

Abstract
Withdrawal anxiety is a significant factor contributing to continued alcohol abuse in alcoholics. This anxiety is extensive, long-lasting, and develops well after the obvious physical symptoms of acute withdrawal. The neurobiological mechanisms underlying this prolonged withdrawal-induced anxiety are not well understood. The basolateral amygdala is a major emotional center in the brain and regulates the expression of anxiety. New evidence suggests that increased glutamatergic function in the basolateral amygdala may contribute to withdrawal-related anxiety following chronic ethanol exposure. Recent evidence also suggests that kainate-type ionotropic glutamate receptors are inhibited by intoxicating concentrations of acute ethanol. This acute sensitivity suggests potential contributions by these receptors to the increased glutamatergic function seen during chronic exposure. Therefore we examined the effect of chronic intermittent ethanol (CIE) and withdrawal (WD) on kainate receptor (KAR) mediated synaptic transmission in the basolateral amygdala (BLA) of Sprague Dawley rats. Our study showed that CIE, but not withdrawal, increased synaptic responses mediated by KARs. Interestingly, both CIE and WD occluded KAR-mediated synaptic plasticity. Finally, we found that BLA fEPSP responses were increased during CIE and WD via a mechanism that is independent from glutamate release from presynaptic terminals. Taken together, these data suggest that KARs might also contribute to postsynaptic increases in glutamatergic synaptic transmission during CIE and that the mechanisms responsible for the expression of KAR-dependent synaptic plasticity might be engaged by chronic ethanol exposure and withdrawal.
Keywords: basolateral amygdala, electrophysiology, kainite, anxiety, withdrawal
Introduction
Withdrawal from chronic alcohol exposure causes long-term alterations in numerous behavioral outcomes. While several of these alterations are believed to contribute significantly to treatment efficacy in human alcoholics, withdrawal-related anxiety makes a significant contribution to relapse in humans (Cohn et al., 2003; Linnoila, 1989; Lucht et al., 2002; Verheul et al., 2005). The neurophysiological mechanisms governing these long-term changes in anxiety-related behaviors are not well understood. Importantly, numerous rodent models of chronic ethanol exposure have long-term changes in anxiety-like behavior during withdrawal as a common characteristic (Borlikova et al., 2006; Breese et al., 2005; Kliethermes, 2005; Lack et al., 2007; Santucci et al., 2008; Valdez et al., 2002; Zhao et al., 2007). The neural circuitry and neurophysiological mechanisms governing anxiety-like behavior in rodents have been relatively well-studied. Importantly, it has recently been demonstrated that these same brain areas appear to mediate increased anxiety following withdrawal from chronic ethanol exposure (Funk and Koob, 2007; Knapp et al., 2007; Lack et al., 2007).
The basolateral amygdala (BLA) is a central component of the neural circuitry governing anxiety-related information and is also involved with alcohol withdrawal-related behaviors. The flow of information through the amygdala starts with cortical and thalamic input into the lateral and basolateral nuclei, then proceeds from there through efferent projections to the central nucleus of the amygdala, bed nucleus of the stria terminalis and the nucleus accumbens (De Olmos et al., 1985). Alcohol interacts with neurotransmitter systems mediating both fast excitatory and inhibitory synaptic transmission in the amygdala. Importantly, the balance of these neurotransmitter systems can be disrupted to alter anxiety-related behavior (Lack et al., 2007; Sanders and Shekhar, 1995). Recent work has shown that chronic intermittent ethanol and withdrawal produce a significant up-regulation of the glutamatergic system in the BLA that may ultimately influence anxiety-like behavior (Lack et al., 2007).
There are three major subtypes of ionotropic glutamate receptors: NMDA, AMPA, and kainate-type (KA-R). While we are beginning to understand the effects of alcohol on BLA AMPA and NMDA receptors, very little is known about the effects of alcohol exposure on kainate-type glutamate receptors. These receptors are structurally similar to the better-studied AMPA-type glutamate receptors and are typically found in both pre- and postsynaptic compartments throughout the forebrain (Jin et al., 2006; Weiner et al., 1999; West et al., 2007). Functional KA-Rs can consist of five potential subunit combinations: GLUR5–7 are needed for functional channels; the KA1–2 subunits confer unique pharmacological and biophysical properties to the native receptor (Braga et al., 2004). GLUR5,6 and KA2 subunits are highly expressed in the BLA; and, kainate receptors contribute to glutamatergic excitatory postsynaptic potentials (EPSPs) in this brain region (Li and Rogawski, 1998).
Until recently, the close similarities between AMPA- and kainate-type glutamate receptors had prevented a systematic understanding of the distinct roles these receptors play in various physiological processes. However, the 5 development of selective antagonists allow the separation of AMPA- and KA-receptor mediated responses (reviewed in (Pinheiro and Mulle, 2006)). For example, we now know that KA-Rs contribute to a significant postsynaptic response in the BLA (Li and Rogawski, 1998) and also mediate a form of long-lasting heterosynaptic plasticity in this brain region (Li et al., 2001). Recent evidence also suggests that the acute sensitivity of KA-Rs to ethanol may play a prominent role in regulating plastic changes in BLA synaptic transmission (Lack et al., 2008). Furthermore, our data with the GluR5-selective antagonist UBP296 supports suggestions that GluR5-containing KARs in the BLA are a potential therapeutic target for anxiety related behaviors (Aroniadou-Anderjaska et al., 2007) and provides strong rationale for determining the effects of CIE and WD on KA-Rs in the BLA.
Materials & Methods
Animals
All animal procedures were performed in accordance with protocols approved by Wake Forest University School of Medicine Animal Care and Use Committee and were consistent with the NIH Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (120 – 140g) were obtained from Harlan at the beginning of the experiments described below. All animals were housed in an animal care facility at 23°C with a 12-hour light/dark cycle and given food and water ad libitum. Rats were weighed daily to ensure that at least 80% of their free-feeding weight was maintained during vapor chamber ethanol exposure.
Chronic Ethanol Exposure
Ethanol exposure was accomplished via an ethanol vapor chamber similar to that used in other studies (Lack et al., 2007). Briefly, animals in their home cages were placed into air-tight, Plexiglas enclosures and exposed to either ethanol vapor (~28mg/L) or room air during the light cycle (12 hours/day) for 10 consecutive days. Animals receiving the chronic intermittent treatment were divided into those euthanized while still intoxicated at the end of the last exposure (CIE) and those euthanized 24 hours after the last exposure (WD). Tail blood was taken periodically during the exposure to monitor blood-ethanol concentration and trunk blood was collected on the day of sacrifice for subjects in the CIE group. Blood ethanol levels at the time the CIE animals were sacrificed were 225±32mg/deciliter.
Preparation of Brain Tissue
Animals were anesthetized with isoflurane and euthanized by decapitation. Coronal brain slices (400μm) were prepared as described previously (Lack et al., 2007) using modified aCSF (in mM: 180 Sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2•6H20, 26 NaHCO3, 1.2 NaH2PO4, 10 glucose) containing 100μM ketamine. Slices from all experimental groups were transferred and stored in 0.5 liter of standard oxygenated aCSF solution (in mM: 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 10 glucose, and 2 CaCl2) at room temperature for at least 1 hour and up to 6 hours.
Electrophysiology
Methods for whole-cell ‘blind’ patch-clamp recordings from principle BLA projection neurons within slices were similar to those reported previously (Lack et al., 2007). Electrodes were filled with an intracellular pipette solution containing (in mM): 122 CsOH, 17.5 CsCl, 10 HEPES, 1 EGTA, 5 NaCl, 0.1 CaCl2, 4 Mg-ATP, 0.3 Na-GTP, and 2 QX-314 (Cl); pH adjusted to 7.2 with gluconic acid; osmolarity between 280–290 mmol/kg. Recordings wereΩ made from glutamatergic projection neurons based on their low input resistance (<40MΩ, (Rainnie, 1999)) and large whole-cell capacitance (McCool and Botting, 2000). These neurons represent 90–95% of the neurons in the BLA (McDonald, 1982).
Excitatory postsynaptic currents (EPSCs) were electrically evoked every 20 sec by 0.2 msec square-wave stimulation within the external capsule (EC) (Fig. 1A) using platinum/iridium concentric bipolar stimulating electrodes (FHC, Bowdoinham, ME) with an inner pole diameter of 25 μm and resistance of 8–12MΩ. Compound glutamatergic events were pharmacologically isolated using 10μM bicuculline to inhibit fast GABAergic transmission. In addition, kainate currents were isolated using 50μM DL-2-amino-5-phosphono-pentanoic acid (APV), a NMDA receptor antagonist, and 50 μM GYKI 53655 (1-(4-aminophenyl)-3-methylcarbamyl-4-methyl-3,4-dihydro-7,8-methylenedioxy-5H-2,3-benzodiazepine), an AMPA receptor-specific non-competitive antagonist. Recordings were acquired with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) and digitized with a Digidata 1200B (Axon Instruments).
Figure 1. CIE but not WD increases kainate receptor-mediated synaptic function in the BLA.

A, Slices were stimulated in the external capsule and the recording electrode was placed in the basolateral amygdala as shown (taken from (Paxinos and Watson, 1997)). B, Exemplar traces show the GYKI 53655-resistant, kainate receptor-mediated EPSC in neurons from each treatment group at increasing stimulation intensities. C, The input/output relationship for EPSC area (here, expressed as charge or picoCoulomb) versus stimulation intensity shows significant increases in KA-R EPSC at stimulation intensities >25μA following CIE treatment (n=7 neurons) compared to either CON (n=7) or WD (n=7). One-way ANOVA followed by Newman-Keuls posttest; *, # -- P< 0.05, **, ## -- P<0.01 for CIE compared to CON (*) and WD (#).
Paired pulse ratio
Paired-pulse facilitation (PP) during whole-cell recordings was measured using pairs of electrical stimuli of equal intensity at 25, 50, or 250 msec interpulse intervals. Ratios of the amplitudes of the evoked EPSCs were calculated as the amplitude of the second event minus the amplitude of the first event divided by the amplitude of the first event. All values were expressed as mean ± SEM, and data were subjected to a one way ANOVA and Newman-Kuels post-test with P<0.05 considered statistically significant.
Field EPSP Recordings
Slices were placed in the recording chamber and were continuously superfused in aCSF (2 ml/min) warmed to approximately 30°C (Lack et al., 2008). Electrodes were filled with an extracellular pipette solution containing 150mM NaCl. Ensemble postsynaptic potentials were evoked every 30 sec by brief (0.2 msec) square-wave electrical stimulation within the external capsule (EC) along the lateral border of the BLA. These synaptic responses are referred to as field excitatory postsynaptic potentials (fEPSPs) throughout the manuscript and were completely sensitive to both the AMPA/kainate receptor antagonist DNQX (20 μM) and to tetrodotoxin (1 μM; not shown). For the ATPA-induced plasticity experiments, maximal fEPSP responses were determined in every slice; the stimulus intensity that evoked 50% maximal amplitude was used for the baseline period. All field experiments were recorded in the presence of 10 μM bicuculline methiodide. Baseline fEPSPs were recorded for 10 minutes prior to application of the kainate receptor agonist ATPA ((RS)-2-Amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid; 5 μM) for 15 minutes; the slice was then washed for 40 minutes in drug-free aCSF. fEPSP recordings were acquired with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) in the current-clamp mode and digitized with a Digidata 1200B (Axon Instruments). All values were expressed as mean percent control ± SEM. Data were subjected to a one way ANOVA or a two-way ANOVA (see text) with P<0.05 considered statistically significant.
Input/Output Recordings
Input/output recordings were measured for both KAR-mediated EPSCs and fEPSPs. For both types of recording, input/output values were collected using increasing stimulus intensities for each cell or slice. At each stimulus intensity, EPSC amplitude or fEPSP slope was measured and compared across the treatment conditions. All values were expressed as mean response area (for EPSCs) or percent control (for fEPSPs) ± SEM, and data were subjected to a two-way ANOVA with stimulus intensity and fEPSP slope as the main dependent variables with Bonferroni post-hoc analyses; P<0.05 was considered statistically significant.
Results
CIE, but not WD, increases KA-R function at BLA glutamatergic synapses
Previous research in our lab indicated that kainate receptors are inhibited by acute ethanol (Lack et al., 2008). These results suggested that KA-Rs might then be up-regulated during chronic intermittent exposure and withdrawal in response to this acute sensitivity. Therefore, we determined the contribution made by KA-Rs to glutamatergic synaptic responses during CIE and WD. To examine this, we used electrophysiology to measure the input/output (stimulus-response) relationship for KA-R via stimulation of the external capsule (Li et al., 2001; Li and Rogawski, 1998). Since KA-R mediated responses are small relative to AMPA-mediate synaptic currents, we measured evoked kainate EPSCs using stimulus trains of 3 pulses (100Hz, 10msec inter-stimulus interval) in the presence of a blocker cocktail containing the NMDA receptor antagonist APV, the GABAA receptor antagonist bicuculline, and the non-competitive AMPA receptor antagonist GYKI 53655 (Fig. 1A, C).
Across discrete stimulus intensities, we found that the charge carried by the GYKI-resistant, KAR-mediated synaptic responses were significantly increased in CIE neurons, but not in cells recorded from CON and WD slices (Fig 2B & C). Likewise, the amplitude of the GYKI-resistant KAR synaptic response was significantly greater in CIE neurons than in either CON or WD neurons at both the 25μA stimulus (CIE, 27.3±7.7pA, n=7; CON, 10.1±2.2pA, n=7; WD, 11.2±1.5 pA, n=8; P<0.05 with One-way ANOVA and Newman-Kuels post-test) and the 35μA stimulus intensity (CIE, 56.4±12.2pA; CON, 23.0±4.5pA; WD, 27.55±5.64pA; P<0.05 with One-way ANOVA and Newman-Kuels post-test). These data suggest that the KA receptor contribution to BLA synaptic responses following EC electrical stimulation is increased during CIE relative to CON and returns to CON levels during WD.
Figure 2. CIE and WD occlude ATPA induced synaptic plasticity in the BLA.
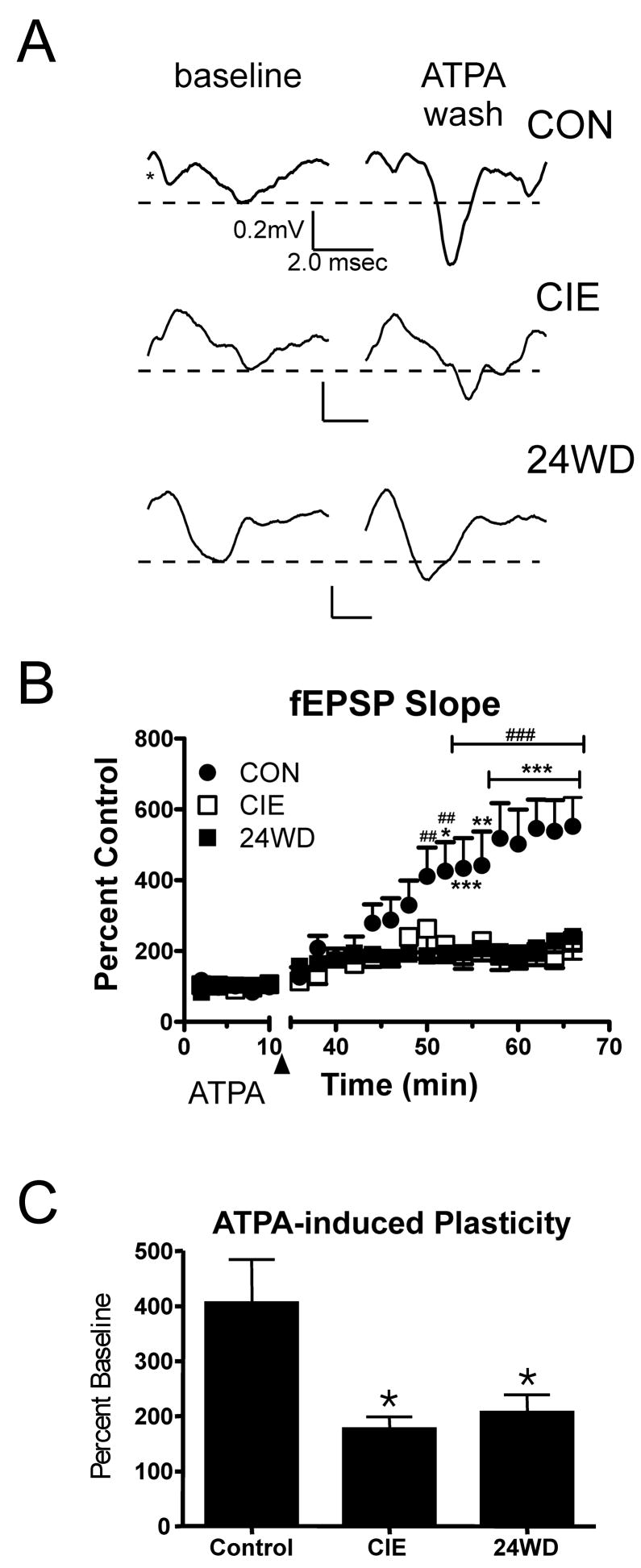
A, Exemplar fEPSP responses are from a single representative slice and averaged from 20 baseline traces (first 10 minutes) and the last 20 traces (last 10 minutes) of wash out. Traces are shown for each condition. Asterisk indicates the approximate position of the stimulation artifact that has been removed here for clarity. B, Time-course illustrates the significant attenuation of ATPA-induced increases in fEPSP slope measured in CIE (n=8) and WD slices (n=10) compared to CON (n=8). The arrowhead indicates the 10 minutes during which 5μM ATPA was perfused on the slices. fEPSP slope from each slice was normalized to the mean CON values measured during the 10min baseline period. * -- P< 0.05, **, ## -- P<0.01, ***, ### -- P<0.001; two-way ANOVA followed by Bonferoni’s test, CON relative to CIE (*) and WD(#). C, Summary of ATPA-induced facilitation of the fEPSP slope. fEPSP slope is expressed as a percent increase during the 10 minute baseline period measured during the last 10 minutes of the washout period. ATPA-induced increases in fEPSP slope were significantly attenuated by the CIE and WD treatments. * -- P<0.05, one-way ANOVA with Newman-Keuls posttest.
CIE AND WD occlude ATPA induced synaptic plasticity in the BLA
BLA KA-Rs have been shown to mediate a form of glutamatergic synaptic plasticity in principal neurons, potentially through recruitment of additional excitatory synapses (Li and Rogawski, 1998). We have recently shown that this synaptic plasticity, induced by the selective agonist ATPA, can be inhibited by both acute application of ethanol and the GluR5 competitive antagonist, UBP 296 ((RS)-1-(2-Amino-2-carboxyethyl)-3-(2-carboxybenzyl)pyrimidine-2,4-dione; (Lack et al., 2008)). Given that CIE, but not WD, increased KA-R synaptic function, we hypothesized that ATPA-induced plasticity would be more readily established in CIE slices relative to both CON and WD. Following a 10min baseline, we applied 5μM ATPA for 15min, followed with a 40 min washout period with drug-free aCSF. ATPA exposure caused a slowly developing increase in the fEPSP slope in CON slices (n=10, Fig. 2A & B). This is consistent with previous findings (Lack et al., 2008; Li et al., 2001). Surprisingly, this time-dependent, ATPA-induced plasticity (Fig. 2B) was significantly attenuated in slices from both CIE (n=8) and WD (n=10) with significant main effects of treatment (F=45.94, P<0.0001, two-way ANOVA) and time (F= 11.73, P<0.0001) with a significant interaction between variables (F=3.48, P<0.0001). Comparing these data from the baseline region (first 10 min) and the last 10min of washout (Fig. 2C), ATPA increased fEPSP slope in CON slices (406.3%±29.11%) but was significantly less effective in slices from both CIE (177.5%±21.33%, P<0.05, One-way ANOVA with Bonferroni’s posttest) and WD animals (207.4%±32.11%, n=10, P<0.05). These data indicate that ATPA-induced synaptic plasticity at EC-BLA glutamatergic synapses is partially occluded by CIE and WD. Since KA-R synaptic function is up-regulated in CIE slices (relative to control, Fig. 1), these data also suggest that the mechanisms involved with the expression of kainate receptor-dependent increases in synaptic function might be affected by these treatments.
CIE and WD increases field EPSP responses in the BLA
Previous studies have shown dramatic increases in glutamatergic function at local BLA synapses after CIE and WD (Lack et al., 2007). This suggests that CIE and WD might occlude ATPA-induced plasticity by engaging expression-associated mechanisms at EC-BLA synapses. To test this, we measured stimulus-response relationships for fEPSPs in BLA slices from CON-, CIE-, and WD- treated rats. We electrically stimulated the external capsule using a range of intensities, from 5μA to 40μA at intervals of 5μA (Fig. 3 A & B). fEPSP slope in CIE and WD slices significantly differed from CON at most stimuli >15μA (Fig. 3A) with two-way ANOVA indicating significant main effects of treatment (F=28.8, P<0.0001) and stimulation intensity (F=19.5, P<0.0001) but no interaction between these factors. These results are complementary to previous findings showing that there is increased AMPA-type glutamate receptor function in the BLA in response to chronic intermittent ethanol treatment and 24 hours of withdrawal (Lack et al., 2007). Importantly, the findings suggest that the occlusion of ATPA-induced plasticity is related to the increased synaptic responses in CIE and WD slices. These ethanol/withdrawal-dependent effects closely parallel the cellular responses that characterize activity-dependent synaptic plasticity in this brain region.
Figure 3. CIE and WD increase fEPSP responses to electrical stimulation of EC to BLA synapses.

A, Input/Output measurements of fEPSP slope versus stimulation intensity show significant increases after CIE and WD treatment across a range of intensities. B, Exemplar traces show the increase in fEPSP slope for a representative control, CIE and WD slice and are averaged from 20 baseline traces (first 10 minutes) and the last 20 traces (last 10 minutes) of wash out. (*, p< 0.05; **, p< 0.01, one-way ANOVA followed by Dunnett’s test, relative to control.)
Increased Synaptic Responses in CIE and WD BLA Does not Involve Pre-synaptic Release-related Mechanisms
fEPSPs represent action potentials within a population of neurons. However, we previously showed that CIE and WD can increase presynaptic function at local BLA glutamatergic synapses (Lack et al., 2007). To examine whether similar effects might be responsible for increased synaptic responsiveness at EC-BLA synapses, we carried out a paired pulse facilitation experiment to measure presynaptic release probability indirectly with postsynaptic responses to pairs of electrical stimuli. We used inter-pulse intervals of 25, 50 and 250msec to determine the ratio of the second synaptic response to the first across times that yield information about release probability (25 and 50msec; (Andreasen and Hablitz, 1994; Katz et al., 1993)) as well as autoreceptor-mediated decreases in presynaptic function (250msec; (Brucato et al., 1992)). We compared the ratios of the CIE and WD treated slices to those of control slices. We found no significant difference (one-way ANOVA) between paired pulse ratios in the CIE (n=10) or 24WD (n=10) neurons compared to CON (n=10) at any of the intervals tested (Fig. 4 A & B). This suggests that the increased response at EC to BLA synapses seen in CIE and WD with the fEPSP measures does not involve a significant change in presynaptic release probability.
Figure 4.

Increased excitation of EC to BLA synapses was not associated with a change in paired-pulse ratio (PPR). A, Representative traces of paired-pulse 50 responses for each treatment group (P>0.05, one-way ANOVA). B, Bar graphs summarizing the effect of CIE and WD on the amplitude of pairs of KA EPSCs evoked at an inter-pulse interval of 25, 50, and 250msec.
Discussion
Our research suggests that kainate receptors in the BLA, like NMDA and AMPA receptors, actively participate in the overall increase in glutamatergic function in response to chronic ethanol exposure. Specifically, chronic intermittent ethanol increases KA-R-mediated synaptic responses in the BLA. In fact, this ethanol-dependent increase in KA-R function during CIE might contribute to glutamatergic synaptic plasticity during either CIE or WD. This is consistent with previous findings that repeated ethanol withdrawal causes synaptic strengthening in the amygdala and occludes BLA long-term plasticity (Stephens et al., 2005). Saturation of LTP has been shown to impair fear learning (Moser et al., 1998) but leads to generalized increases in anxiety-like behavior. We have recently shown that BLA-dependent processes contribute to this withdrawal-associated anxiety (Lack et al., 2007). Therefore, occlusion of ATPA-induced synaptic plasticity by CIE and WD may suggest that KA-R mediated plasticity could contribute to withdrawal-related anxiety.
CIE, but not WD, increases KA-R synaptic function in the BLA
Previous research in our lab showed that BLA kainate receptors and kainate receptor-mediated synaptic plasticity are inhibited by acute ethanol (Lack et al., 2008). This suggests that the up-regulation of KAR-mediated synaptic responses in chronic ethanol-exposed neurons arises from this acute sensitivity. Although the mechanism responsible for this up-regulation is not currently known, it was apparently transient since KAR-mediated synaptic responses returned to baseline following twenty four hours of withdrawal. This rapid return would suggest that changes in gene or protein expression levels are less-likely potential mechanisms. Unfortunately, there is little guidance in the literature with respect to the effects of chronic ethanol/withdrawal on kainate receptors. Previous findings have been inconsistent and shown either no effect of chronic ethanol on kainate receptor subunit protein levels (Chandler et al., 1999; Ferreira et al., 2001) or up-regulation during withdrawal (Carta et al., 2002). These contrasting findings likely reflect the diversity of experimental approaches (e.g. native versus in vitro systems) and ethanol exposures.
The behavioral consequences of increased kainate receptor-mediated synaptic function during chronic intermittent ethanol exposure are not precisely defined. However, we have previously shown that anxiety-like behavior is not increased in CIE animals (Lack et al., 2007). This suggests two possible interpretations: 1) CIE-dependent increases in KAR function have no behavioral relevance or; 2) KAR-dependent contributions during CIE are not behaviorally manifest until withdrawal. Supporting this latter interpretation, ATPA microinjection into ethanol-naïve BLA increases anxiety-like behavior (Lack et al., 2008). Since AMPA-, NMDA- and KAR-dependent synaptic function is increased in both CIE and WD animals, the absence of any significant increase in anxiety-like behavior in CIE animals presumably reflects ethanol-sensitive contributions by other neurotransmitter systems.
CIE and WD occlude ATPA induced synaptic plasticity in the BLA
The KA-R agonist ATPA has been shown to increase synaptic strength in BLA principal neurons via an unknown, calcium-dependent mechanism (Li et al., 2001). However, in the CA1 region of the hippocampus, long-term ATPA application led to an enduring increase in the number of glutamatergic synapses in that region (Vesikansa et al., 2007). Using 5μM ATPA, a concentration that does not affect AMPA receptors (Stensbol et al., 1999), we were able to replicate the ATPA-induced synaptic plasticity in the current work. We have recently shown that this facilitation can be blocked by GluR5 antagonist UBP 296 and by acute ethanol (Lack et al., 2008). In the present study, both CIE and WD occluded ATPA-induced synaptic plasticity in the BLA. This was an unexpected finding considering the function of the KA-Rs was elevated in CIE neurons and went back to baseline during WD. One possible explanation is that extrasynaptic KAR, and not the synaptic receptors measured in Figure 1, are responsible for ATPA-induced plasticity. However, the initiation of synaptic plasticity at EC-BLA synapses by low-frequency electrical stimulation is dependent on synaptic activation of kainate receptors; and, this process parallels to ATPA-induced plasticity in this same brain region (Li et al., 2001). An alternative explanation is that KAR may be important only for the initiation of the synaptic plasticity at some undefined point during the chronic intermittent ethanol exposure. Regardless, we have recently shown that AMPA receptors are functionally up-regulated during both CIE and WD as well (Lack et al., 2007). Since AMPA receptors have been shown to maintain both NMDAR-dependent and NMDAR-independent LTP in the hippocampus (Grover, 1998) and in the BLA (Maren, 2005; McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997), our findings suggest that chronic intermittent ethanol engages the mechanisms related to the expression/maintenance of synaptic plasticity, ultimately resulting in the occlusion of ATPA-induced plasticity at the time of our measures. This hypothesis parallels previous findings showing repeated ethanol/withdrawal leading to the occlusion of NMDA-dependent forms of synaptic plasticity in the basolateral amygdala (Stephens et al., 2005).
CIE and WD increase fEPSP responses at EC to BLA synapses
An alternative explanation for the occlusion of ATPA-induced plasticity during CIE and WD would be that these treatments have down-regulated or blocked the mechanisms required for the expression of this type of ‘pharmacological LTP’. If this were the case, we would expect that the mechanisms associated with the expression of plasticity (e.g. AMPA receptors) would not be engaged by the treatments. However, our previous work has shown dramatic increases in AMPA receptor function after CIE and WD (Lack et al., 2007). Alternatively, the responses of BLA neurons themselves might be reduced during CIE and WD such that the increased AMPA receptor function was negated by reduced neuronal excitability. However, fEPSP responses measured following activation of the EC/BLA synapses were increased after CIE and WD. Thus, both glutamatergic synaptic transmission and the neurophysiological responses of BLA neurons to that transmission are increased during CIE and WD. These findings are inconsistent with any treatment-dependent inhibition of the mechanisms responsible for the expression of APTA-related plasticity.
CIE and WD do not affect presynaptic release probability at EC to BLA glutamatergic synapses
Because our ATPA-fEPSP data suggested that the mechanisms responsible for the expression of synaptic plasticity were engaged by CIE and WD, we examined whether there was any treatment-specific alteration in presynaptic release of glutamate. This hypothesis was supported by our previous data that showed an increase in glutamate release from ‘local’ glutamatergic synapses during CIE and WD (Lack et al., 2007). However, unlike at local glutamatergic synapses, we found no evidence of an increase in presynaptic probability of glutamate release at EC/BLA synapses following CIE and WD. The EC-BLA synapses contain cortical inputs for many divergent brain regions including insular cortices (McDonald and Mascagni, 1996), the neighboring entorhinal cortex (McDonald and Mascagni, 1997), and executive-control areas like the anterior cingulate (McDonald and Mascagni, 1996). The ‘local stimulus’ approach used in the Lack et al. 2007 study would have engaged these inputs as well as glutamatergic synapses arising from thalamus (LeDoux et al., 1991), hippocampus (Kishi et al., 2006), limbic and parietal cortex (McDonald and Mascagni, 1996), and intra-amygdala projections (Savander et al., 1995). The contrasting results found in the current study and in the Lack et al. 2007 study suggest that the neural circuitry contributing to distinct glutamatergic release pathways in the BLA might be differentially sensitive to chronic ethanol exposure and withdrawal.
Conclusions
In summary, the current work provides the first evidence of facilitation of kainate receptor synaptic function by chronic ethanol. This facilitation is contrasted with the occlusion of ATPA-mediated synaptic plasticity during both CIE and withdrawal. The overall increase in neurophysiologic responses during CIE and WD indicates that this occlusion results from a CIE/WD-dependent recruitment of the cellular mechanisms that govern the expression of synaptic plasticity within the BLA. This interpretation is supported by previous findings showing diminished activity-dependent synaptic plasticity in the BLA following alcohol exposure/withdrawal (Stephens et al., 2005). With respect to downstream efferent areas like the central amygdala and the bed nucleus of the stria terminalis, the precise ramifications of this alcohol-dependent recruitment are not yet know. However, we hypothesize that the increased responsiveness of BLA neurons (evidenced by the fEPSP data) might be manifested as increases in glutamatergic transmission in these areas as well. This hypothesis is consistent with the increased anxiety-like behavior following ethanol exposure/withdrawal (Lack et al., 2007). Likewise, the alcohol-dependent recruitment of mechanisms responsible for the expression of synaptic plasticity in the BLA is also consistent with the attenuation of amygdala-dependent fear learning following multiple withdrawals (Stephens et al., 2001). The present findings illustrate additional neurophysiological mechanisms by which chronic ethanol and withdrawal influence anxiety-related neural circuitry.
Acknowledgments
Funded by NIH/NIAAA awards: R56 AA014445 & R01 AA016671 (BAM), F31 AA016442 (AKL), F31 AA017576 (MRD), and T32 AA007565 (DTC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreasen M, Hablitz JJ. Paired-pulse facilitation in the dentate gyrus: a patch-clamp study in rat hippocampus in vitro. J Neurophysiol. 1994;72:326–336. doi: 10.1152/jn.1994.72.1.326. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Qashu F, Braga MF. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: implications for epilepsy and anxiety disorders. Amino Acids. 2007;32:305–315. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Borlikova GG, Le Merrer J, Stephens DN. Previous experience of ethanol withdrawal increases withdrawal-induced c-fos expression in limbic areas, but not withdrawal-induced anxiety and prevents withdrawal-induced elevations in plasma corticosterone. Psychopharmacology (Berl) 2006;185:188–200. doi: 10.1007/s00213-005-0301-3. [DOI] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1-and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology. 2005;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucato FH, Morrisett RA, Wilson WA, Swartzwelder HS. The GABAB receptor antagonist, CGP-35348, inhibits paired-pulse disinhibition in the rat dentate gyrus in vivo. Brain Res. 1992;588:150–153. doi: 10.1016/0006-8993(92)91355-i. [DOI] [PubMed] [Google Scholar]
- Carta M, Olivera DS, Dettmer TS, Valenzuela CF. Ethanol withdrawal upregulates kainate receptors in cultured rat hippocampal neurons. Neurosci Lett. 2002;327:128–132. doi: 10.1016/s0304-3940(02)00399-3. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Norwood D, Sutton G. Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcohol Clin Exp Res. 1999;23:363–370. [PubMed] [Google Scholar]
- Cohn TJ, Foster JH, Peters TJ. Sequential studies of sleep disturbance and quality of life in abstaining alcoholics. Addict Biol. 2003;8:455–462. doi: 10.1080/13556210310001646439. [DOI] [PubMed] [Google Scholar]
- De Olmos J, Alheid GF, Beltamino CA. Amygdala. In: Paxinos G, editor. The Rat Nervous System. San Diego: Academic Press; 1985. pp. 223–335. [Google Scholar]
- Ferreira VM, Frausto S, Browning MD, Savage DD, Morato GS, Valenzuela CF. Ionotropic glutamate receptor subunit expression in the rat hippocampus: lack of an effect of a long-term ethanol exposure paradigm. Alcohol Clin Exp Res. 2001;25:1536–1541. [PubMed] [Google Scholar]
- Funk CK, Koob GF. A CRF(2) agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res. 2007;1155:172–178. doi: 10.1016/j.brainres.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM. Evidence for postsynaptic induction and expression of NMDA receptor independent LTP. J Neurophysiol. 1998;79:1167–1182. doi: 10.1152/jn.1998.79.3.1167. [DOI] [PubMed] [Google Scholar]
- Jin XT, Pare JF, Raju DV, Smith Y. Localization and function of pre-and postsynaptic kainate receptors in the rat globus pallidus. Eur J Neurosci. 2006;23:374–386. doi: 10.1111/j.1460-9568.2005.04574.x. [DOI] [PubMed] [Google Scholar]
- Katz PS, Kirk MD, Govind CK. Facilitation and depression at different branches of the same motor axon: evidence for presynaptic differences in release. J Neurosci. 1993;13:3075–3089. doi: 10.1523/JNEUROSCI.13-07-03075.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Tsumori T, Yokota S, Yasui Y. Topographical projection from the hippocampal formation to the amygdala: a combined anterograde and retrograde tracing study in the rat. J Comp Neurol. 2006;496:349–368. doi: 10.1002/cne.20919. [DOI] [PubMed] [Google Scholar]
- Kliethermes CL. Anxiety-like behaviors following chronic ethanol exposure. Neurosci Biobehav Rev. 2005;28:837–850. doi: 10.1016/j.neubiorev.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Overstreet DH, Angel RA, Navarro M, Breese GR. The amygdala regulates the antianxiety sensitization effect of flumazenil during repeated chronic ethanol or repeated stress. Alcohol Clin Exp Res. 2007;31:1872–1882. doi: 10.1111/j.1530-0277.2007.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Ariwodola OJ, Chappell AM, Weiner JL, McCool BA. Ethanol inhibition of kainate receptor-mediated excitatory neurotransmission in the rat basolateral nucleus of the amygdala. Neuropharmacology. 2008;55:661–668. doi: 10.1016/j.neuropharm.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol. 2007;98:3185–3196. doi: 10.1152/jn.00189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Farb CR, Romanski LM. Overlapping projections to the amygdala and striatum from auditory processing areas of the thalamus and cortex. Neurosci Lett. 1991;134:139–144. doi: 10.1016/0304-3940(91)90526-y. [DOI] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology. 1998;37:1279–1286. doi: 10.1016/s0028-3908(98)00109-9. [DOI] [PubMed] [Google Scholar]
- Linnoila MI. Anxiety and alcoholism. J Clin Psychiatry. 1989;50(Suppl):26–29. [PubMed] [Google Scholar]
- Lucht M, Jahn U, Barnow S, Freyberger HJ. The use of a symptom checklist (SCL-90-R) as an easy method to estimate the relapse risk after alcoholism detoxification. Eur Addict Res. 2002;8:190–194. doi: 10.1159/000066131. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- McCool BA, Botting SK. Characterization of strychnine-sensitive glycine receptors in acutely isolated adult rat basolateral amygdala neurons. Brain Res. 2000;859:341–351. doi: 10.1016/s0006-8993(00)02026-6. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a Golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F. Cortico-cortical and cortico-amygdaloid projections of the rat occipital cortex: a Phaseolus vulgaris leucoagglutinin study. Neuroscience. 1996;71:37–54. doi: 10.1016/0306-4522(95)00416-5. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F. Projections of the lateral entorhinal cortex to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1997;77:445–459. doi: 10.1016/s0306-4522(96)00478-2. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Moser EI, Krobert KA, Moser MB, Morris RG. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 3. London: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Rainnie DG. Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J Neurophysiol. 1999;82:69–85. doi: 10.1152/jn.1999.82.1.69. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52:701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- Santucci AC, Cortes C, Bettica A, Cortes F. Chronic ethanol consumption in rats produces residual increases in anxiety 4 months after withdrawal. Behav Brain Res. 2008;188:24–31. doi: 10.1016/j.bbr.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Savander V, Go CG, LeDoux JE, Pitkanen A. Intrinsic connections of the rat amygdaloid complex: projections originating in the basal nucleus. J Comp Neurol. 1995;361:345–368. doi: 10.1002/cne.903610211. [DOI] [PubMed] [Google Scholar]
- Stensbol TB, Slok FA, Trometer J, Hurt S, Ebert B, Kjoller C, Egebjerg J, Madsen U, Diemer NH, Krogsgaard-Larsen P. Characterization of a new AMPA receptor radioligand, [3H]2-amino-3-(3-carboxy-5-methyl-4-isoxazolyl)propionic acid. Eur J Pharmacol. 1999;373:251–262. doi: 10.1016/s0014-2999(99)00269-1. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Brown G, Duka T, Ripley TL. Impaired fear conditioning but enhanced seizure sensitivity in rats given repeated experience of withdrawal from alcohol. Eur J Neurosci. 2001;14:2023–2031. doi: 10.1046/j.0953-816x.2001.01824.x. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Ripley TL, Borlikova G, Schubert M, Albrecht D, Hogarth L, Duka T. Repeated ethanol exposure and withdrawal impairs human fear conditioning and depresses long-term potentiation in rat amygdala and hippocampus. Biol Psychiatry. 2005;58:392–400. doi: 10.1016/j.biopsych.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- Verheul R, Lehert P, Geerlings PJ, Koeter MW, van den Brink W. Predictors of acamprosate efficacy: results from a pooled analysis of seven European trials including 1485 alcohol-dependent patients. Psychopharmacology (Berl) 2005;178:167–173. doi: 10.1007/s00213-004-1991-7. [DOI] [PubMed] [Google Scholar]
- Vesikansa A, Sallert M, Taira T, Lauri SE. Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J Physiol. 2007;583:145–157. doi: 10.1113/jphysiol.2007.133975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Dunwiddie TV, Valenzuela CF. Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. Mol Pharmacol. 1999;56:85–90. doi: 10.1124/mol.56.1.85. [DOI] [PubMed] [Google Scholar]
- West PJ, Dalpe-Charron A, Wilcox KS. Differential contribution of kainate receptors to excitatory postsynaptic currents in superficial layer neurons of the rat medial entorhinal cortex. Neuroscience. 2007;146:1000–1012. doi: 10.1016/j.neuroscience.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Weiss F, Zorrilla EP. Remission and resurgence of anxiety-like behavior across protracted withdrawal stages in ethanol-dependent rats. Alcohol Clin Exp Res. 2007;31:1505–1515. doi: 10.1111/j.1530-0277.2007.00456.x. [DOI] [PubMed] [Google Scholar]