and Zou17 tested their simplex minimization method on a benchmark of size similar to the one employed to validate FlexE. When only the best ranking pose and a threshold of 2 Å are considered, the algorithm was able to correctly reproduce the crystallographic pose of 72 of the considered 87 ligands (82.7 %). It should be noticed that, in order to focus on the influence of the receptor flexibility, the ligands were treated as rigid bodies. A semi flexible ligand approach (namely, multiple conformations of each ligand were pregenerated, each conformer was docked in the structural ensemble as a rigid body, and the results were merged before ranking) was attempted for the cAPK ensemble. The results turned out only marginally less accurate than in the single ligand conformation approach. The whole procedure was reported to be computationally very efficient, with a calculation time comparable to that of a single receptor run. FITTED 1.0 was tested on six structural ensembles accounting for five targets of pharmaceutical interest, five to nine structures composing each target ensemble, and a collection of 33 ligands. Based on the standard success criterion, both the semiflexible and the fully flexible FITTED 1.0 implementations accuracies turned out to be 73%. This approach is very time consuming and, as the authors acknowledged in a recent review,50 too slow to be truly competitive.
In this study we demonstrated that the proposed 4D protocol is both fast and accurate. Figure 5 shows that even if cognate conformers are excluded from the conformer ensembles, the 4D protocol improves the docking performance by as much as 21%. The basic 4D docking paradigm could be used as a starting point to develop more advanced protocols that will fully exploit the possibilities offered by the 4D grid implementation: different weights could be assigned to specific regions of the receptor conformers, omission models could be generated on the fly,2 and, in principle, even individual ligand moieties could be assigned different 4D coordinates, effectively experiencing chimerical grid potentials.
Figure 5.
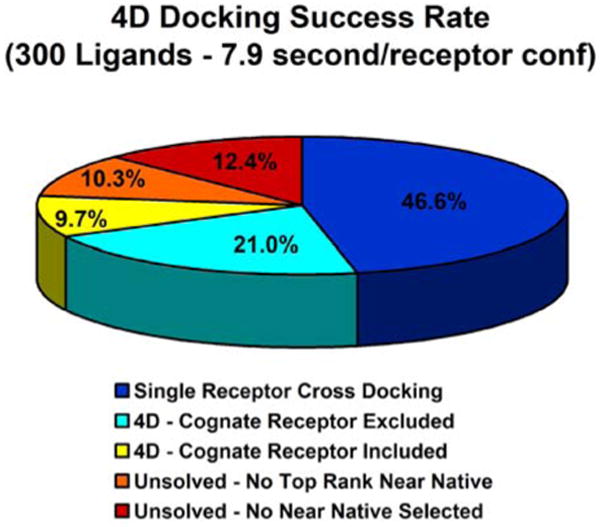
4D docking results summary. The fraction of ligands that can be correctly reproduced by both single rigid conformer cross-docking and 4D docking, the fraction that can be correctly reproduced only by 4D docking without including the cognate receptor in the structural ensemble, and the fraction that can be correctly reproduced only by 4D docking and only when the cognate receptor is included in the structural ensemble are reported in blue, cyan blue, and yellow, respectively. The fraction of ligands which can be correctly sampled but not ranked in the top position and the fraction that cannot be selected at all, are reported in orange and red, respectively.
We exploited the X-ray structures deposited in the RCSB as a source of receptor plasticity to test our method. The resulting benchmark is, at the best of our knowledge, the only clean set employed in the validation of an ensemble docking protocol as well as one including the highest number of different targets and structures. However, our method can be easily applied to structural ensembles that include receptor conformations generated by computational means, such as, and not limited to, normal mode generated structures,51 and ligand-guided binding pocket variants.52
Conclusions
Here we presented a new 4D docking protocol that considers multiple receptor conformers an extra dimension of the search space. The procedure dramatically improved the accuracy of ligand docking to a flexible receptor yet was shown to be almost as fast as a single conformer docking. The 4D docking performance was evaluated on a large pharmaceutically relevant benchmark and compared to the explicit docking to multiple receptor conformers.
4D docking provides natural benefits for three kinds of applications. First, predicting a docking pose for a particular ligand to a flexible receptor. Second, virtual ligand screening that benefits from the adequate consideration of the receptor flexibility. Third, ligand specificity profiling of one ligand versus multiple flexible receptors.21, 49, 53 All three applications can be simplified with the 4D docking protocol. Moreover, virtual ligand screening and ligand specificity profiling can be accelerated committing parallel computational resources to explore different ligands and receptors, rather than iterate the same calculation over multiple conformers of the same protein.
Supplementary Material
The complete list of PDB structures employed in the study, the detailed results of the ligand docking simulations for each ensemble, the complete description of the cluster analysis exercise on the structural ensemble before assembling the 4D grids. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank George Nicola for reviewing the manuscript. This work was supported by NIH/NIGMS grants 5-R01-GM071872-02 and 1-R01-GM074832-01A1.
Abbreviations
- 4D Docking
Four-dimensional Docking
- ICM
Internal Coordinate Mechanics
- MRC
Multiple Receptor Conformers
- RMSD
Root Mean Square Deviation
Bibliography
- 1.Teague SJ. Implications of protein flexibility for drug discovery. Nat Rev Drug Discov. 2003;2:527–41. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 2.Bottegoni G, Kufareva I, Totrov M, Abagyan R. A new method for ligand docking to flexible receptors by dual alanine scanning and refinement (SCARE) J Comput Aided Mol Des. 2008;22:311–25. doi: 10.1007/s10822-008-9188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–25. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 5.Teodoro ML, Kavraki LE. Conformational flexibility models for the receptor in structure based drug design. Current Pharmaceutical Design. 2003;9:1635–1648. doi: 10.2174/1381612033454595. [DOI] [PubMed] [Google Scholar]
- 6.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Current Opinion in Structural Biology. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Damm KL, Carlson HA. Exploring Experimental Sources of Multiple Protein Conformations in Structure-Based Drug Design. J Am Chem Soc. 2007;129:8225–8235. doi: 10.1021/ja0709728. [DOI] [PubMed] [Google Scholar]
- 8.Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6:447–52. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 9.Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol. 2000;57:213–8. [PubMed] [Google Scholar]
- 10.Knegtel RMA, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. Journal of Molecular Biology. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 11.Shoichet KD, Kuntz ID, Bodian DL. Molecular docking using shape descriptors. Journal of Computational Chemistry. 1992;13:380–397. [Google Scholar]
- 12.Garrett M, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- 13.Sotriffer CA, Dramburg I. “In situ cross-docking” to simultaneously address multiple targets. J Med Chem. 2005;48:3122–5. doi: 10.1021/jm050075j. [DOI] [PubMed] [Google Scholar]
- 14.Zentgraf M, Fokkens J, Sotriffer CA. Addressing protein flexibility and ligand selectivity by “in situ cross-docking”. ChemMedChem. 2006;1:1355–9. doi: 10.1002/cmdc.200600073. [DOI] [PubMed] [Google Scholar]
- 15.Claussen H, Buning C, Rarey M, Lengauer T. FlexE: efficient molecular docking considering protein structure variations. J Mol Biol. 2001;308:377–95. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 16.Rarey M, Kramer B, Lengauer T, Klebe G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. Journal of Molecular Biology. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 17.Huang SY, Zou X. Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 18.Ewing TJA, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. Journal of Computer-Aided Molecular Design. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 19.Barril X, Morley SD. Unveiling the Full Potential of Flexible Receptor Docking Using Multiple Crystallographic Structures. J Med Chem. 2005;48:4432–4443. doi: 10.1021/jm048972v. [DOI] [PubMed] [Google Scholar]
- 20.Corbeil CR, Englebienne P, Moitessier N. Docking ligands into flexible and solvated macromolecules. 1. Development and validation of FITTED 1.0. J Chem Inf Model. 2007;47:435–49. doi: 10.1021/ci6002637. [DOI] [PubMed] [Google Scholar]
- 21.Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, McCammon JA. Ensemble-Based Virtual Screening Reveals Potential Novel Antiviral Compounds for Avian Influenza Neuraminidase. J Med Chem. 2008;51:3878–3894. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK. Testing a Flexible-receptor Docking Algorithm in a Model Binding Site. Journal of Molecular Biology. 2004;337:1161–1182. doi: 10.1016/j.jmb.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Grünberg R, Leckner J, Nilges M. Complementarity of Structure Ensembles in Protein-Protein Binding. Structure. 2004;12:2125–2136. doi: 10.1016/j.str.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Eisenstein M. Introducing a 4th Dimension to Protein-Protein Docking. Structure. 2004;12:2095–2096. doi: 10.1016/j.str.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucl Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LSL. The Universal Protein Resource (UniProt) Nucl Acids Res. 2005;33:D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boeckmann B, Blatter MC, Famiglietti L, Hinz U, Lane L, Roechert B, Bairoch A. Protein variety and functional diversity: Swiss-Prot annotation in its biological context. Comptes Rendus Biologies. 2005;328:882–899. doi: 10.1016/j.crvi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Abagyan R, Orry A, Raush E, Budagyan L, Totrov M. ICM Manual 3.5. Molsoft LCC: La Jolla, CA; 2007. [Google Scholar]
- 29.Kleywegt GJ, Harris MR, Zou Jy, Taylor TC, Wahlby A, Jones TA. The Uppsala Electron-Density Server. Acta Crystallographica Section D. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 30.An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics. 2005;4:752–61. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, Murray CW. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem. 2007;50:726–41. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 32.Matthieu Schapira MTRA. Prediction of the binding energy for small molecules, peptides and proteins. Journal of Molecular Recognition. 1999;12:177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 33.Totrov M, Abagyan R. The contour-buildup algorithm to calculate the analytical molecular surface. J Struct Biol. 1996;116:138–43. doi: 10.1006/jsbi.1996.0022. [DOI] [PubMed] [Google Scholar]
- 34.Abagyan R, Frishman D, Argos P. Recognition of distantly related proteins through energy calculations. Proteins. 1994;19:132–40. doi: 10.1002/prot.340190206. [DOI] [PubMed] [Google Scholar]
- 35.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 36.Totrov M, Abagyan R. Protein-Ligand docking as an energy optimization problem. In: Raffa RB, editor. Drug-receptor thermodynamics : introduction and applications. Wiley; Chichester; New York: 2001. pp. 603–624. [Google Scholar]
- 37.Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- 38.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem. 2003;46:3045–59. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 39.Totrov M, Abagyan R. Derivation of sensitive discrimination potential for virtual screening. RECOMB '99 Proceedings of the Third Annual International Conference on Computational Molecular Biology; Lyon (France). 1999; New York: Lyon (France): ACM Press; 1999. pp. 37–38. [Google Scholar]
- 40.Tremblay AM, Giguere V. The NR3B subgroup: an ovERRview. Nucl Recept Signal. 2007;5:e009. doi: 10.1621/nrs.05009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greschik H, Flaig R, Renaud JP, Moras D. Structural Basis for the Deactivation of the Estrogen-related Receptor {gamma} by Diethylstilbestrol or 4-Hydroxytamoxifen and Determinants of Selectivity. J Biol Chem. 2004;279:33639–33646. doi: 10.1074/jbc.M402195200. [DOI] [PubMed] [Google Scholar]
- 42.Zuercher WJ, Gaillard S, Orband-Miller LA, Chao EYH, Shearer BG, Jones DG, Miller AB, Collins JL, McDonnell DP, Willson TM. Identification and Structure-Activity Relationship of Phenolic Acyl Hydrazones as Selective Agonists for the Estrogen-Related Orphan Nuclear Receptors ERRβ and ERRγ. J Med Chem. 2005;48:3107–3109. doi: 10.1021/jm050161j. [DOI] [PubMed] [Google Scholar]
- 43.Jordan VC, Collins MM, Rowsby L, Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. 1977;75:305–16. doi: 10.1677/joe.0.0750305. [DOI] [PubMed] [Google Scholar]
- 44.Chao EY, Collins JL, Gaillard S, Miller AB, Wang L, Orband-Miller LA, Nolte RT, McDonnell DP, Willson TM, Zuercher WJ. Structure-guided synthesis of tamoxifen analogs with improved selectivity for the orphan ERRgamma. Bioorg Med Chem Lett. 2006;16:821–4. doi: 10.1016/j.bmcl.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 45.Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8:341–350. doi: 10.1038/nrc2368. [DOI] [PubMed] [Google Scholar]
- 46.Cowan-Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, Rheinberger P, Centeleghe M, Fabbro D, Manley PW. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallographica Section D. 2007;63:80–93. doi: 10.1107/S0907444906047287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–6. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole J, Murray CW, Willem J, Nissink M, Taylor RD, Taylor R. Comparing protein-ligand docking programs is difficult. Proteins: Structure, Function, and Bioinformatics. 2005;60:325–332. doi: 10.1002/prot.20497. [DOI] [PubMed] [Google Scholar]
- 49.Cavasotto CN, Orry AJW, Abagyan RA. The Challenge of Considering Receptor Flexibility in Ligand Docking and Virtual Screening. Current Computer - Aided Drug Design. 2005;1:423–440. [Google Scholar]
- 50.Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol. 2008;153(1):S7–26. doi: 10.1038/sj.bjp.0707515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–40. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 52.Bisson WH, Cheltsov AV, Bruey-Sedano N, Lin B, Chen J, Goldberger N, May LT, Christopoulos A, Dalton JT, Sexton PM, Zhang XK, Abagyan R. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proceedings of the National Academy of Sciences. 2007;104:11927–11932. doi: 10.1073/pnas.0609752104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao S, Sanschagrin PC, Greenwood JR, Repasky MP, Sherman W, Farid R. Improving database enrichment through ensemble docking. J Comput Aided Mol Des. 2008;22:621–7. doi: 10.1007/s10822-008-9182-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The complete list of PDB structures employed in the study, the detailed results of the ligand docking simulations for each ensemble, the complete description of the cluster analysis exercise on the structural ensemble before assembling the 4D grids. This material is available free of charge via the Internet at http://pubs.acs.org.