

Abstract
The human cytomegalovirus (HCMV)-encoded viral G protein-coupled receptor pUS28 contributes to an array of biological effects, including cell migration and proliferation. Using FIX-BAC (bacterial artificial chromosome, derived from the HCMV clinical isolate VR1814) and lambda red recombination techniques, we generated HCMV recombinants expressing amino-terminally FLAG-tagged versions of wild-type pUS28 (FLAG–US28/WT), G-protein coupling deficient pUS28 (FLAG–US28/R129A) and chemokine-binding domain deficient pUS28 (FLAG–US28/ΔN). Infection with the FLAG–US28/R129A virus failed to induce inositol phosphate accumulation, indicating that G-protein coupling is essential for pUS28 signalling to phospholipase C-β (PLC-β) during HCMV infection. The FLAG–US28/ΔN virus induced about 80 % of the level of PLC-β signalling induced by the FLAG–US28/WT virus, demonstrating that the N-terminal chemokine-binding domain is not required for pUS28-induced PLC-β signalling in infected cells. The data presented here are the first to describe the functional analyses of several key pUS28 mutants in HCMV-infected cells. Elucidating the mechanisms by which pUS28 signals during infection will provide important insights into HCMV pathogenesis.
INTRODUCTION
The human cytomegalovirus (HCMV) encodes fourteen putative G protein-coupled receptors (GPCRs), which constitute about 7 % of its genome, implying that these receptors serve an important role in HCMV biology (Browne et al., 1992; Chee et al., 1990; Rigoutsos et al., 2003; Welch et al., 1991). One of these GPCRs, pUS28, stimulates smooth muscle cell migration and promotes cellular proliferation, but is not required for viral replication in vitro (Bodaghi et al., 1998; Maussang et al., 2006; Streblow et al., 1999; Vieira et al., 1998). Rodent cytomegaloviruses encode the related GPCRs, pM33 and pR33, which like HCMV pUS28 are not essential for viral replication in vitro (Beisser et al., 1998; Davis-Poynter et al., 1997). However, animal studies indicate that the pM33 and pR33 GPCRs are critical for viral dissemination in vivo (Beisser et al., 1998; Davis-Poynter et al., 1997). It is therefore likely that pUS28 plays a similar role in viral dissemination within an infected human host. More detailed analyses of the biological and signalling activities of pUS28 are required to gain a better understanding regarding how this protein contributes to viral pathogenesis.
pUS28 exhibits significant signalling activity, including the ability to activate the phospholipase C-β (PLC-β), the tyrosine kinase c-Src and the small G protein RhoA (Billstrom et al., 1998; Casarosa et al., 2001; Gao & Murphy, 1994; McLean et al., 2004; Melnychuk et al., 2004; Minisini et al., 2003; Streblow et al., 2003; Waldhoer et al., 2002). pUS28 shares significant homology to C-C chemokine receptors and, accordingly, can bind C-C chemokines, including CCL5/RANTES, CCL2/MCP-1 and CCL4/MIP-1β (Bodaghi et al., 1998; Kledal et al., 1998; Kuhn et al., 1995; Neote et al., 1993). Interaction of C-C chemokines with pUS28 requires the presence of a 6 aa segment between amino acids 11 and 16, as pUS28 mutants deleted for this region are unable to bind chemokine (Casarosa et al., 2005). Although these chemokines bind with high affinity to pUS28, their roles in signalling remain unclear as some pUS28 signalling pathways appear to be chemokine-dependent, while others appear to be chemokine-independent (Billstrom et al., 1998; Casarosa et al., 2001; Melnychuk et al., 2004; Minisini et al., 2003; Streblow et al., 2003). HCMV-infected cells secrete CCL5/RANTES and CCL2/MCP-1, which further complicates issues regarding chemokines and pUS28 signalling activity, as the secreted chemokines could bind to pUS28 and activate it in an autocrine manner (Bodaghi et al., 1998; Michelson et al., 1997; Randolph-Habecker et al., 2002; Taylor & Bresnahan, 2006). Thus, it remains unclear if the chemokine-independent signalling activity exhibited by pUS28 in HCMV-infected cells is truly due to pUS28 alone or is the result of an interaction between pUS28 and a secreted chemokine.
The current understanding of the pUS28 domains required for signalling comes from studies using transient over-expression systems to express pUS28 mutants (Casarosa et al., 2003, 2005; Miller et al., 2003; Mokros et al., 2002; Waldhoer et al., 2003). pUS28 activity has therefore been assessed by overexpressing pUS28 mutants in the absence of an HCMV infection. However, pUS28 activity may be influenced by viral or cellular gene products (e.g. host cell chemokines) expressed in HCMV-infected cells (Bodaghi et al., 1998; Michelson et al., 1997; Randolph-Habecker et al., 2002; Streblow et al., 1999). Additionally, it is still unclear how the expression levels of pUS28 in many heterologous systems compare with those in HCMV-infected cells and could represent non-physiological levels of pUS28 expression. It is now appreciated that over-expression of signalling proteins has led to spurious conclusions about their functions; it is therefore important to study the effects of pUS28 mutations at physiologically relevant cellular concentrations.
The experiments reported here use HCMV viral recombinants expressing mutant pUS28 proteins to investigate the role of G-protein coupling (using the FLAG–US28/R129A recombinant virus) and chemokine-binding (using the FLAG–US28/ΔN recombinant virus) in pUS28-mediated signal transduction in HCMV-infected cells.
METHODS
Cell culture
Human foreskin fibroblasts (HFF) and embryonic lung fibroblasts (MRC-5) were purchased from ATCC and maintained in Dulbecco’s modified Eagle’s medium (Mediatech) supplemented with 10 % Fetal Clone III serum (HyClone) and 1 % penicillin/streptomycin (Mediatech). Cells were grown at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2 and were used between passages 3 and 20.
Construction of HCMV US28 recombinants
Recombineering at the US28 locus was performed using the red recombinase plasmid pKD46 and the Flp recombinase plasmid pCP20 as described previously (Datsenko & Wanner, 2000). Escherichia coli harbouring HCMV FIX-BAC (bacterial artificial chromosome) was obtained from G. Hahn (Hahn et al., 2002). For ΔUS28 FIX-BAC, PCR primers with homology to the UTRs of US28 were used to amplify an FLP recognition target (FRT)-flanked kanamycin (Kan) resistance gene, the PCR fragment was recombined into parental FIX-BAC and screened for recombination by growth in Kan. For FLAG–US28/ΔN FIX-BAC, the Kan cassette was first removed from ΔUS28 FIX-BAC using the Flp recombinase. Primers with homology to untranslated regions (UTRs) of US28 were then used to amplify FLAG–US28/ΔN, with an FRT–KAN–FRT cassette following the stop codon, and this PCR fragment was recombined into ΔUS28 FIX-BAC. Recombination was screened by growth in Kan and the FRT–KAN–FRT cassette was later removed using the Flp recombinase. For FLAG–US28/WT and FLAG–US28/R129A, BACs were manipulated using a two-step method. In step one, PCR primers with homology to the UTRs of the US28 were used to amplify a cassette containing a Kan resistance gene and LacZ fragment which was then recombined into FIX-BAC to knockout US28. Recombinants were selected based on Kan resistance and blue colour upon exposure to X-Gal. In the second step, PCR products containing FLAG–US28/WT or FLAG–US28/R129A were recombined into the US28 locus, removing the Kan/LacZ cassette. Recombinants were selected by Kan sensitivity and white colour when plated on X-Gal-containing medium. Two independent recombinant BACs were made for each mutant used in this study. Recombination at the US28 locus was verified by PCR of BAC DNA isolated from E. coli using a standard mini-prep and amplified using primer sets with homology external to the site of recombination at the US28 locus, within the FLAG sequence or the UL146 gene. The sequences of each recombinant, including the US28 locus and regions surrounding the recombination site, were confirmed by automated ABI DNA sequencing (University of Cincinnati). For reconstitution of recombinant viruses, 2 × 105 MRC-5 cells were plated in six-well plates and transfected with 2 μg FIX-BAC DNAs using either SuperFect (Qiagen) or Transit IT (Mirus) lipid transfection reagents according to the manufacturer’s protocol. Ten days post-transfection, MRC-5 cells were transferred to T-25 flasks and fed with fresh medium every 3–4 days. After the appearance of virus-associated cytopathic effects (CPE), infected MRC-5 cells were mixed with uninfected HFFs and cultured until the CPE reached 100 %. Supernatants containing recombinant viruses were used for the generation of virus stocks.
Inositol phosphate accumulation
HFFs were seeded in 12-well plates at a density of 1.5 × 105 cells per well and either mock-infected or infected with HCMV recombinants at an m.o.i. of 0.03, 0.1, 0.3, 1 or 3. Twenty-four hours post-infection, virus-containing medium was removed and replaced with serum-free modified Eagle’s medium (MEM; Mediatech) containing 1 μCi ml−1 (74 kBq ml−1) of [3H]myoinositol (PerkinElmer Life Sciences). Forty-eight hours post-infection, medium was removed and replaced with serum-free medium containing 20 mM LiCl for 2 h. Reactions were stopped by aspirating medium, adding 1 ml of 0.4 M perchloric acid, and cooling undisturbed at 4 °C for 5 min. Supernatant (800 μl) was neutralized with 400 μl of 0.72 M KOH/0.6 M KHCO3 and subjected to centrifugation. Supernatant (1 ml) was diluted with 3 ml distilled H2O and applied to freshly prepared Dowex columns (AG1-X8; Bio-Rad). Columns were washed two times with distilled H2O; total inositol phosphates were eluted with 4.0 ml of 0.1 M formic acid, 1 M ammonium formate and eluates containing accumulated inositol phosphates were counted in a liquid scintillation counter. Neutralized supernatant (50 μl) was counted in a liquid scintillation counter to measure total incorporated [3H]myoinositol. Data are expressed as accumulated inositol phosphate over total incorporated [3H]myoinositol.
Immunoprecipitation and Western blotting
HFFs were seeded in 100 mm dishes at a density of 2.0 × 106 cells per plate and either mock-infected or infected with HCMV recombinants at an m.o.i. of 3. Forty-eight hours post-infection, cells were lysed in 1 ml RIPA buffer [150 mM NaCl, 10 mM Tris, 5 mM EDTA, 0.1 % SDS, 1.0 % DOC, 1.0 % Triton X-100 and Complete protease inhibitors (Roche)]. Clarified lysate was saved as whole-cell extracts (50 μl) or incubated with anti-FLAG M2-agarose beads (Sigma) to immunoprecipitate pFLAG–US28 proteins. Beads were washed twice with lysis buffer and eluted using 50 μl Laemmli sample buffer. Whole-cell extracts or FLAG immunoprecipitates were separated by SDS-PAGE and subjected to Western blotting using antibodies directed against the FLAG epitope (sc-805; Santa Cruz), HCMV IE proteins (MAB810; Chemicon) or HCMV pUL44 (a kind gift from John D. Shanley, University of Connecticut, CT). Reactive proteins were detected using the appropriate secondary antibodies in an enhanced chemiluminescence system (ECL; Amersham Biosciences).
Radioligand binding
HFFs were seeded in 12-well plates at a density of 1.5 × 105 cells per well and either mock-infected or infected with HCMV recombinants at an m.o.i. of 3. Forty-eight hours post-infection, cells were pre-incubated in the absence or presence of 14 nM unlabelled CCL5/RANTES for 15 min in ice-cold binding buffer (50 mM HEPES, 1 mM CaCl2, 5 mM MgCl2 and 0.5 % BSA). 125I-labelled CCL5/RANTES (Perkin Elmer) was then added to a final concentration of 28 pM and incubated for 3 h at 4 °C. Cells were washed three times in ice-cold binding buffer supplemented with 500 mM NaCl, lysed in 500 mM NaOH and specific binding was evaluated using a liquid scintillation counter.
FACS analysis of cell surface expression
HFFs were seeded in 12-well plates at a density of 1.5 × 105 cells per well and either mock-infected or infected with HCMV recombinants at an m.o.i. of 3. Forty-eight hours post-infection, cells were dislodged by trypsinization, washed twice in ice-cold PBS and stained for 2 h at 4 °C in M2-biotin (Sigma) diluted 1: 100 in FACS staining buffer (PBS supplemented with 0.5 % BSA). Cells were washed twice with ice-cold PBS and stained for 2 h at 4 °C in streptavidin-phycoerythrin (PE) (BD biosciences) diluted 1: 100 in FACS staining buffer. Cells were washed twice with ice-cold PBS and analysed using the FL2 channel on a FACSCalibur flow cytometer (BD Biosciences).
RESULTS
Construction of US28/R129A and US28/ΔN mutant viruses
In order to investigate the pUS28 domains involved in signalling at physiological levels in the context of an HCMV infection, HCMV FIX-BAC containing the genome of the clinical isolate VR1814 was used to create site-directed mutants of US28 by lambda red-mediated recombination (Borst et al., 1999; Britt et al., 2004; Datsenko & Wanner, 2000; Hahn et al., 2002; Oppenheim et al., 2004; Wagner & Koszinowski, 2004). Using this methodology, we constructed ΔUS28, FLAG–US28/WT, FLAG–US28/R129A and FLAG–US28/ΔN recombinant viruses. pUS28/R129A and pUS28/ΔN mutants are particularly informative as transient over-expression studies demonstrate that they fail to activate G-proteins and bind chemokines, respectively. US28 was knocked out by inserting an FRT–Kan–FRT cassette into the US28 locus, creating ΔUS28 FIX-BAC (Fig. 1, top panel). The Kan cassette was excised using the Flp recombinase and this construct was used to create FLAG–US28/ΔN FIX-BAC, which lacks amino acids 2–16 (Fig. 1, middle panel). To create FLAG–US28/WT and FLAG–US28/R129A FIX-BACs, US28 was first knocked out by recombining a Kan/LacZ cassette into the US28 locus. After confirmation of the Kan/LacZ insertion, the cassette was then replaced by recombination with either FLAG–US28/WT or FLAG–US28/R129A sequences (Fig. 1, lower panel).
Fig. 1.
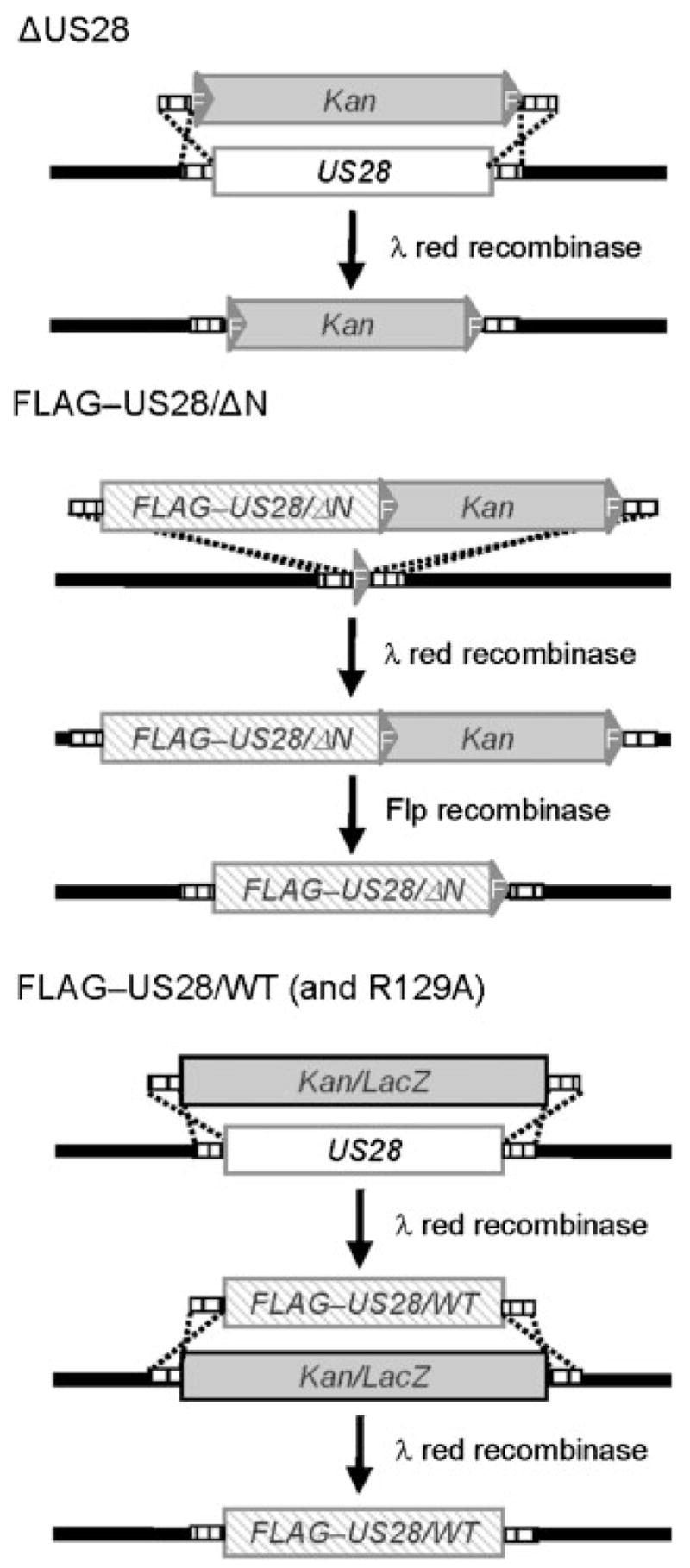
Deletion and modification of US28 in the HCMV genome. A schematic representation of strategies used to manipulate US28 in the FIX-BAC genome is illustrated. Lambda phage red recombination was used to delete the US28 gene (ΔUS28, upper panel), introduce a FLAG-tagged US28 gene deleted for the chemokine binding domain located between amino acids 2 and 16 (FLAG–US28/ΔN, middle panel), introduce a FLAG-tagged wild-type US28 gene (FLAG–US28/WT, lower panel), or introduce a FLAG-tagged US28 gene containing a R129A mutation in the G-protein coupling motif (FLAG–US28/R129A, lower panel). The Kan or Kan/LacZ selectable markers were removed from the final constructs with lambda phage red or Flp recombinases as indicated.
BAC DNA was isolated from E. coli and analysed by PCR for the expected recombination events using primers homologous to the 5′ and 3′ UTRs of US28 (Fig. 2, top panel). Each of the recombinant-generated PCR product is consistent with the predicted size for each mutant. The recombinant FIX-BACs were then subjected to additional PCR analyses using primer pairs that specifically amplify recombinants containing the FLAG epitope (Fig. 2, middle panel) or amplify the UL146 region as a positive control (Fig. 2, lower panel). Each of the FLAG epitope-containing FIX-BACs generated the expected PCR product with the FLAG-specific primers, and all FIX-BACs generated the expected fragment for the UL146 region. Next, the entire US28 region in each recombinant was verified by sequencing and the overall integrity of the FIX-BACs was monitored by restriction analyses (data not shown). Each HCMV-FIX recombinant was reconstituted by transfecting FIX-BAC DNAs into MRC-5 fibroblasts, and the resulting supernatants were used to create virus stocks in human foreskin fibroblasts (HFFs) (Borst et al., 1999). Virus stocks were titred using a standard plaque assay and it was determined that each mutant grew with similar kinetics to the parental HCMV-FIX (data not shown). We have engineered two completely independent clones for each US28 variant to confirm that the phenotypes observed are due to the targeted US28 mutation and not due to second site mutations elsewhere in the genome.
Fig. 2.

PCR analyses of HCMV US28 recombinants. FIX-BAC DNAs isolated from E. coli were used as templates in all PCR reactions. Primers with homology to 5′and 3′sequences flanking the US28 coding region were used to amplify the US28 locus (upper panel) by PCR. The primer with homology to the 5′ flanking sequence was used in combination with a FLAG-specific primer to verify the addition of an N-terminal FLAG epitope (middle panel). Amplification of the UL146 gene serves as a positive PCR control (lower panel).
Characterization of pFLAG–US28/R129A and pFLAG–US28/ΔN expression and chemokine binding in HCMV-infected cells
With the recombinant viruses in hand, we characterized the expression and chemokine-binding profiles of pFLAG–US28/WT, pFLAG–US28/R129A and pFLAG–US28/ΔN in HCMV-infected cells. The incorporation of the N-terminal FLAG epitope into pUS28 allows us to easily analyse the kinetics of US28 expression and enables us to determine if mutant proteins such as pFLAG–US28/R129A and pFLAG–US28/ΔN are expressed at levels equivalent to pFLAG–US28/WT. There is some discrepancy in the literature regarding the kinetics of US28 expression, as RT-PCR analyses suggest that it may be expressed as early as 4 h post-infection, while Northern and Western blot analyses suggest that US28 is expressed beginning 24 and 48 h post-infection (Bodaghi et al., 1998; Boomker et al., 2006; Minisini et al., 2003; Mokros et al., 2002; Zipeto et al., 1999). We infected HFFs with the FLAG–US28/WT recombinant virus at an m.o.i. of 3 and harvested protein extracts at 6, 12, 24 and 48 h post-infection. pFLAG–US28/WT was immunoprecipitated from infected cell extracts using anti-FLAG agarose beads and analysed by Western blot with FLAG polyclonal antibody. We included the immunoprecipitation step prior to Western blot analyses to enrich for pUS28 protein. In our experiments, pFLAG–US28/WT protein could be detected as early as 6 h post-infection and rapidly increases in quantity between 24 and 48 h post-infection (Fig. 3). US28 expression reaches maximal levels between 48 and 72 h post-infection (data not shown). For reference, whole-cell extracts were analysed by Western blot for immediate early and early proteins (IE1/IE2 and pUL44) (Fig. 3) (Loh et al., 1999). Therefore, while a small fraction of US28 may be expressed with immediate-early kinetics at 6 and 12 h post-infection, the majority of pUS28 protein is expressed with early kinetics at 24–48 h post-infection as observed previously (Mokros et al., 2002).
Fig. 3.

Kinetics of pUS28 expression in HCMV FLAG–US28/WT-infected cells. The pFLAG–US28 protein was immunoprecipitated from HCMV FLAG–US28/WT-infected HFFs (using anti-FLAG M2 agarose beads) and analysed by Western blotting using an α-FLAG polyclonal antibody. Overexposure of the blot enables pFLAG–US28 expression to be detected at 6 h post-infection (upper panels). Whole-cell lysates from the same time points were separated by SDS-PAGE and analysed by Western blotting using an α-IE1/IE2 antibody (lower middle panel) or an α-UL44 antibody (lower panel). Results shown are representative of three independent experiments.
Each recombinant virus was used to infect HFFs at an m.o.i. of 3 and pUS28 expression was analysed at 48 h post-infection as described above (Fig. 4a, upper panel). pFLAG–US28/WT and pFLAG–US28/R129A exhibit identical levels of expression and run at the same molecular mass by SDS-PAGE, while pFLAG–US28/ΔN is expressed at a similar level but runs at a lower molecular mass. pFLAG–US28/ΔN was expected to migrate faster since it is deleted for amino acids 2–16. Surprisingly, pFLAG–US28/WT and pFLAG–US28/R129A appear as doublets, while pFLAG–US28/ΔN migrates as a single band. This suggests that pUS28 may be post-translationally modified at the amino terminus, or that the pUS28 amino terminus is required for modification somewhere else in the protein. This was not examined further, but may be the result of O-linked glycosylation, as proposed by Casarosa et al. (2005). Extracts prepared from cells infected with each mutant were analysed for IE1/2 expression as a control to demonstrate equivalent infection with each recombinant virus (Fig. 4a, lower panel).
Fig. 4.

CCL5/RANTES binding to pFLAG–US28/R129A and pFLAG–US28/ΔN in infected cells. (a) Immunoprecipitation followed by Western blotting was performed as described in Fig. 3 to detect expression of the various forms of pFLAG–US28 encoded by HCMV parent, HCMV FLAG–US28/WT, HCMV ΔUS28, HCMV FLAG–US28/R129A and HCMV FLAG–US28/ΔN (upper panel). Whole-cell lysates from the same samples were subjected to Western blotting using an α-IE1/IE2 antibody (lower panel). Results shown are representative of three independent experiments. (b) HFFs infected as described in (a) were incubated with 28 pM [125I]CCL5/RANTES in the absence or presence of 14 nM unlabelled RANTES to discriminate between specific and non-specific binding. The data shown represent specific binding of [125I]CCL5/RANTES as assessed by liquid scintillation chromatography and are derived from three independent experiments performed in duplicate. (c) HFFs were infected with HCMV ΔUS28, HCMV FLAG–US28/WT, HCMV FLAG–US28/R129A or HCMV FLAG–US28/ΔN for 48 h. Surface expression of pFLAG–US28 on infected cells was detected by staining with FLAG-specific M2-biotin, followed by streptavidin-PE and analysed by FACS. The histograms shown are representative of at least four independent experiments performed in duplicate.
The ability of each pUS28 mutant to bind to C-C chemokines during HCMV infection of HFFs was then assessed using [125I]CCL5/RANTES binding experiments. Uninfected cells and cells infected with the ΔUS28 virus exhibited negligible amounts of [125I]CCL5/RANTES binding, while cells infected with FLAG–US28/WT virus exhibited chemokine-binding equivalent to the parental HCMV-FIX virus (Fig. 4b). Cells infected with the FLAG–US28/R129A virus bound CCL5/RANTES at 51 % of the levels exhibited by the FLAG–US28/WT virus. In contrast, cells infected with the FLAG–US28/ΔN virus were completely defective in [125I]CCL5/RANTES binding. Importantly, the results with the pFLAG–US28/ΔN mutant in HCMV-infected cells are consistent with ligand binding experiments performed in transfected cells expressing pUS28/ΔN and indicate that amino acids 2–16 of pUS28 play an essential role in chemokine binding (Casarosa et al., 2003, 2005).
We next sought to investigate the ability of the pUS28 mutants to accumulate on the cell surface in HCMV-infected HFFs. Although pUS28 undergoes constitutive internalization and is largely localized to intracellular vesicles, we are able to detect cell surface expression of pFLAG–US28/WT using anti-FLAG antibodies in FACS experiments (Fig. 4c, upper panel). pFLAG–US28/R129A exhibited similar levels of cell surface expression (116±22 %) in comparison with pFLAG–US28/WT (Fig. 4c, middle panel). pFLAG–US28/ΔN, however, was partially defective in its ability to accumulate on the cell surface (16±5 %) in comparison with pFLAG–US28/WT (Fig. 4c, lower panel). Since pUS28 does undergo constitutive internalization, it is unclear if the decreased cell surface expression of pFLAG–US28/ΔN is due to faster internalization kinetics or perhaps becomes partially trapped as it traffics to the plasma membrane.
pUS28 signalling to PLC-β in HCMV-infected cells requires G-protein coupling, but not chemokine binding
pUS28 is a potent activator of PLC-β, resulting in high levels of inositol phosphate (InsP) accumulation; however, the domains in pUS28 required for this activity in HCMV-infected cells remain unknown.
Therefore, we utilized the HCMV-FIX US28 recombinant viruses to address issues regarding the influence of G-protein coupling and chemokine binding on pUS28-stimulated PLC-β signalling in HCMV-infected cells. HFFs were uninfected or infected at various m.o.i. with the parental HCMV-FIX or with the FLAG–US28/WT recombinant and PLC-β signalling was assessed at 48 h post-infection by measuring the accumulation of total inositol phosphates (InsP). The FLAG–US28/WT recombinant exhibited a dose-dependent induction of InsP accumulation indistinguishable from that of the parental virus (Fig. 5a). This indicated that the N-terminal FLAG epitope did not affect pUS28 signalling and we therefore utilized the FLAG–US28/WT recombinant as the control virus for all subsequent experiments. We also compared signalling in ΔUS28-infected cells with FLAG–US28/WT-infected cells (Fig. 5b) and, similar to the results of Minisini et al. (2003), we observed no InsP accumulation with the ΔUS28 virus. These data confirm that the InsP signalling we observe in HCMV-FIX-infected cells is solely due to pUS28 and provides the basis for our analyses of InsP signalling induced by the pFLAG–US28 mutants.
Fig. 5.
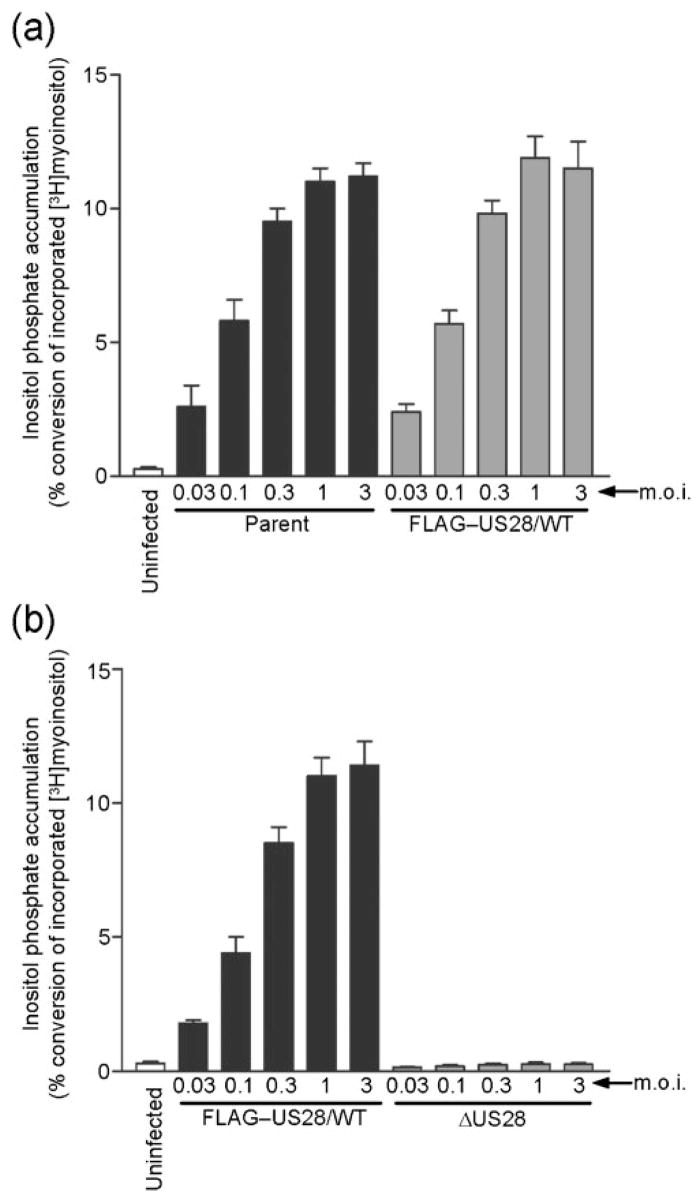
Characterization of PLC-βsignalling in cells infected with HCMV FLAG–US28/WT or HCMV ΔUS28. HFFs were infected with increasing m.o.i. (0.03 to 3.0 p.f.u. per cell) using HCMV parent or HCMV FLAG–US28/WT (a), and HCMV FLAG–US28/WT or HCMV ΔUS28 (b). Medium containing 1 μCi ml−1 (37 kBq ml−1) [3H]myoinositol was added at 24 h post-infection and accumulated InsPs were determined at 48 h post-infection using anion-exchange chromatography. The data represent four independent experiments performed in duplicate and are presented as a ratio of accumulated inositol phosphates to total 3H incorporation.
A highly conserved feature among G protein-coupled receptors is the presence of an aspartate-arginine-tyrosine (DRY) motif in the second intracellular loop. The DRY motif is necessary for the exchange of GDP for GTP on the Gα subunit of the heterotrimeric complex, and thus is critical for GPCR-mediated signalling. This highly conserved motif is present in many of the herpesviral GPCRs, including pUS28, and is necessary for pUS28 signalling to PLC-β in transfected cells (Pleskoff et al., 2005; Waldhoer et al., 2003). HFFs infected with FLAG–US28/WT or FLAG–US28/R129A viruses were assessed for pUS28-stimulated InsP signalling at 48 h post-infection (Fig. 6a). Cells infected with the FLAG–US28/R129A virus exhibited very low levels of InsP accumulation, indistinguishable from ΔUS28 infection at each m.o.i. tested (P<0.05). These results indicate that this conserved DRY motif is critical for pUS28 activation of PLC-β signalling in infected cells and indicate that pUS28 action is mediated by traditional, G protein-dependent events.
Fig. 6.

pUS28 requires the G-protein coupling motif, but not the chemokine binding domain for signalling through the PLC-β pathway in HCMV-infected cells. HFFs were infected with increasing m.o.i. (0.03 to 3.0 p.f.u. per cell) using HCMV FLAG–US28/WT or HCMV FLAG–US28/R129A (a), and HCMV FLAG–US28/WT or HCMV FLAG–US28/ΔN (b). Medium containing 1 μCi ml−1 [3H]myoinositol was added at 24 h post-infection and accumulated InsPs were determined at 48 h post-infection using anion-exchange chromatography. The data represent at least four independent experiments performed in duplicate and are presented as a ratio of accumulated inositol phosphates to total 3H incorporation.
Addition of exogenous CCL5/RANTES to HCMV-infected cells does not enhance pUS28-stimulated PLC-β activity, suggesting that pUS28 activates PLC-β constitutively (Minisini et al., 2003). However, HCMV-infected cells are known to produce CCL5/RANTES and CCL2/MCP-1, and therefore the potential effects of these chemokines in facilitating pUS28-induced PLC-β signalling in an autocrine fashion have not been directly examined (Bodaghi et al., 1998; Michelson et al., 1997; Randolph-Habecker et al., 2002). The chemokine-binding domain deficient FLAG–US28/ΔN recombinant virus provides an important reagent to directly examine the potential effects of chemokine binding on pUS28 signalling activity. HFFs were uninfected or infected with the FLAG–US28/ΔN virus and signalling was compared with cells infected with the FLAG–US28/WT virus at 48 h post-infection (Fig. 6b). Cells infected with the FLAG–US28/ΔN virus exhibited 43 % of the InsP accumulation induced by the FLAG–US28/WT virus at an m.o.i. of 0.03 (P<0.05) and 78 % of the InsP accumulation induced by the FLAG–US28/WT virus at an m.o.i. of 3.0 (P<0.05). These results indicate that, in infected cells, chemokine binding to pUS28 is not required for high levels of pUS28-stimulated signalling through the PLC-β pathway.
DISCUSSION
Our studies are the first to explore pUS28 function in HCMV-infected cells using recombinant viruses expressing site-directed mutants of pUS28. Using these recombinant viruses, we demonstrated that pUS28-induced PLC-β signalling in HCMV-infected cells is dependent on G-protein coupling, but not dependent on chemokine binding. Prior to the studies presented here, our understanding of the functional domains present in the pUS28 protein has come from experiments performed by transiently overexpressing pUS28 mutants in HCMV-negative cell lines (Casarosa et al., 2003, 2005; Miller et al., 2003; Pleskoff et al., 2005; Waldhoer et al., 2003). While these studies laid the foundation for understanding US28 function, they do not take into account the interaction of pUS28 with the rest of the viral gene products. Moreover, HCMV infection may cause physiological changes in the host cell such as altered expression of G-protein regulators, which in turn could potentially affect pUS28 signalling activity. Utilizing these recombinant viruses, our results are quite similar to those reported using heterologous expression systems, suggesting that other viral gene products do not in fact influence pUS28 signalling through the PLC-β pathway. However, future experiments may reveal differences in pUS28 activity between infection and overexpression studies when different pUS28-specific signalling pathways are analysed.
Our analysis of pUS28 wild-type and mutant proteins during HCMV infection allows us to correlate many important biological properties, including PLC-β signalling, chemokine binding and localization to the plasma membrane. Cells infected with the FLAG–US28/R129A virus failed to signal through PLC-β and bound 51 % of the CCL5/RANTES in comparison with the FLAG–US28/WT virus. FACS analysis revealed that pFLAG–US28/R129A and pFLAG–US28/WT are similarly expressed on the plasma membrane, indicating that both proteins should equally be exposed to chemokine. The modest decrease in chemokine binding observed in FLAG–US28/R129A-infected cells is likely due to a conformational change in pUS28/R129A, which alters the ligand affinity as has been observed for several GPCRs with mutations in their DRY box (Bennett et al., 2000; Chung et al., 2002; Rhee et al., 2000). pFLAG–US28/ΔN signalling to PLB-β remains quite robust (78 % compared with pFLAG–US28/WT at an m.o.i. of 3.0). However, pFLAG–US28/ΔN was completely defective in CCL5/RANTES binding, which is expected from studies reported in Casarosa et al. (2005). Interestingly, pFLAG–US28/ΔN is partially defective in its steady-state cell-surface expression in comparison with pFLAG–US28/WT. Therefore, we attribute the slight defect in pFLAG–US28/ΔN-induced PLC-β signalling to the reduced steady state levels of pFLAG–US28/ΔN compared with pFLAG–US28/WT.
The ability of pUS28 to signal so potently through PLC-β in the absence of chemokine binding is an important diversion from events occurring with most cellular GPCRs. While there are several reports of agonist independent signalling for cellular receptors, this signalling is usually far more conservative than that exhibited by pUS28 (Carroll et al., 2001; Quitterer et al., 1996; Seifert & Wenzel-Seifert, 2002). Although pUS28-dependent PLC-β signalling does not require chemokine-binding, several other pUS28-dependent signalling pathways, including RhoA and c-Src, do appear to require chemokine (Melnychuk et al., 2004; Minisini et al., 2003; Streblow et al., 2003). Our mutants will be particularly interesting in future studies aimed at understanding how pUS28 activates molecules such as RhoA and c-Src on a more mechanistic level. It is tempting to speculate that pUS28 may be coupled to Gq/PLC-β in a chemokine-independent manner and then switch to G12/RhoA in the presence of chemokine.
HCMV recombinants expressing mutant pUS28 proteins will allow us to continue to investigate pUS28 signalling in the context of viral infection and will facilitate the analyses of biological activities of pUS28 including cellular proliferation and migration.
Acknowledgments
We would like to thank G. Hahn for FIX-BAC DNA, B. Wanner for recombination plasmids and J. Shanley for UL44 antibody (Datsenko & Wanner, 2000; Hahn et al., 2002; Loh et al., 1999). We would also like to thank N. Sawtell, O. Schneider, J. Sherrill, J. Stringer, R. Thompson and T. Thompson for critical evaluation of this manuscript. This work was supported by an American Heart Association Predoctoral Fellowship 0615196B awarded to M. P. M. S. and by National Institutes of Health Grant R01 AI058159 and March of Dimes Grant 5-FY-04-17 awarded to W. E. M.
References
- Beisser PS, Vink C, Van Dam JG, Grauls G, Vanherle SJ, Bruggeman CA. The R33 G protein-coupled receptor gene of rat cytomegalovirus plays an essential role in the pathogenesis of viral infection. J Virol. 1998;72:2352–2363. doi: 10.1128/jvi.72.3.2352-2363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett TA, Maestas DC, Prossnitz ER. Arrestin binding to the G protein-coupled N-formyl peptide receptor is regulated by the conserved “DRY” sequence. J Biol Chem. 2000;275:24590–24594. doi: 10.1074/jbc.C000314200. [DOI] [PubMed] [Google Scholar]
- Billstrom MA, Johnson GL, Avdi NJ, Worthen GS. Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J Virol. 1998;72:5535–5544. doi: 10.1128/jvi.72.7.5535-5544.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodaghi B, Jones TR, Zipeto D, Vita C, Sun L, Laurent L, Arenzana-Seisdedos F, Virelizier JL, Michelson S. Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J Exp Med. 1998;188:855–866. doi: 10.1084/jem.188.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomker JM, Verschuuren EA, Brinker MG, de Leij LF, The TH, Harmsen MC. Kinetics of US28 gene expression during active human cytomegalovirus infection in lung-transplant recipients. J Infect Dis. 2006;193:1552–1556. doi: 10.1086/503779. [DOI] [PubMed] [Google Scholar]
- Borst EM, Hahn G, Koszinowski UH, Messerle M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J Virol. 1999;73:8320–8329. doi: 10.1128/jvi.73.10.8320-8329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt WJ, Jarvis M, Seo JY, Drummond D, Nelson J. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J Virol. 2004;78:539–543. doi: 10.1128/JVI.78.1.539-543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne H, Churcher M, Minson A. Identification, characterization and deletion analysis of HCMV gene products with homology to G protein-coupled receptors. The Seventeenth Annual International Herpesvirus Workshop; 1992. p. 275. [Google Scholar]
- Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, et al. BAY36–7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol. 2001;59:965–973. [PMC free article] [PubMed] [Google Scholar]
- Casarosa P, Bakker RA, Verzijl D, Navis M, Timmerman H, Leurs R, Smit MJ. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem. 2001;276:1133–1137. doi: 10.1074/jbc.M008965200. [DOI] [PubMed] [Google Scholar]
- Casarosa P, Menge WM, Minisini R, Otto C, van Heteren J, Jongejan A, Timmerman H, Moepps B, Kirchhoff F, et al. Identification of the first nonpeptidergic inverse agonist for a constitutively active viral-encoded G protein-coupled receptor. J Biol Chem. 2003;278:5172–5178. doi: 10.1074/jbc.M210033200. [DOI] [PubMed] [Google Scholar]
- Casarosa P, Waldhoer M, LiWang PJ, Vischer HF, Kledal T, Timmerman H, Schwartz TW, Smit MJ, Leurs R. CC and CX3C chemokines differentially interact with the N terminus of the human cytomegalovirus-encoded US28 receptor. J Biol Chem. 2005;280:3275–3285. doi: 10.1074/jbc.M407536200. [DOI] [PubMed] [Google Scholar]
- Chee MS, Satchwell SC, Preddie E, Weston KM, Barrell BG. Human cytomegalovirus encodes three G protein-coupled receptor homologues. Nature. 1990;344:774–777. doi: 10.1038/344774a0. [DOI] [PubMed] [Google Scholar]
- Chung DA, Wade SM, Fowler CB, Woods DD, Abada PB, Mosberg HI, Neubig RR. Mutagenesis and peptide analysis of the DRY motif in the alpha2A adrenergic receptor: evidence for alternate mechanisms in G protein-coupled receptors. Biochem Biophys Res Commun. 2002;293:1233–1241. doi: 10.1016/S0006-291X(02)00357-1. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Poynter NJ, Lynch DM, Vally H, Shellam GR, Rawlinson WD, Barrell BG, Farrell HE. Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J Virol. 1997;71:1521–1529. doi: 10.1128/jvi.71.2.1521-1529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JL, Murphy PM. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J Biol Chem. 1994;269:28539–28542. [PubMed] [Google Scholar]
- Hahn G, Khan H, Baldanti F, Koszinowski UH, Revello MG, Gerna G. The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol. 2002;76:9551–9555. doi: 10.1128/JVI.76.18.9551-9555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kledal TN, Rosenkilde MM, Schwartz TW. Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Lett. 1998;441:209–214. doi: 10.1016/s0014-5793(98)01551-8. [DOI] [PubMed] [Google Scholar]
- Kuhn DE, Beall CJ, Kolattukudy PE. The cytomegalovirus US28 protein binds multiple CC chemokines with high affinity. Biochem Biophys Res Commun. 1995;211:325–330. doi: 10.1006/bbrc.1995.1814. [DOI] [PubMed] [Google Scholar]
- Loh LC, Keeler VD, Shanley JD. Sequence requirements for the nuclear localization of the murine cytomegalovirus M44 gene product pp50. Virology. 1999;259:43–59. doi: 10.1006/viro.1999.9700. [DOI] [PubMed] [Google Scholar]
- Maussang D, Verzijl D, van Walsum M, Leurs R, Holl J, Pleskoff O, Michel D, van Dongen GA, Smit MJ. Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:13068–13073. doi: 10.1073/pnas.0604433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean KA, Holst PJ, Martini L, Schwartz TW, Rosenkilde MM. Similar activation of signal transduction pathways by the herpesvirus-encoded chemokine receptors US28 and ORF74. Virology. 2004;325:241–251. doi: 10.1016/j.virol.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Melnychuk RM, Streblow DN, Smith PP, Hirsch AJ, Pancheva D, Nelson JA. Human cytomegalovirus-encoded G protein-coupled receptor US28 mediates smooth muscle cell migration through Gα 12. J Virol. 2004;78:8382–8391. doi: 10.1128/JVI.78.15.8382-8391.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson S, Dal Monte P, Zipeto D, Bodaghi B, Laurent L, Oberlin E, Arenzana-Seisdedos F, Virelizier JL, Landini MP. Modulation of RANTES production by human cytomegalovirus infection of fibroblasts. J Virol. 1997;71:6495–6500. doi: 10.1128/jvi.71.9.6495-6500.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WE, Houtz DA, Nelson CD, Kolattukudy PE, Lefkowitz RJ. G-protein-coupled receptor (GPCR) kinase phosphorylation and beta-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J Biol Chem. 2003;278:21663–21671. doi: 10.1074/jbc.M303219200. [DOI] [PubMed] [Google Scholar]
- Minisini R, Tulone C, Luske A, Michel D, Mertens T, Gierschik P, Moepps B. Constitutive inositol phosphate formation in cytomegalovirus-infected human fibroblasts is due to expression of the chemokine receptor homologue pUS28. J Virol. 2003;77:4489–4501. doi: 10.1128/JVI.77.8.4489-4501.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokros T, Rehm A, Droese J, Oppermann M, Lipp M, Hopken UE. Surface expression and endocytosis of the human cytomegalovirus-encoded chemokine receptor US28 is regulated by agonist-independent phosphorylation. J Biol Chem. 2002;277:45122–45128. doi: 10.1074/jbc.M208214200. [DOI] [PubMed] [Google Scholar]
- Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- Oppenheim AB, Rattray AJ, Bubunenko M, Thomason LC, Court DL. In vivo recombineering of bacteriophage λ by PCR fragments and single-strand oligonucleotides. Virology. 2004;319:185–189. doi: 10.1016/j.virol.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Pleskoff O, Casarosa P, Verneuil L, Ainoun F, Beisser P, Smit M, Leurs R, Schneider P, Michelson S, Ameisen JC. The human cytomegalovirus-encoded chemokine receptor US28 induces caspase-dependent apoptosis. FEBS J. 2005;272:4163–4177. doi: 10.1111/j.1742-4658.2005.04829.x. [DOI] [PubMed] [Google Scholar]
- Quitterer U, AbdAlla S, Jarnagin K, Müller-Esterl W. Na+ ions binding to the bradykinin B2 receptor suppress agonist-independent receptor activation. Biochemistry. 1996;35:13368–13377. doi: 10.1021/bi961163w. [DOI] [PubMed] [Google Scholar]
- Randolph-Habecker JR, Rahill B, Torok-Storb B, Vieira J, Kolattukudy PE, Rovin BH, Sedmak DD. The expression of the cytomegalovirus chemokine receptor homolog US28 sequesters biologically active CC chemokines and alters IL-8 production. Cytokine. 2002;19:37–46. doi: 10.1006/cyto.2002.0874. [DOI] [PubMed] [Google Scholar]
- Rhee MH, Nevo I, Levy R, Vogel Z. Role of the highly conserved Asp-Arg-Tyr motif in signal transduction of the CB2 cannabinoid receptor. FEBS Lett. 2000;466:300–304. doi: 10.1016/s0014-5793(00)01094-2. [DOI] [PubMed] [Google Scholar]
- Rigoutsos I, Novotny J, Huynh T, Chin-Bow ST, Parida L, Platt D, Coleman D, Shenk T. In silico pattern-based analysis of the human cytomegalovirus genome. J Virol. 2003;77:4326–4344. doi: 10.1128/JVI.77.7.4326-4344.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Streblow DN, Soderberg-Naucler C, Vieira J, Smith P, Wakabayashi E, Ruchti F, Mattison K, Altschuler Y, Nelson JA. The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell. 1999;99:511–520. doi: 10.1016/s0092-8674(00)81539-1. [DOI] [PubMed] [Google Scholar]
- Streblow DN, Vomaske J, Smith P, Melnychuk R, Hall L, Pancheva D, Smit M, Casarosa P, Schlaepfer DD, Nelson JA. Human cytomegalovirus chemokine receptor US28-induced smooth muscle cell migration is mediated by focal adhesion kinase and Src. J Biol Chem. 2003;278:50456–50465. doi: 10.1074/jbc.M307936200. [DOI] [PubMed] [Google Scholar]
- Taylor RT, Bresnahan WA. Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J Virol. 2006;80:920–928. doi: 10.1128/JVI.80.2.920-928.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, Schall TJ, Corey L, Geballe AP. Functional analysis of the human cytomegalovirus US28 gene by insertion mutagenesis with the green fluorescent protein gene. J Virol. 1998;72:8158–8165. doi: 10.1128/jvi.72.10.8158-8165.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Koszinowski UH. Mutagenesis of viral BACs with linear PCR fragments (ET recombination) Methods Mol Biol. 2004;256:257–268. doi: 10.1385/1-59259-753-X:257. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Kledal TN, Farrell H, Schwartz TW. Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J Virol. 2002;76:8161–8168. doi: 10.1128/JVI.76.16.8161-8168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M, Casarosa P, Rosenkilde MM, Smit MJ, Leurs R, Whistler JL, Schwartz TW. The carboxyl terminus of human cytomegalovirus-encoded 7 transmembrane receptor US28 camouflages agonism by mediating constitutive endocytosis. J Biol Chem. 2003;278:19473–19482. doi: 10.1074/jbc.M213179200. [DOI] [PubMed] [Google Scholar]
- Welch AR, McGregor LM, Gibson W. Cytomegalovirus homologs of cellular G protein-coupled receptor genes are transcribed. J Virol. 1991;65:3915–3918. doi: 10.1128/jvi.65.7.3915-3918.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipeto D, Bodaghi B, Laurent L, Virelizier JL, Michelson S. Kinetics of transcription of human cytomegalovirus chemokine receptor US28 in different cell types. J Gen Virol. 1999;80:543–547. doi: 10.1099/0022-1317-80-3-543. [DOI] [PubMed] [Google Scholar]