

Summary
Integrin-mediated adhesion to substratum is required for cyclin D1 induction in mesenchymal cells, but we show here that the induction of cyclin D1 persists despite blockade of ECM-integrin signaling in MCF10A mammary epithelial cells. E-cadherin-mediated cell-cell adhesion also supports cyclin D1 induction in these cells, and the combined inhibition of both E-cadherin and integrin adhesion is required to prevent the expression of cyclin D1 mRNA and protein. Our previous studies described a pro-proliferative effect of E-cadherin in MCF10A cells, mediated by Rac, and we now show that Rac is required for cyclin D1 mRNA induction by both E-cadherin and integrin engagement. The levels of p21Cip1 and p27Kip1, Cdk inhibitors that are also targets of integrin signaling, are not affected by E-cadherin-mediated cell-cell adhesion. Finally, we show that the increased expression of cyclin D1 mRNA associated with E-cadherin-dependent cell-cell adhesion is causally linked to an increased entry into S phase. Our results identify Rac signaling to cyclin D1 as a crucial pro-proliferative effect of E-cadherin-mediated cell-cell adhesion.
Keywords: G1 phase, Cell cycle, Proliferation
Introduction
Progression through the cell cycle is controlled by the activity of cyclin-dependent kinases (Cdks) (Sherr, 1994; Sherr and Roberts, 1999). Cyclin D1 is the major D type cyclin in many cell types and is usually the first cyclin to be induced when cells enter G1 phase from quiescence (G0). Once expressed, cyclin D1 binds to Cdk4 or Cdk6 to form an active holoenzyme that phosphorylates the retinoblastoma 1 (Rb1) protein. Phosphorylation of Rb1 allows for the dissociation of associated E2Fs which then promote transcription of downstream E2F cell cycle targets including cyclin E and cyclin A. The cyclin-D1–Cdk4/6 complex also promotes G1 phase cell-cycle progression by titrating Cdk inhibitors, p21Cip1 and p27Cip1, and thereby contributing to the activation of cyclin-E–Cdk2 complexes that further phosphorylate Rb1. Tight control of cyclin D1 gene expression is therefore a crucial issue in the regulation of G1-phase progression.
In fibroblasts, the induction of cyclin D1 mRNA requires coordinated signaling by growth factor receptor tyrosine kinases (RTKs) and integrins. For example, in the presence of growth factors, integrin-mediated cell adhesion to the extracellular matrix (ECM) leads to a sustained activation of ERKs (also known as MAPKs) that is required for cyclin D1 gene expression (Welsh et al., 2001; Villanueva et al., 2007). RTKs and integrins also regulate the activation of Rac (RAC1), and integrin signaling additionally controls the coupling of Rac to its downstream targets (del Pozo et al., 2000). Although cyclin D1 is induced downstream of activated Rac (Joyce et al., 1999; Klein et al., 2007; Page et al., 1999), endogenous Rac signaling to cyclin D1 is not readily detected in fibroblasts because the pathway is inhibited by Rho (Welsh et al., 2001).
Epithelial cells have more complex adhesion systems than fibroblasts. In addition to integrin-mediated adhesion to the ECM, epithelial cells rely on adherens junctions for tissue integrity and function, and E-cadherin plays a major role in mediating these adherens junctions in many epithelial cell types. E-cadherin is a transmembrane protein that mediates cell-cell adhesion by calcium-dependent homophilic binding through its extracellular domain (Gumbiner, 1996). β-catenin binding to the cytoplasmic domain of E-cadherin acts as a link to the actin cytoskeleton (Drees et al., 2005; Knudsen et al., 1995; Nieset et al., 1997; Yamada et al., 2005). A current hypothesis suggests that cadherin-mediated binding of β-catenin may affect catenin-dependent transcription of LEF-regulated genes (Gottardi et al., 2001; Sadot et al., 1998). Interestingly, the cyclin D1 gene can be regulated by β-catenin and LEF (Shtutman et al., 1999; Tetsu and McCormick, 1999), raising the possibility that the formation of E-cadherin adherens junctions might control the expression of cyclin D1 by sequestering β-catenin. However, E-cadherin can also regulate Rac activity (Nakagawa et al., 2001; Noren et al., 2001; Liu et al., 2006) and therefore has the potential to regulate Rac-dependent induction of cyclin D1.
We recently reported that E-cadherin stimulates Rac-GTP loading and promotes cell proliferation in a Rac-dependent manner in MCF10A cells (Liu et al., 2006). However, the pro-proliferative target(s) of E-cadherin within the G1 phase cyclin-Cdk network remained undefined. We now describe the effects of E-cadherin on the G1-phase cyclins and Cdk inhibitors, link Rac signaling to cyclin D1 mRNA, and assess the relative effects of integrin-mediated cell-substratum adhesion and cadherin-mediated cell-cell adhesion on these events in MCF10A mammary epithelial cells. Our results reveal clear differences between the adhesion requirements for cyclin D1 gene expression and S-phase entry in mesenchymal and epithelial cells.
Results
Cyclin D1 gene expression persists in MCF10A cells despite loss of cell-substratum adhesion
Our initial studies examined the induction of cyclin D1 when quiescent MCF10A cells were stimulated with mitogens on dishes coated with collagen (monolayer cultures) or agarose (suspension cultures), the latter being an experimental condition that blocks integrin clustering and downstream signaling. Although cyclin D1 levels were reduced by blocking cell adhesion to the substratum, cyclin D1 was still induced relative to G0 in these suspended epithelial cells (Fig. 1A). Cell adhesion to ECM is thought to control cyclin D1 expression, at least in part, by regulating ERK activity (Assoian and Schwartz, 2001), but we found that cyclin D1 mRNA was induced in suspended MCF10A cells even though ERK activity was absent (Fig. 1B). These results are in striking contrast to our previous studies with human and mouse fibroblasts where blocking integrin-mediated adhesion to the substratum strongly inhibited cyclin D1 induction as well as the activation of ERKs (Bohmer et al., 1996; Zhu et al., 1996; Roovers et al., 1999; Welsh et al., 2001).
Fig. 1.
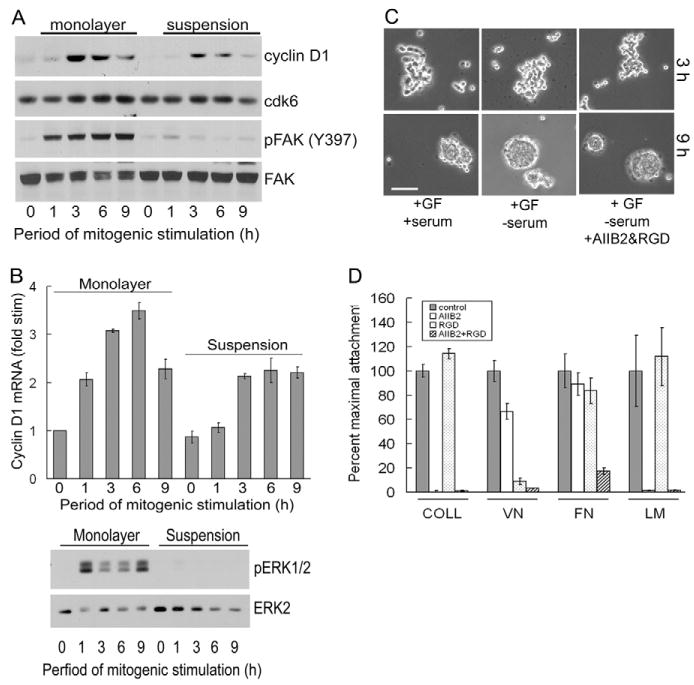
Blockade of integrin-mediated adhesion does not prevent cyclin D1 gene expression in MCF10A cells. (A) Serum-starved MCF10A cells were trypsinized, reseeded on dishes coated with collagen or agarose, and stimulated with 10% FBS and growth factor cocktail. Cell lysates were analyzed by western blotting for cyclin D1, Cdk6, phosphoY397-FAK, and FAK. (B) The experiment in A was repeated except that collected cells were analyzed for cyclin D1 mRNA by QPCR and western blotting using antibodies to dually phosphorylated ERK and total ERK. QPCR results show mean ± s.e.m. of two experiments after normalizing the levels of cyclin D1 mRNA to the level in serum-starved cells. (C) Quiescent MCF10A cells were preincubated in suspension with vehicle or AIIB2 β1-integrin-blocking antibody and RGD peptide. The cells were then plated on agarose-coated dishes and incubated for 3 and 9 hours with either 10% FBS and growth factor cocktail (GF), growth factor cocktail in serum-free medium, or growth factor cocktail in serum-free medium with AIIB2 and RGD. The suspended cells were collected by attachment to poly-L-lysine-coated coverslips and visualized by phase-contrast microscopy. Bar, 210 μm. (D) Attachment of MCF10A cells to collagen (COLL), vitronectin (VN), fibronectin (FN) or laminin-1 (LM) in the absence and presence of AIIB2 and/or RGD as described in the Materials and methods. The results are plotted as percentage maximal attachment relative to the positive control (no inhibitors) for each matrix protein.
Growth factor signaling was required for cyclin D1 induction in both the adherent and suspended MCF10A cells (Fig. 1A,B). Moreover, the autophosphorylation of FAK (also known as PTK2) at Y397 was strongly reduced by cell detachment in the suspended MCF10A cells (Fig. 1A), minimizing the possibility that residual integrin signaling was responsible for the cyclin D1 expressed in the suspended cells.
We noticed that MCF10A cells began to form loose aggregates after ∼3 hours in suspension and then proceeded to form spheroid-like structures by 9 hours (Fig. 1C, first column). The same effects were seen when the cells were incubated in serum-free medium (Fig. 1C, second column), indicating that serum-derived matrix proteins were not responsible for the aggregation. Nevertheless, we directly inhibited any residual integrin signaling by treating the cells with a β1-integrin-blocking antibody AIIB2 and the RGD peptide. The individual inhibitors had the largely expected effects on integrin-mediated adhesion, and the combination of both inhibitors abolished attachment to collagen, laminin, vitronectin and fibronectin (Fig. 1D). However, cell aggregation in suspension was not diminished by treatment with the integrin inhibitors (Fig. 1C, third column). Taken together, the results of Fig. 1 indicated that another adhesion system was responsible for the tight cell aggregation in suspended MCF10A cells, and that this system might also be responsible for the ERK-independent expression of cyclin D1.
E-cadherin- and integrin-mediated adhesion jointly regulate cyclin D1 gene expression in MCF10A cells
In addition to integrins, numerous cell-cell adhesion systems including cadherins may be responsible for the formation of spheroids. We therefore analyzed the MCF10A cell aggregates that formed in suspension culture for the expression and localization of E-cadherin and β-catenin by immunofluorescence microscopy. Both E-cadherin and β-catenin localized to the cell-cell interface (Fig. 2A). Thus, E-cadherin-mediated cell-cell adhesion was intact in the suspended cell aggregates. Knock-down of E-cadherin with small interfering RNA (siRNA) (Fig. 2B) inhibited the formation of tight spheroids in suspended MCF10A cells (Fig. 2C), and largely phenocopied the effect of disrupting cadherin-mediated adhesions by calcium chelation with EGTA (Fig. 2D). These results show that E-cadherin-mediated cell-cell adhesion is responsible, at least in large part, for the formation of spheroids in nonadherent MCF10A and suggest that E-cadherin might be responsible for the integrin-independent expression of cyclin D1.
Fig. 2.
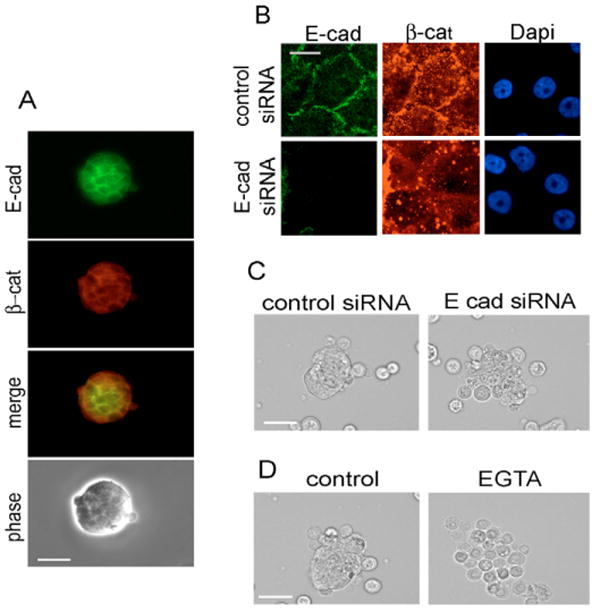
E-cadherin-mediated adhesion in suspended MCF10A cells. (A) Quiescent MCF10A cells were trypsinized, reseeded on dishes coated with agarose, stimulated with 10% FBS and growth factor cocktail for 9 hours, and analyzed by phase-contrast or epifluorescence microscopy for E-cadherin (E-cad) and β-catenin (β-cat). (B) MCF10A cells were transfected with control or E-cadherin siRNA, rendered quiescent by serum and growth factor starvation, trypsinized, reseeded on collagen-coated dishes, stimulated with 10% FBS and growth factor cocktail for 9 hours, and analyzed by epifluorescence microscopy for E-cadherin, β-catenin and DAPI-stained nuclei. (C) Cells prepared as in B were seeded on agarose-coated dishes with 10% FBS and growth factor cocktail for 9 hours, collected on poly-L-lysine-coated coverslips, and analyzed by phase-contrast microscopy. (D) Quiescent MCF10A cells were trypsinized and pre-incubated in suspension in the absence and presence of 2 mM EGTA (30 minutes at 37°C) in DMEM-F12 with 1 mg/ml heat-inactivated fatty acid-free BSA. The pre-treated cells were directly replated on agarose-coated dishes, and stimulated with 10% FBS and growth factor cocktail for 9 hours. The cells were then collected on poly-L-lysine-coated coverslips and analyzed by phase-contrast microscopy. Bars, 210 μm.
We transfected MCF10A cells with either control or E-cadherin siRNA and then cultured them in monolayer or suspension. Western blotting for E-cadherin or Y397-phosphorylated FAK confirmed that these treatments were effective inhibitors of E-cadherin and integrin signaling, respectively. Inhibition of E-cadherin signaling in monolayer cultures (Fig. 3A,B; E-cadherin siRNA; Mono) or integrin signaling in suspension cultures (Fig. 3A,B; control siRNA; Susp) partially inhibited the induction of cyclin D1 mRNA and protein, though the magnitude of the effects varied somewhat between experiments. By contrast, the inhibition of both E-cadherin and integrin signaling (knock-down of E-cadherin in suspended MCF10A cells) consistently resulted in a strong inhibition of cyclin D1 induction (Fig. 3A,B; E-cadherin siRNA in Susp). Similar results were obtained with two distinct E-cadherin siRNAs (supplementary material Fig. S1) and a dominant-negative E-cadherin adenovirus lacking the cytoplasmic domain (supplementary material Fig. S2). Interestingly, we did not see E-cadherin-dependent changes in the levels of p21 or p27, the Cdk inhibitors that are often targeted by extracellular signals in G1 phase (Fig. 3C). Moreover, ERK activity was not affected by knock-down of E-cadherin (Fig. 3C), indicating that cadherin induction of cyclin D1 operated through a distinct signaling pathway.
Fig. 3.
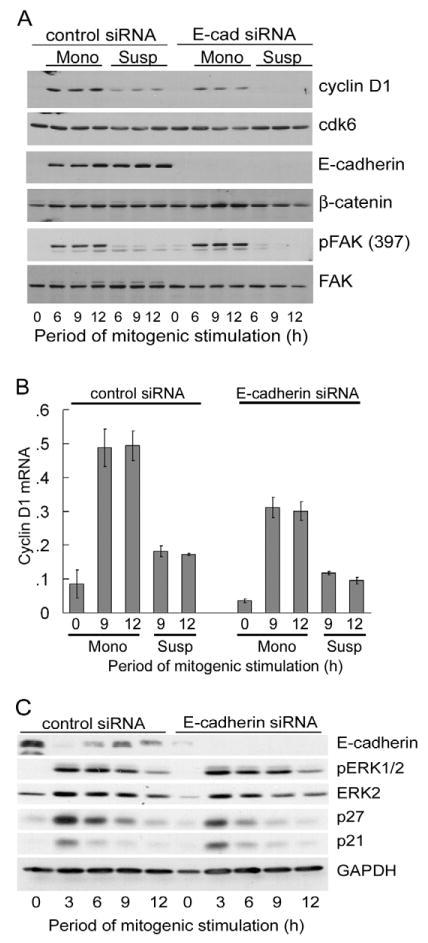
E-cadherin-mediated cell-cell adhesion and cell-substratum signaling coordinately regulate cyclin D1 expression in MCF10A cells. MCF10A cells were transfected with control or E-cadherin (E-cad) siRNA, rendered quiescent by serum and growth factor starvation, trypsinized, and reseeded in a monolayer (Mono) or in suspension (Susp) on collagen- or agarose-coated dishes, respectively. The cells were stimulated with 10% FBS and growth factor cocktail. (A) Samples were collected at the indicated time points and analyzed by western blotting for cyclin D1, Cdk6, E-cadherin, β-catenin, phosphoY397-FAK and FAK. (B) The experiment in A was repeated with cells seeded in monolayer (on collagen-coated dishes containing glass coverslips) (Liu et al., 2006) or in suspension (on agarose-coated dishes) at 2×104 cells/cm2. Collected cells were analyzed by QPCR for cyclin D1 mRNA. (C) Cells seeded on collagen-coated dishes and stimulated with 10% FBS and growth factor cocktail were collected and analyzed by western blotting for E-cadherin, p21Cip1, p27Kip1, dually phosphorylated ERK, total ERK and GAPDH (loading control).
We conclude that cyclin D1 is the major G1 phase pro-proliferative target of E-cadherin in MCF10A cells and that integrins and E-cadherins are each sufficient to induce cyclin D1. The combined inhibition of both adhesion systems results in the near complete loss of cyclin D1 mRNA and protein expression. Thus, E-cadherin- and integrin-mediated adhesion account for essentially all the adhesion dependency of cyclin D1 mRNA expression in MCF10A cells. Additionally, we found that knockdown of E-cadherin did not affect cyclin D1 protein stability in suspended MCF10A cells (Fig. 4A), nor could E-cadherin knockdown reduce the dose-dependent expression of an ectopically expressed cyclin D1 coding domain (Fig. 4B). Thus, the change in steady-state cyclin D1 protein levels associated with knock-down of E-cadherin largely reflects the stimulatory effect of E-cadherin on cyclin D1 mRNA.
Fig. 4.
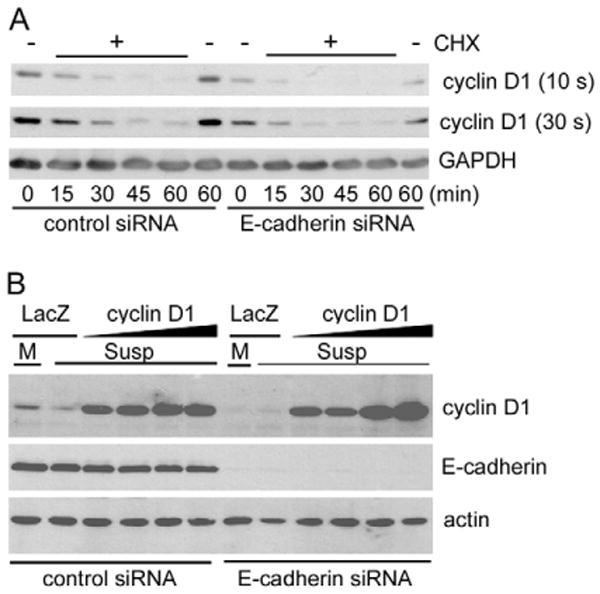
Cyclin D1 half-life is not regulated by E-cadherin. MCF10A cells were transfected with control or E-cadherin siRNA during serum and growth factor starvation. (A) The starved cells were trypsinized, reseeded in suspension on agarose-coated dishes and stimulated with 10% FBS and growth factor cocktail for 6 hours to induce cyclin D1. Cycloheximide (CHX) was then added, and the cells were collected every 15 minutes for 1 hour. Collected cells were analyzed by western blotting using anti-cyclin D1 and anti-GAPDH (loading control). The cyclin D1 blot was exposed for 10 and 30 seconds (s) so that all bands could be readily detected and easily compared between samples (e.g. compare 10 s ‘control siRNA’ with 30 s ‘E-cadherin siRNA’). (B) MCF10A cells were transfected with control or E-cadherin siRNA and then infected with β-galactosidase (LacZ) (20 MOI) or cyclin D1 adenoviruses (2.5-20 MOI) during serum and growth factor starvation. The cells were trypsinized, reseeded in monolayer [M; β-galactosidase control only] or suspension (Susp) on dishes coated with collagen or agarose, respectively. The cells were stimulated with 10% FBS and growth factor cocktail for 9 hours. Cell lysates were analyzed by western blotting for cyclin D1, E-cadherin, and actin (loading control).
E-cadherin-mediated cyclin D1 gene expression in MCF10A cells is dependent on Rac
Others have reported that E-cadherin-mediated adhesion stimulates Rac activity (Nakagawa et al., 2001; Noren et al., 2001). We recently reported that E-cadherin also stimulates the activation of Rac in MCF10A cells, and we linked this effect to E-cadherin stimulation of MCF10A cell proliferation (Liu et al., 2006). Since the results in Fig. 3 show that E-cadherin stimulates cyclin D1 mRNA levels independent of changes in ERK activity, we asked if the stimulatory effect of E-cadherin on cyclin D1 mRNA levels was dependent on Rac. Indeed, we found that expression of a dominant-negative construct, N17-Rac, inhibited cyclin D1 mRNA expression in a dose-dependent manner (Fig. 5A). As an independent approach to inhibiting Rac signaling, we infected MCF10A cells with an adenovirus encoding β2-chimerin, a Rac-specific GTPase activating protein (Caloca et al., 2003; Yang et al., 2005). β2-chimerin inhibited cyclin D1 mRNA expression in a dose-dependent manner (Fig. 5B) that was similar to that of the N17-Rac adenovirus.
Fig. 5.
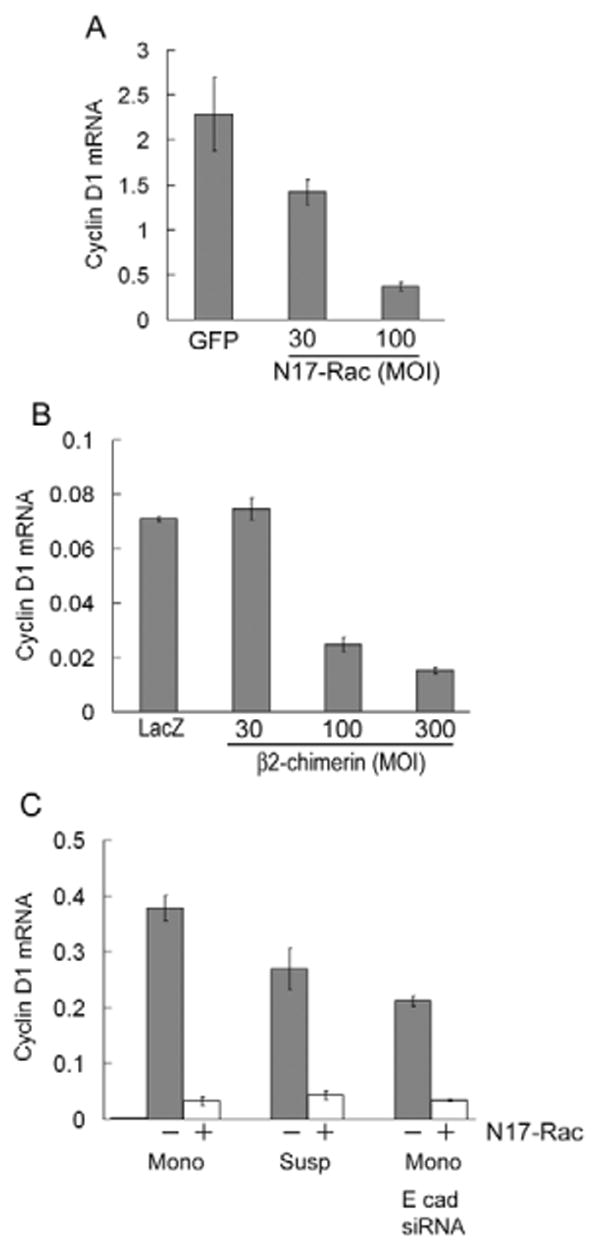
Cell-cell- and cell-substratum-dependent expression of cyclin D1 mRNA is mediated by Rac. MCF10A cells were infected with adenoviruses encoding GFP, β-galactosidase (LacZ), N17Rac or β2-chimerin and serum starved. Control viruses were used at the highest MOI of the test virus. The infected, serum-starved cells were plated on collagen-coated dishes and stimulated with 10% FBS and growth factor cocktail. (A) Cells were mitogen stimulated for 9 hours, and the effect of N17-Rac on cyclin D1 mRNA was determined by QPCR. (B) Cells were mitogen stimulated for 9 hours, and the effect of β2-chimerin on cyclin D1 mRNA was determined by QPCR. (C) MCF10A cells were transfected with control or E-cadherin siRNA and then infected with β-galactosidase (LacZ) or N17-Rac adenovirus during serum- and growth factor-starvation. Quiescent, transfected/infected cells were trypsinized, reseeded in monolayer (Mono) or suspension (Susp) on collagen- or agarose-coated dishes, respectively. The cells were stimulated with 10% FBS and growth factor cocktail for 12 hours. Total RNA was isolated and analyzed for cyclin D1 mRNA by QPCR.
Moreover, we found that inhibition of Rac signaling effectively blocked the induction of cyclin D1 mRNA after mitogen stimulation of suspended MCF10A cells (where E-cadherin is regulating cyclin D1 mRNA; Fig. 5C, Susp) and after mitogen stimulation of adherent MCF10A cells expressing E-cadherin siRNA (where integrins are regulating cyclin D1 mRNA; Fig. 5C, E-cad siRNA). Consistent with previous studies (del Pozo et al., 2000; Liu et al., 2006), Rac-GTP levels were lowered by incubating cells in suspension or by knocking down E-cadherin (not shown). However, Rac signaling to cyclin D1 can also be regulated downstream of Rac-GTP loading (Welsh et al., 2001). The exact mechanisms by which E-cadherin and integrins regulate Rac-dependent induction of cyclin D1 mRNA remain to be fully determined.
These inhibitory effects of N17-Rac and β2-chimerin on cyclin D1 mRNA were also seen when examining cyclin D1 protein (Fig. 6A,B). Similarly, knock-down of Rac1 with siRNA inhibited cyclin D1 expression (Fig. 6C). ERK activity was minimally affected by Rac inhibition (Fig. 6A) or Rac1 knock-down (Fig. 6C). Thus, in addition to their complementary effects on cyclin D1 gene expression, E-cadherin- and integrin-dependent adhesions share a common Rac-dependent signaling pathway to cyclin D1 mRNA in MCF10A cells. Nevertheless, we cannot exclude the possibility that ERK activity has some role in the integrin-dependent induction of cyclin D1 mRNA in MCF10A cells because there is a partial inhibition of cyclin D1 mRNA and protein expression when integrin-dependent ERK activity is blocked by incubating MCF10A cells in suspension (refer to Fig. 1).
Fig. 6.
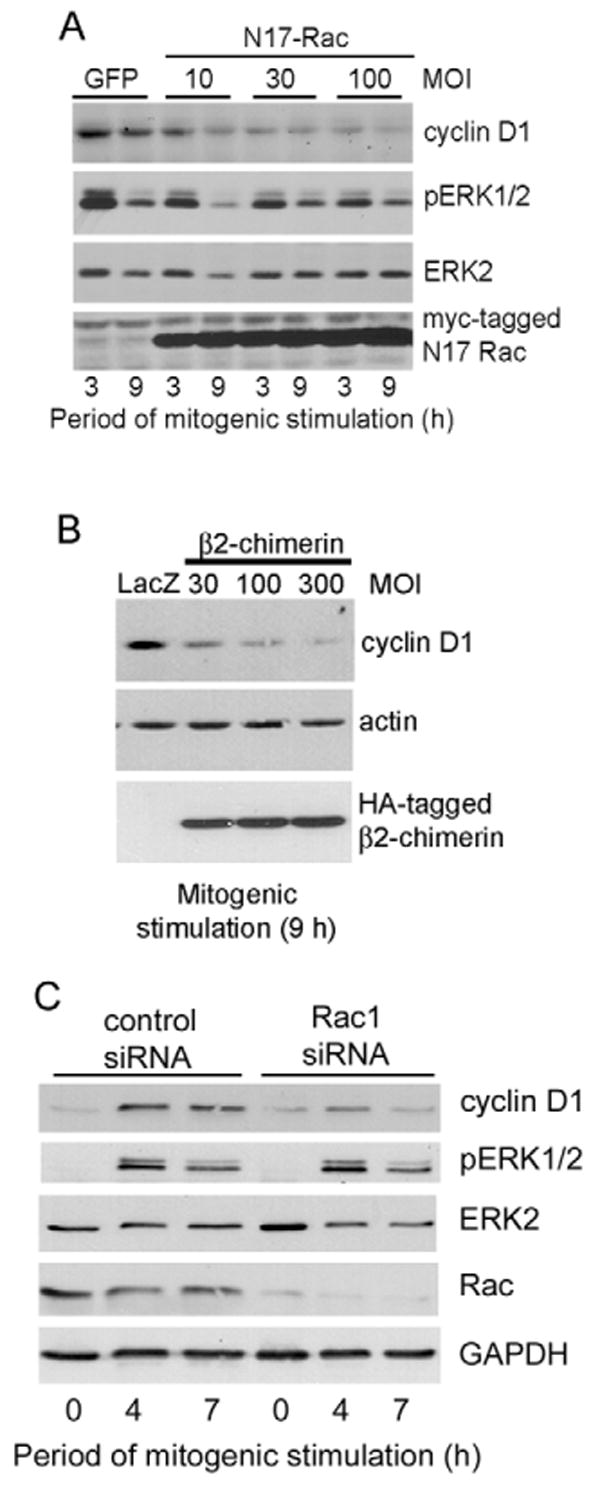
Rac inhibition blocks cyclin D1 expression. (A,B) MCF10A cells were infected with adenoviruses encoding GFP, β-galactosidase (LacZ), N17-Rac, or β2-chimerin and serum starved. Control viruses were used at the highest MOI of the test virus. The infected, serum-starved cells were plated on collagen-coated dishes, stimulated with 10% FBS and growth factors for 9 hours. Collected cells were analyzed by western blotting with the antibodies shown. (C) MCF10A cells were transfected with irrelevant control or Rac1 siRNAs and serum starved. The cells were plated on collagen-coated dishes, stimulated with 10% FBS and growth factor cocktail for the times shown, and analyzed by western blotting using antibodies to cyclin D1, dually phosphorylated ERK, total ERK, Rac and GAPDH (loading control).
E-cadherin-dependent expression of cyclin D1 contributes to efficient S-phase entry
Our previous studies documented the proliferative effect of E-cadherin-dependent adhesions by plating cells at different densities (Liu et al., 2006). MCF10A cells plated at low density attached and spread on the substrate but the large majority of cells fail to form cell-cell adhesions. By contrast, plating cells at an intermediate density allowed for attachment, spreading, and efficient formation of cell-cell adhesions (Liu et al., 2006). These cell-cell adhesions were mediated by E-cadherin and associated with increased S-phase entry, typically ∼1.5-3 fold. We exploited this system as a complementary approach to studying the link between E-cadherin, cyclin D1, and S-phase entry. We found that the increased incorporation of BrdU seen in MCF10A cells plated at a density that allowed for the formation of cell-cell adhesions (Fig. 7A, control siRNA) was associated with an increased expression of cyclin D1 mRNA (Fig. 7B, control siRNA). Knock-down of E-cadherin attenuated the contact-dependent increases in both BrdU incorporation and cyclin D1 gene expression (Fig. 7A,B, E-cad siRNAs). Conversely, ectopic expression of cyclin D1 released cells from their requirement for cell-cell contact in order to synthesize DNA (Fig. 7C). Taken together, our combined data show that the pro-mitogenic effect of E-cadherin in MCF10A cells can be explained by a Rac-dependent induction of cyclin D1 mRNA.
Fig. 7.
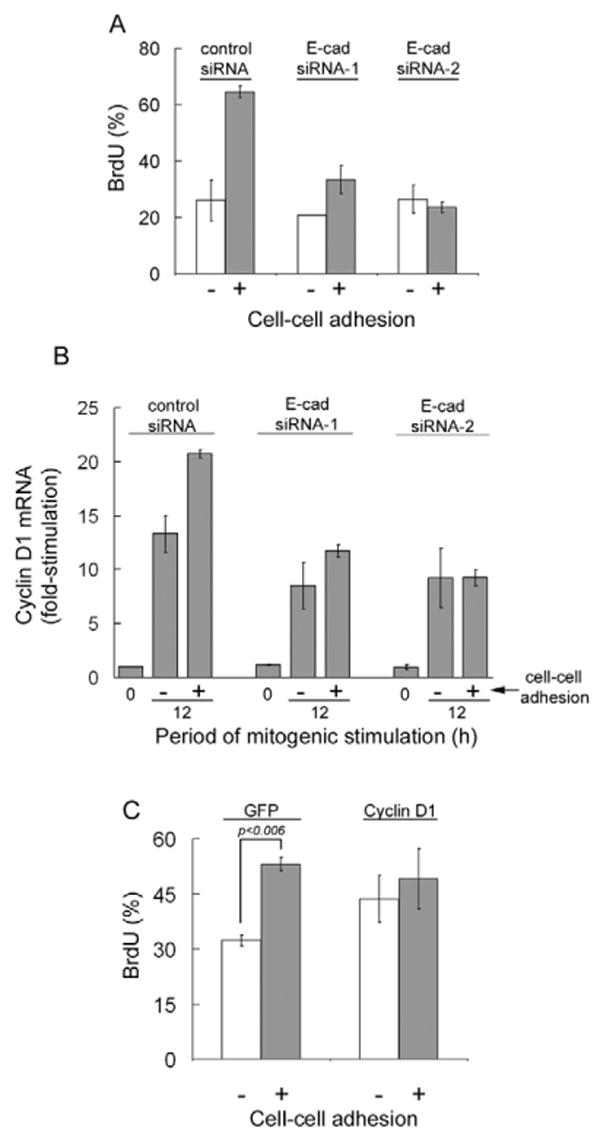
Causal relationship between the stimulatory effects of E-cadherin-mediated cell-cell adhesion on cyclin D1 gene expression and S-phase entry. (A) Serum-starved control and E-cadherin (E-cad) siRNA-transfected MCF10A cells were trypsinized, reseeded in full maintenance medium (see Materials and Methods) on collagen-coated, 25-mm glass coverslips at 2×104 cells/cm2 (with cell-cell adhesion) or 2×103 cells/cm2 (without cell-cell adhesion) as described by Liu et al. (Liu et al., 2006). BrdU was added when the cells were seeded and stimulated; the number of BrdU-labeled nuclei (relative to DAPI-stained nuclei) was determined after 24 hours. (B) Duplicate samples were collected at 12 hours and analyzed for cyclin D1 mRNA by QPCR. Results were calculated relative to the level of cyclin D1 mRNA in serum-starved cells expressing control siRNA. (C) Serum-starved MCF10A cells infected with Ad-GFP or Ad-cyclin D1 (100 MOI) were treated and analyzed as in A. Results for each panel show means ± s.e.m. of two experiments.
We consistently noticed a relatively small inhibitory effect of the E-cadherin siRNAs on cyclin D1 gene expression in the absence of cell-cell adhesion (Fig. 7B). Although we cannot exclude the possibility of small off-target effects of these two siRNAs, a likely explanation is that these siRNAs are targeting the small percentage of MCF10A cells that form cell-cell adhesions even at low seeding densities.
Discussion
Our findings characterize the cell cycle regulatory mechanism by which E-cadherin promotes S-phase entry in MCF10A cells. By experimentally isolating the individual effects of cell-substratum and cell-cell adhesion, we show that E-cadherin-mediated cell-cell adhesion is sufficient to cooperate with growth factor signaling and stimulate cyclin D1 gene expression. These results reveal a striking difference between epithelial and fibroblastic cells in their requirements for cyclin D1 induction. Although both cell types require growth factor and adhesion receptor signaling to induce cyclin D1, fibroblasts depend on integrin-mediated adhesion whereas the adhesion requirement in these epithelial cells can be satisfied by signaling through either integrins or E-cadherin. Thus, our studies identify a cell type in which integrin signaling is dispensable for cyclin D1 induction.
Consistent with the redundant effects of integrins and E-cadherins on cyclin D1 expression in MCF10A cells, we find that both of these adhesion receptors use a Rac-dependent signaling pathway to induce cyclin D1 mRNA. We and others have previously shown that both ERK and Rac can induce cyclin D1 gene expression, but most of the studies with the Rac pathway have relied on overexpression of an activated Rac allele (Gjoerup et al., 1998; Joyce et al., 1999; Klein et al., 2007; Page et al., 1999; Westwick et al., 1997). We previously showed that endogenous Rac can induce cyclin D1 mRNA in fibroblasts, but this pathway is only seen upon deliberate inhibition of Rho (Welsh et al., 2001). By contrast, the results described here show that endogenous Rac signaling plays a major role in regulating cyclin D1 gene expression in MCF10A mammary epithelial cells. Neither E-cadherin nor Rac1 knock-down strongly affected ERK activity, consistent with our previous studies indicating that Rac can stimulate cyclin D1 gene expression independently of ERK (Welsh et al., 2001).
E-cadherin has long been thought of as a tumor- and growth-suppressor. Loss or downregulation of E-cadherin has been found in several types of cancer (Hajra and Fearon, 2002), and re-expression of functional E-cadherin reduced invasiveness of epithelial tumor cells and arrested tumor metastasis in late stage tumor progression in transgenic mice (Perl et al., 1998; Vleminckx et al., 1991). E-cadherin appears to play a role in contact inhibition of proliferation as cells reach confluence in cell-culture (Kandikonda et al., 1996; Takahashi and Suzuki, 1996).
Despite the evidence that E-cadherin suppresses proliferation, other studies support a proliferative role for E-cadherin. Consistent with our data, inhibition of E-cadherin-mediated adhesion with a dominant negative E-cadherin adenovirus inhibited proliferation in keratinocytes (Zhu and Watt, 1996). In contrast to most cancer types, more than 85% of ovarian carcinomas have elevated E-cadherin levels, and suppression of E-cadherin function decreases proliferation (Reddy et al., 2005). E-cadherin expression is also present in aggressive inflammatory breast cancer and some derivative metastases (Charafe-Jauffret et al., 2004; Kowalski et al., 2003). In the context of normal developing tissues, loss of E-cadherin leads to inhibition of proliferation in the blastocyst, the mammary gland and the hair follicle (Boussadia et al., 2002; Ohsugi et al., 1997; Tinkle et al., 2004). This link between cadherin engagement and proliferative capacity may exist to provide a growth advantage to epithelial cells appropriately positioned within their native tissues, and to support proliferation in early development when extracellular matrix is poorly organized. It would be interesting to know the degree to which Rac-dependent regulation of cyclin D1 underlies these diverse pro-proliferative effects of E-cadherin.
Materials and Methods
Cell culture
MCF10A cells were maintained in 1:1 low glucose Dulbecco's modified Eagle's medium (DMEM):Ham's F12 nutrient medium supplemented with 10 mM Hepes buffer pH 7.4, 5% horse serum (Invitrogen), growth factor cocktail [20 ng/ml epithelial growth factor (EGF; BD Biosciences), 10 μg/ml insulin (Sigma), 0.5 μg/ml hydrocortisone (Sigma) and 100 ng/ml cholera toxin (List Biologicals)] and 1% gentamicin (Invitrogen). For cell cycle time-course experiments, MCF10A cells were seeded at 2-3×106 cells per 100-mm dish or at 1×106 per 60-mm dish and allowed to attach and spread overnight in maintenance medium before synchronization by serum and growth factor starvation in 1:1 low glucose DMEM-Ham's F12 nutrient medium, 10 mM Hepes pH 7.4, with 1 mg/ml BSA and gentamicin for 2 days. Unless noted otherwise, the quiescent MCF10A cells were trypsinized and suspended in fresh starvation medium for 30 minutes at 37°C prior to reseeding onto 100-mm or 60-mm dishes coated with 1.27 μg/cm2 collagen (monolayer) or 1% agarose (suspension). The cells were stimulated with 10% FBS and growth factor cocktail. For sample collection pre- or post-stimulation, cells were washed once with cold PBS, 1 mM NaVO5, scraped, split into separate tubes for protein and mRNA analysis, collected by centrifugation, quick frozen, and stored at −80°C prior to analysis.
To assess cell aggregation in suspension, quiescent MCF10A cells (1×106) were seeded in 150-mm agarose-coated dishes and stimulated in 20 ml 10% FBS (unless indicated) and growth factor cocktail in DMEM-F12. At selected times, a 1 ml aliquot of each suspension culture was seeded into a 35-mm dish containing coverslips coated with 25 μg/ml poly-L-lysine (Sigma). Cells were allowed to attach for 5 minutes. The coverslips were gently washed with cold PBS, fixed with 3.7% formaldehyde in PBS, and examined by phase-contrast microscopy or immunofluorescence microscopy using a 4× objective.
Epifluorescence and phase-contrast microscopy
Quiescent MCF10A were seeded in 100-mm dishes containing autoclaved glass coverslips coated with collagen and then stimulated with 10% FBS and growth factor cocktail in DMEM-F12. Cells were fixed in 3.7% formaldehyde, briefly incubated with 50 mM NH4Cl in PBS, and then blocked and permeabilized simultaneously with 0.5% Triton X-100 in PBS with 0.2% BSA and 2% goat serum for at least 30 minutes at room temperature. Anti-E-cadherin mouse monoclonal antibody (Invitrogen, 13-700) was diluted 1:500 (4 μg/ml) in fresh blocking buffer without Triton X-100 and 0.1 ml was incubated on the coverslips for 1 hour. Alexa Fluor 488 donkey anti-mouse IgG diluted 1:200 was combined with anti-β-catenin rabbit polyclonal antibody (sc-7199; Santa Cruz Biotechnology) diluted 1:100 (2 μg/ml) in fresh blocking buffer without Triton X-100 and incubated on the coverslips for 1 hour. Finally, Alexa Fluor 594 donkey anti-rabbit IgG was diluted 1:500 in PBS and added to the coverslips for 1 hour. In some cases E-cadherin was stained alone, followed by staining of F-actin with 1.5 units/ml Rhodamine-phalloidin diluted in PBS. The coverslips were washed three times in PBS between each incubation, and all samples were stained with DAPI to visualize nuclei. Images were obtained by epifluorescence microscopy, captured using a Hamamatsu digital CCD camera and analyzed with Openlab Imaging System software. To measure S-phase entry, mitogen-stimulated cells were seeded in the presence of BrdU (Amersham) for 24 hours, fixed and analyzed for BrdU incorporation by immunofluorescence as described previously (Liu et al., 2006). Micrograph figures were assembled in Photoshop (Adobe).
Western blot analysis
Western blotting used standard procedures and was performed as described previously (Welsh et al., 2001) using 30 μg of total cellular protein and the following antibodies: p21 (sc-6246; Santa Cruz Biotechnology), p27 (610241; BD Transduction Laboratories), Cdk6 (sc-177; Santa Cruz Biotechnology), actin (sc-8432; Santa Cruz Biotechnology), phospho-FAK Y397 (44-624; Biosource), FAK (610087; BD Transduction Laboratories), E-cadherin (13-1700; Invtrogen), β-catenin (sc-7199; Santa Cruz Biotechnology), Rac (05-389; Upstate Biotechnology), and GAPDH (sc-25778; Santa Cruz Biotechnology), T202/Y204-phosphorylated (active) ERK (9101; Cell Signaling) and ERK (610030; BD Transduction Laboratories). Rabbit polyclonal cyclin D1 was either prepared in our laboratory using recombinant cyclin D1 as the immunogen or purchased from Upstate Biotechnology (06-137). Anti-mouse IgG-HRP (na931; Amersham) and protein A-HRP (na9120; Amersham) were used as secondary antibodies. Western blot figures were assembled in Photoshop (Adobe).
Attachment assay
To determine whether integrin inhibitors were sufficient to block adhesion to ECM proteins, trypsinized quiescent cells were suspended (0.5-1×105 cells/ml serum and growth factor-free medium) and incubated in suspension (30 minutes at 37°C) with β1-integrin blocking antibody AIIB2 (a kind gift from David Boettiger, University of Pennsylvania, Philadelphia, PA) or 3 mM RGD peptide (Calbiochem). The cells (3×105) were then seeded in duplicate in 24-well dishes coated with collagen (1.27 μg/cm2), laminin (1.27 μg/cm2), fibronectin (0.318 μg/cm2) or vitronectin (0.635 μg/cm2), in the presence of the growth factor cocktail without serum, and allowed to attach for 45 minutes (collagen, laminin and fibronectin) or 1 hour (vitronectin). Unattached cells were removed by three washes with ice-cold PBS. Attached cells were stained with 0.5% crystal violet in 20% methanol for 3 hours and then washed five times with water. The stain was eluted from attached cells with 200 μl 1 M sodium citrate in 50% ethanol for 30 minutes, and the absorbance was determined at 595 nm. Means ± s.d. were calculated from duplicate samples after subtracting the non-specific absorbance seen in the absence of cells.
Quantitative real-time RT-PCR (QPCR)
Total RNA was extracted from cell pellets with 0.5 ml of TRIzol (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed on 10 ng/μl total RNA from each sample using Applied Biosystems reverse transcription reagents (N8080234) (5.5 mM, MgCl2, 2 mM dNTP, 2.5 μM random hexamers, 0.4 U/μl RNase inhibitor and 1.25 U/μl multiscribe reverse transcriptase) according to the manufacturer's instructions. 2.5 μl duplicate aliquots of cDNA for each sample were then subjected to 40 amplification cycles of PCR (Applied Biosystems Prism 7000 sequence detection system) using Taqman universal PCR master mix. To analyze levels of cyclin D1 mRNA, the PCR reaction mixture contained 300 nM forward primer (5′-TGTTCGTGGCCTCTAAGATGAAG-3′), 300 nM reverse primer (5′-AGGTTCCACTTGAGCTTGTTCAC-3′) and 250 nM TAMRA probe (5′-6FAM-AGCAGCTCCATTTGCAGCAGCTCCT-TAMRA-3′). In some experiments the analysis for cyclin D1 was duplexed with Cdk4 (control transcript), in which case the PCR reaction also contained 150 nM Cdk4 forward primer (5′-ACAAGTGGTGGAACAGTCAAGCT-3′), 200 nM reverse primer (5′-GCATATGTGGACTGCAGAAGAACT-3′) and 150 nM TAMRA probe (5′-VIC-ATGGCACTTACACCCGTGGTTGTTACACTCT-TAMRA-3′). To determine the levels of 18S rRNA, the PCR reaction mixture contained 150 nM forward primer (5′-CCTGGTTGATCCTGCCAGTAG-3′), 150 nM reverse primer (5′-CCGTGC-GTACTTAGACATGCA-3′) and 125 nM minor-groove binder (MGB) probe (VIC-5′-TGCTTGTCTCAAAGATTA-3′ minor-groove-binder non-fluorescent quencher). RNA expression was quantified from a standard curve using ABI Prism 7000 sequence detection system software. QPCR results show the levels of cyclin D1 mRNA normalized to 18S rRNA or Cdk4 mRNA and were expressed as the mean ± s.d. of duplicate PCR reactions unless noted otherwise. Control transcript levels did not vary reproducibly from any of the treatments or conditions present during the experiment.
siRNA transfection and adenoviral infection
For RNA interference (RNAi) experiments, MCF10A cells were seeded in 35- or 100-mm culture dishes (0.5-1×108 cells/cm2) in maintenance medium without antibiotics the day before transfection. Cells were washed three times with serum- and growth factor-free Optimem (Invitrogen) and then transfected with siRNA oligonucleotides (Ambion) for human E-cadherin (150 nM 5′-GAGUGAAUUUUGAAGAUUGtt-3′; ID#44988 or 200 nM 5′-GCACGUACACAGCCCUAAUtt-3′; ID#146381), human Rac1 (150 nM 5′-GAAUAUAUCCCUACUGUCUtt-3′; ID#45358), or irrelevant control (targeting mouse LIM kinase 1; 5′-GGUAUUGACAGGGAUCUGAtt-3′; ID#156130) in the presence of 10 μl Lipofectamine 2000 (Invitrogen) per well. The cultures were incubated in this serum-free medium for 2 days.
Adenoviruses were titred with Adeno-X rapid titer kit (BD Biosciences). For infection, MCF10A cells were seeded at 2×106 cells per 100-mm dish and allowed to attach and spread the day before infection. Cells were washed and serum- and growth factor-starved for 8 hours prior to overnight infection. Control viruses were used at the highest MOI of the test virus. Cells were incubated in fresh, serum-free medium for an additional 24 hours before use in experimentation. When adenoviral infection was combined with RNAi, the cells were transfected with siRNA as described above, and the adenovirus was added for 16 hours, beginning 24 hours after starvation. The adenovirus was then removed and the medium was replaced with fresh serum-free medium for a total starvation time of 48 hours.
Analysis of cyclin D1 stability
Quiescent MCF10A cells that had been transfected with control or E-cadherin siRNA oligonucleotides were trypsinized, reseeded (∼3×106) in 100-mm dishes coated with agarose, and stimulated with 10% FBS and growth factors for 6 hours to allow for the expression of cyclin D1. Cycloheximide (10 μg/ml) was then added to the culture medium. Samples were collected every 15 minutes for 1 hour and analyzed for cyclin D1 and GAPDH by western blotting.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/2/226/DC1
Acknowledgments
We thank Jeffrey Albrecht (Hennepin County Medical Center, Minneapolis, MN), Marcelo Kazanietz (University of Pennsylvania) and Anne Ridley (University of London) for adenoviruses. This work was supported by NIH grants CA72639 to R.K.A., HL73305 to C.S.C. and CA078731 to V.M.W. A.K.F. was supported by pre-doctoral training grant R25-CA-101871. W.F.L. was supported by the National Science Foundation.
References
- Assoian RK, Schwartz MA. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev. 2001;111:48–53. doi: 10.1016/s0959-437x(00)00155-6. [DOI] [PubMed] [Google Scholar]
- Bohmer R, Scharf E, Assoian R. Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage-dependent expression of cyclin D1. Mol Biol Cell. 1996;7:101–111. doi: 10.1091/mbc.7.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. Ecadherin is a survival factor for the lactating mouse mammary gland. Mech Dev. 2002;115:53–62. doi: 10.1016/s0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Caloca MJ, Wang H, Kazanietz MG. Characterization of the Rac-GAP (Rac-GTPase-activating protein) activity of β2-chimaerin, a ‘non-protein kinase C’ phorbol ester receptor. Biochem J. 2003;375:313–321. doi: 10.1042/BJ20030727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Tarpin C, Bardou VJ, Bertucci F, Ginestier C, Braud AC, Puig B, Geneix J, Hassoun J, Birnbaum D, et al. Immunophenotypic analysis of inflammatory breast cancers: identification of an ‘inflammatory signature’. J Pathol. 2004;202:265–273. doi: 10.1002/path.1515. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. α-Catenin is a molecular switch that binds E cadherin-β-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoerup O, Lukas J, Bartek J, Willumsen BM. Rac and Cdc42 are potent stimulators of E2F-dependent transcription capable of promoting retinoblastoma susceptibility gene product hyperphosphorylation. J Biol Chem. 1998;273:18812–18818. doi: 10.1074/jbc.273.30.18812. [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Wong E, Gumbiner BM. E cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–1060. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Hajra KM, Fearon ER. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer. 2002;34:255–268. doi: 10.1002/gcc.10083. [DOI] [PubMed] [Google Scholar]
- Joyce D, Bouzahzah B, Fu M, Albanese C, D'Amico M, Steer J, Klein JU, Lee RJ, Segall JE, Westwick JK, et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem. 1999;274:25245–25249. doi: 10.1074/jbc.274.36.25245. [DOI] [PubMed] [Google Scholar]
- Kandikonda S, Oda D, Niederman R, Sorkin BC. Cadherin-mediated adhesion is required for normal growth regulation of human gingival epithelial cells. Cell Adhes Commun. 1996;4:13–24. doi: 10.3109/15419069609010760. [DOI] [PubMed] [Google Scholar]
- Klein EA, Yang C, Kazanietz MG, Assoian RK. NfkappaB independent signaling to the cyclin D1 gene by Rac. Cell Cycle. 2007;6:1115–1121. doi: 10.4161/cc.6.9.4147. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Soler AP, Johnson KR, Wheelock MJ. Interaction of α-actinin with the cadherin/catenin cell-cell adhesion complex via α-catenin. J Cell Biol. 1995;130:67–77. doi: 10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski PJ, Rubin MA, Kleer CG. E cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5:R217–R222. doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WF, Nelson CM, Pirone DM, Chen CS. E cadherin engagement stimulates proliferation via Rac1. J Cell Biol. 2006;173:431–441. doi: 10.1083/jcb.200510087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Fukata M, Yamaga M, Itoh N, Kaibuchi K. Recruitment and activation of Rac1 by the formation of E cadherin-mediated cell-cell adhesion sites. J Cell Sci. 2001;114:1829–1838. doi: 10.1242/jcs.114.10.1829. [DOI] [PubMed] [Google Scholar]
- Nieset J, Redfield A, Jin F, Knudsen K, Johnson K, Wheelock M. Characterization of the interactions of α-catenin with α-actinin and βcatenin/plakoglobin. J Cell Sci. 1997;110:1013–1022. doi: 10.1242/jcs.110.8.1013. [DOI] [PubMed] [Google Scholar]
- Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Ohsugi M, Larue L, Schwarz H, Kemler R. Cell junctional and cytoskeletal organization in mouse blastocysts lacking E cadherin. Dev Biol. 1997;185:261–271. doi: 10.1006/dbio.1997.8560. [DOI] [PubMed] [Google Scholar]
- Page K, Li J, Hodge JA, Liu PT, Vanden Hoek TL, Becker LB, Pestell RG, Rosner MR, Hershenson MB. Characterization of a Rac1 signaling pathway to cyclin D (1) expression in airway smooth muscle cells. J Biol Chem. 1999;274:22065–22071. doi: 10.1074/jbc.274.31.22065. [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Ren C, Lindgren P, Boman K, Shen Y, Lundin E, Ottander U, Rytinki M, Liu K. Formation of E cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol Endocrinol. 2005;19:2564–2578. doi: 10.1210/me.2004-0342. [DOI] [PubMed] [Google Scholar]
- Roovers K, Davey G, Zhu X, Bottazzi ME, Assoian RK. α5β1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol Biol Cell. 1999;10:3197–3204. doi: 10.1091/mbc.10.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadot E, Simcha I, Shtutman M, Ben-Ze'ev A, Geiger B. Inhibition of β-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci USA. 1998;95:15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Suzuki K. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E cadherin-mediated cell-cell adhesion. Exp Cell Res. 1996;226:214–222. doi: 10.1006/excr.1996.0221. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Tinkle CL, Lechler T, Pasolli HA, Fuchs E. Conditional targeting of E cadherin in skin: insights into hyperproliferative and degenerative responses. Proc Natl Acad Sci USA. 2004;101:552–557. doi: 10.1073/pnas.0307437100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Yung Y, Walker JL, Assoian RK. ERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targets. Mol Biol Cell. 2007;18:1457–1463. doi: 10.1091/mbc.E06-10-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–957. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, Der CJ. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Liu Y, Leskow FC, Weaver VM, Kazanietz MG. Rac-GAP-dependent inhibition of breast cancer cell proliferation by β2-chimerin. J Biol Chem. 2005;280:24363–24370. doi: 10.1074/jbc.M411629200. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM. Expression of a dominant negative cadherin mutant inhibits proliferation and stimulates terminal differentiation of human epidermal keratinocytes. J Cell Sci. 1996;109:3013–3023. doi: 10.1242/jcs.109.13.3013. [DOI] [PubMed] [Google Scholar]
- Zhu X, Ohtsubo M, Bohmer R, Roberts J, Assoian R. Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin Ecdk2, and phosphorylation of the retinoblastoma protein. J Cell Biol. 1996;133:391–403. doi: 10.1083/jcb.133.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/2/226/DC1