

Abstract
Acute SIV infection is characterized by explosive infection of memory CD4 T cells in peripheral and mucosal tissues. Interestingly, relatively few memory CD4 T cells are infected until as late as day 7−8 after challenge. However, by day 10 post infection (pi) most of the memory CD4 T cells are infected and carry viral DNA. The rapidity with which infection expands within 2−3 days to encompass virtually the entire memory CD4 T cell compartment suggests significant alterations in the susceptibility of memory CD4 T cells to infection during this period. The mechanism(s) underlying this increased permissiveness to infection is not known. Here we show that IL-15 secretion significantly correlates with the upregulated expression of CD4 on memory CD4 T cells that is associated with increased permissiveness to SIV infection. Activation and proliferation of memory CD8, but not memory CD4 T cells, preceded the amplification of viral infection. Though memory CD4 T cells did not express normal activation markers, they displayed a significant upregulation in the density of CD4 but not CCR5 expression between day 7 and 10 pi that correlated with increased plasma IL-15 levels and infection in these cells. Culture of purified CD4 T cells with IL-15 and/or SIV was associated with a significant increase in the expression of CD4 and infection of these sorted cells. Our results demonstrate that IL-15 contributes to the increased susceptibility of memory CD4 T cells to SIV during the early phase of acute SIV infection.
Keywords: HIV, SIV, Mucosa, CD4 T cell, Gut, Intestine, IL-15R, IL-2R, Acute, Activation
Introduction
Acute SIV infection is characterized by a massive loss of memory CD4 T cells from both mucosal and peripheral tissues(1-3). These cells are primarily lost due to viral infection that peaks at day 10 pi. Interestingly, the kinetics of infection appears to be independent of the route of infection; both intravenous and vaginal challenge display similar kinetics of infection and loss(1-3).
A detailed analysis of the kinetics of viral infection shows that very few memory CD4 T cells in the mucosa or periphery are infected at day 7 pi, whereas most of the memory CD4 T cells were infected at day 10 pi. Li et al(1) using SIV specific riboprobes demonstrated that the level of infection in the vaginal mucosa significantly increased between day 7 and 10 pi. Additionally, Mattapallil et al(2) using a quantitative PCR assay for SIV-gag DNA showed that SIV-gag copies in either the jejunum or periphery increased from ∼1−10 × 103 copies of SIV-gag / 105 memory CD4 T cells at day 7 pi to ∼2 − 2.5 × 105 copies of SIV-gag / 105 memory CD4 T cells at day 10 pi. Single cell analysis revealed that there were ∼2 copies of SIV DNA / cell(2). This suggests that <5% of memory CD4 T cells were infected at day 7 pi, whereas by day 10 pi most of the memory CD4 T cells carried viral DNA.
What drives this massive amplification of viral infection within a short period of 2−3 days? It is generally believed that immune activation associated with acute infection likely plays a major role. Little is known, however, about the exact mechanisms behind this process. We have previously shown that most memory CD4 T cells express similar levels of CCR5 at day 7 and 10 pi(2), suggesting that factors other than SIV coreceptor expression play a role in making the memory CD4 T cells highly permissive to infection during this period.
Previous studies have shown that acute immune activation was associated with increased proliferation and activation of peripheral CD8 T cells(4, 5). The preferential activation and proliferation of memory CD8 T cells during acute SIV infection suggests that specific cytokines likely activate memory CD8 T cells. Cytokines such as IL-7 have been shown to induce homeostatic proliferation of both naïve and memory CD8 T cells, whereas IL-15 primarily supports homeostatic and infection-mediated proliferation and survival of memory CD8 T cells under physiological conditions(6-11). Recent studies(11) have shown that IL-15 has an effect on the proliferation of effector memory CD4 T cells in rhesus macaques. We hypothesized that immune activation during acute SIV infection is likely associated with production of IL-15 that makes memory CD4 T cells more susceptible to infection. We tested this hypothesis using experimental SIV infection of rhesus macaques by evaluating the kinetics of Ki-67 and HLA-DR expression on T cells and measuring plasma IL-15 levels and SIV infection in memory CD4 T cells. Additionally, we evaluated the expression of IL-15Rα and IL-2Rβ on T cells to better delineate the role of IL-15 in acute immune activation and amplification of viral infection. Finally, we performed in vitro culture experiments to directly assess the ability of IL-15 to increase SIV infection in CD4 T cells.
Materials and Methods
Animals & infection
Eight colony-bred healthy Mamu*A01- rhesus macaques (Macaca mulatta) of Indian origin housed at Advanced Bioscience Laboratories Inc., MD were used in the longitudinal study. Animals were housed in accordance with American Association for Accreditation of Laboratory Animal Care guidelines and were seronegative for SIV, simian retrovirus and simian T-cell leukemia virus type-1. All animal care and procedures were reviewed and approved by Institutional Animal Care and Use Committee. Animals were infected with 100 animal infectious doses of uncloned pathogenic SIVmac251 intravenously; peripheral blood, rectal biopsies, and plasma samples were collected longitudinally at various time points prior to and after challenge. Additionally, archival blood and mucosal samples were obtained from 12 SIVmac251 infected animals for analysis.
Blood and Tissue samples
PBMC were isolated by density gradient centrifugation, whereas rectal biopsies were processed using percoll gradients as previously described(2). Plasma viral loads were determined by real-time RT-PCR assay as previously described(12).
Antibodies and flow cytometry
All antibodies except for CD4-Qdot605 (courtesy of Mario Roederer) used in this study were obtained from BD Biosciences (San Diego, CA). All reagents were validated and titrated using rhesus macaque PBMC. For phenotypic analysis and sorting of CD4 T cell subsets, freshly isolated cells were labeled simultaneously with the following combinations of antibodies: CD3-Cy7APC, CD8-Alexa-700, CD4-PE or CD4-Qdot605, HLA-DR-TRPE (Texas Red-PE), CD95-APC and CD28-Cy5-PE. T cell activation was determined by labeling freshly isolated cells using the above panel. After fixing and permeabilizing, the cells were stained with Ki-67-FITC. To determine IL-15Rα and IL-2Rβ expression, cells were labeled with anti-IL-15Rβ (R&D Systems) and IL-2Rβ (R&D Systems) along with anti-CD3, CD4, CD8, CCR5 and CD95. Labeled cells were fixed with 0.5% paraformaldehyde, and analyzed using a modified Becton Dickinson Aria Sorter. One to two million total events were collected for analysis.
To determine which subsets supported viral infection, naïve and memory CD4 T cells (discriminated based on CD28 and CD95(13) expression) were sorted into tubes and subjected to qPCR assay for measuring SIV-gag DNA as described previously(2, 14).
For in vitro culture experiments, cells were labeled with anti-CD14-FITC, CD20-FITC and CD8-FITC and resuspended in warm culture media. Cells negative for these markers representing CD4 T cells were sorted into warm media. After harvesting, cells were labeled with anti-CD3-Cy7-APC, CD4-PE, CD8-Cy5-PE, CD95-APC, and CD28-FITC.
qPCR assay for SIV-gag DNA
CD4 T-cell associated viral DNA was measured by a quantitative PCR assay for SIV gag as previously described(2, 14) using SIV gag primers and probe as described by Lifson et al(15). The assay was calibrated using a cell line that carried a single copy of proviral SIV DNA as described previously(2).
ELISA for IL-15, IL-7 and IL-2
Commercial ELISA kits were used to measure IL-15, IL-7 (huIL-15 and IL-7 kits from R & D Systems) and IL-2 (Monkey IL-2 OptEIA kit from BD Biosciences) in the plasma as per manufacturer's instructions. Each sample was set up in duplicates. The detection limit for IL-15 was 2 pg/ml, IL-7 was 0.1 pg/ml, whereas the limit of detection for IL-2 was 7.8 − 15.6 U/ml. ELISA for IL-2 was performed two times using undiluted plasma, and once using diluted plasma.
In vitro culture assay with rIL-15 and SIVmac251
To determine if IL-15 upregulates CD4 expression on CD4 T cells, and increases the permissibility to SIV infection we sorted CD4 T cells by negative selection (CD14−CD20−CD8−) from peripheral blood of 8 healthy rhesus macaques and cultured them in the presence or absence of IL-15 and/or SIVmac251. The cells were not labeled with either CD3 or CD4 to avoid activating them through these molecules. Sorted cells were >99% pure (Suppl. Fig. 1a), and ∼ 8 × 105 to 1 × 106 of CD4 T cells were obtained from each animal. Since IL-15 can stimulate cells in trans, we used highly purified populations of total CD4 T cells (mix of naïve and memory) to avoid any bystander effect of cytokines released from other cells. Cells were cultured for 7 days in the presence or absence of 5 ng / ml (n = 4) of recombinant simian IL-15 (National Center for Research Resource for Non-human Primate Immune Reagents) and/or 1000 TCID50 of SIVmac251. Previous studies(10, 16) had used 5 ng / ml in long-term culture studies.
Sorted cell cultures were set up after dividing cells from each animal (n = 4) equally as follows: (1) Cells only (2) Cells + IL-15 (3) Cells + IL-15 + IL-15Rα-IgG. An IL-15Rα - immunoglobulin-G (IL-15Rα-IgG) fusion protein (National Center for Research Resource for Non-human Primate Immune Reagents) that specifically blocked the effect of simian IL-15 was used in these experiments at a concentration of 10 ug /ml. Cells were harvested after 7 days, and labeled with CD3/CD4/CD8/CD95/CD28 to determine the density of CD4 expression on memory CD4 T cells.
Additionally, purified CD4 T cells obtained from 4 additional animals were used to determine if IL-15 increased infection in purified CD4 T cells, and to determine if this effect could be blocked with IL-15Rα-IgG. Sorted cell cultures were set up after dividing cells from each animal (n = 4) equally as follows: (1) Cells + SIVmac251 (2) Cells + SIVmac251 + IL-15 (3) Cells + SIVmac251 + IL-15 + IL-15Rα-IgG . Cells were harvested, washed and directly used in a qPCR assay for SIV-gag DNA.
Production of soluble recombinant MamuIL-15Rα-IgG
Recombinant IL-15Ra-IgG fusion protein was produced at the National Center for Research Resource for Non-human Primate Immune Reagents as per procedures described below. The coding sequence of MamuIL-15Rα was cloned and sequenced from rhesus macaque of Indian origin as well as other species of macaques (cynomolgus and pig-tailed) (Genbank accession numbers FJ222743-FJ222748) (http://pathology.emory.edu/Villinger/index.htm). Initially cDNA was reverse transcribed from total mRNA isolated from PBMC with an IL-15Rα specific primer IL-15Rα17 (GTA AAA TGG CAC TGA GTT GAG). Subsequently, standard PCR was performed using high fidelity Taq polymerase (Roche) with primers IL-15Rα19cor2 (GACCATGGAATCACGTGCCCTCCCCCAGTGTCCG TGGAACACGCA) and IL-15Rα3 (GCAGAGAGGCTCCTTCACTCC). The fusion of the IL-15 antagonist, rMamuIL-15Rα-IgG, was performed using a strategy similar to the one described previously for the construction of a PD-1-IgG(17). The Fc portion of the IgG was mutated at positions corresponding to aa 235 and 331 (L235A and P331S) to inactivate potential binding of the fusion protein to complement, and to Fc receptors respectively, effectively inactivating complement and cell mediated cytotoxicity. The schematic representation of the recombinant protein and nucleotide sequence encoding the mature protein with corresponding amino acid sequence is shown in Supplementary Fig. 3.
Briefly, the extracellular domain of IL-15Rα (181 aa) amplified with primers pMT-IL15Rα (CCGGATCCATCACGTGCCCTCCCCCAGTGT) and IL15Rα-IgG2r (ACGTAGATCTACCGACGGTGGATGTAGAGATAGCC), and the IgG2 amplified with primers IL15Rα-IgGf (GGCTATCTCTACATCCACCGTCGGTAGATCTACGT) and IgG6ae (TATGACGTCGAATTCTCATTTACCCGGAGACACGGAGA) were concatamerized by another round of PCR using the primers pMT-IL15Rα and IgG6ae, and the overlap created in the previous amplifications. The gene coding for this fusion construct was subcloned into the pMT-BIP vector designed to produce soluble excreted protein from Schneider-2 (S-2) insect cells in spinner cultures (Invitrogen) using BamHI and EcoRI. All constructs wee verified by sequencing. The protein released in the supernatant of theses culture at a concentration of 7−10 mg/L was then purified by passage over a Protein-G-sepharose capture column and eluted with diluted acetic acid (pH 2.8). The ∼50 kDa purified protein was then dialyzed extensively against PBS and tested for purity, presence of endotoxin, protein content and biological activity. The protein appeared >90% pure and devoid of any significant endotoxin activity. Blocking activity was tested against recombinant MamuIL-15 using the HT-2 indicator cell line as a readout(18). IL-15Rα-IgG (556 ug / ml) was serially diluted and used in the blocking assay. One inhibitory Unit was the equivalent amount of IL-15Rα-IgG needed to inactivate the activity of 1 Unit of IL-15 bioactivity as measured in the HT-2 proliferation assay.
Data analysis
Flow cytometric data was analyzed using FlowJo version 8.6 (Tree Star, Inc., Ashland, OR). Statistical analysis was performed with GraphPad Prism Version 4.0 software (GraphPad Prism Software, Inc. San Diego, CA). The p values shown in all the figures are not corrected for multiple comparisons.
Results
Few memory CD4 T cells in either the mucosa or periphery are infected at day 7 pi
We first evaluated the kinetics of infection by measuring levels of SIV RNA in plasma, and SIV-gag DNA in total memory CD4 T cells (CD95+CD28+ & CD95+CD28−) sorted from peripheral blood and rectal or jejunal biopsies during the first 2−3 weeks of infection. Plasma viral loads (Fig. 1 a) peaked around day 14 pi and declined thereafter. Fewer than 5% of memory CD4 T cells in either the mucosa or periphery carried viral DNA at day 7 pi (Fig. 1b) confirming previous reports(1, 2). On average, at day 7 pi there were 6 − 9 × 103 copies of SIV gag DNA /105 memory CD4 T cells. However, by day 10 pi, the frequency of SIV-gag DNA copies significantly increased to ∼1.9 to 2.5 × 105 copies / 105 memory CD4 T cells suggesting that most memory CD4 T cells were infected in both the mucosa and periphery by day 10 pi. We have previously shown that there are ∼ 2 copies of SIV gag DNA / infected memory CD4 T cell at day 10 pi(2).
Figure 1. Kinetics of viral infection, and CD4 T cell Dynamics.
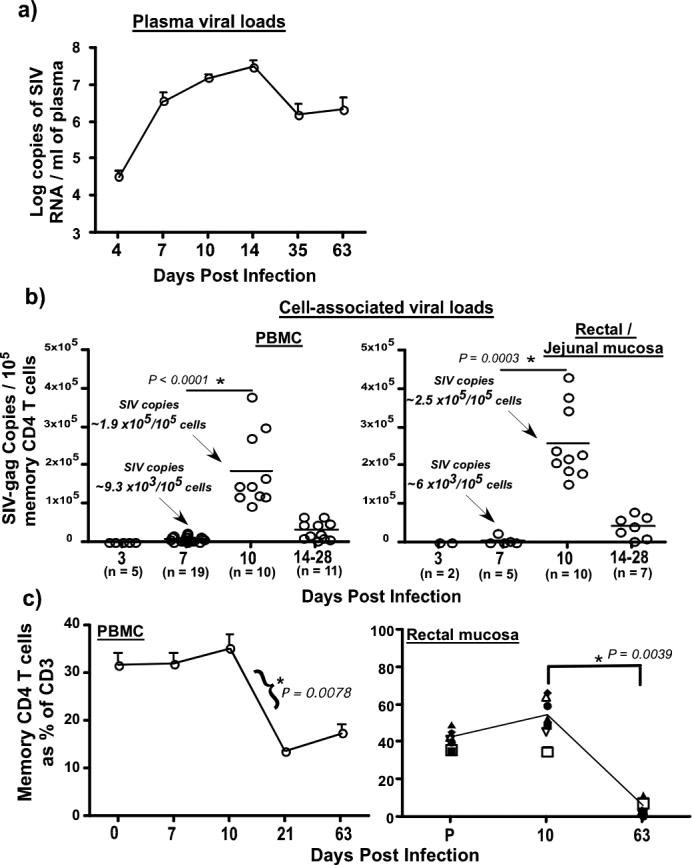
(a) Longitudinal analysis (n = 8) of plasma viral loads during acute SIV infection. Limit of detection is 30 copies /ml of plasma (b) Kinetics of cell-associated viral loads in memory CD4 T cells from peripheral blood and mucosa of SIV infected rhesus macaques. Samples include those collected from the 8 animals followed longitudinally, and archival samples collected from different animals at various time points. The frequency of SIV-gag copies was determined in sorted subsets of memory CD4 T cells using a quantitative PCR assay for SIV-gag DNA. Statistical analysis was performed used Mann-Whitney U test. (c) Longitudinal dynamics of memory CD4 T cells in peripheral blood (n = 8) and rectal mucosa (n = 8). Naïve and memory CD4 T cells were discriminated on the basis of CD28 and CD95 expression with all memory CD4 T cells expressing CD95. Massive loss of memory CD4 T cells occurs by day 14 pi. Very low level of infection is seen at day 7 pi that significantly increased by day 10 pi in both tissues. Statistical analysis was performed using Wilcoxon matched pairs test. Error bars represent standard error. (P= pre-infection samples collected at −d28; ns = not significant).
Following peak infection by day 10 pi, we observed a massive loss of SIV infected cells by day 14 pi that coincided with the loss of memory CD4 T cells (Fig. 1c). The similar kinetics of viral infection in the mucosa and periphery argues against the hypothesis that mucosal CD4 T cells are preferentially infected as compared to memory CD4 T cells in other tissues. Rather, the explosion of infection between day 7 and 10 pi in both of these tissues suggests that other factors likely cause memory CD4 T cells to become more susceptible to infection.
Acute immune activation precedes amplification of viral infection
To determine if the increased susceptibility of memory CD4 T cells to infection was due to acute immune activation, we evaluated the expression of Ki-67 and HLA-DR on CD4 and CD8 T cells in parallel with SIV infection in memory CD4 T cells. Previous studies have shown that acute SIV infection is characterized by immune activation(4, 5, 19). Our results indicate that acute viral infection leads to a significant increase in the proliferation (Fig. 2a) and activation (Fig. 2b) of memory CD8 T cells as early as day 7 pi, whereas there was no change in either the proliferation or activation of CD4 memory T cells during the first 10 days of infection. This suggests that activation of memory CD4 T cells was not the cause for the apparent increased permissiveness of these cells to SIV between day's 7 and 10 pi. Naïve CD4 and CD8 T cells (CD95−) did not express either Ki-67 or HLA-DR (Suppl. Fig2a & b). Consistent with our results, Kaur et al(5) demonstrated that acute SIV infection was associated with a 3 to 5 fold increase in the Ki-67+CD8 T cells that were activated and had a memory phenotype, whereas there was minimal change in the frequency of Ki-67+CD4 T cells.
Figure 2. Kinetics of immune activation during acute SIV infection.
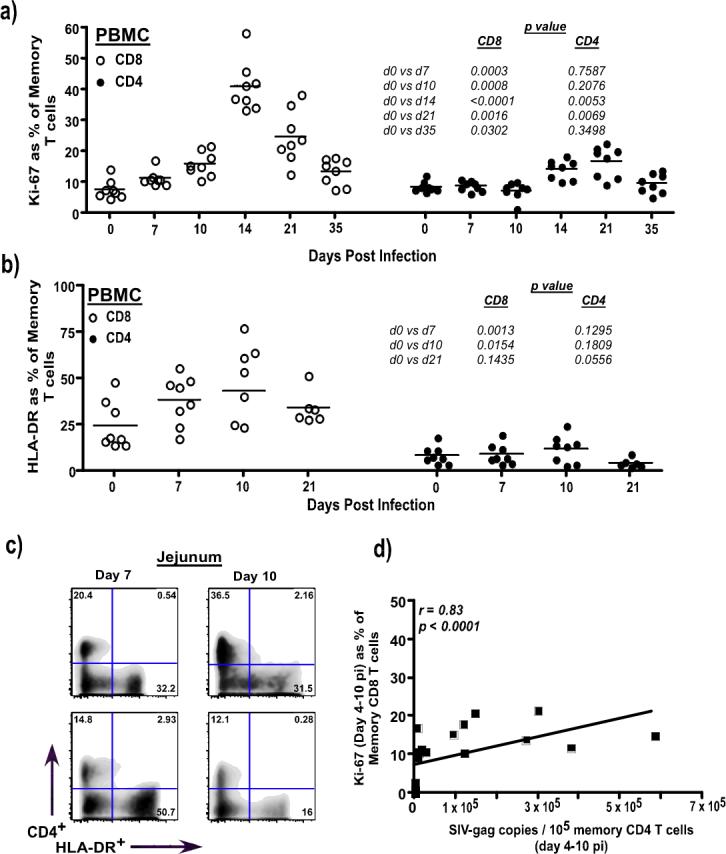
(a) Longitudinal analysis Ki-67 expression in memory CD8 and CD4 T cells from PBMC (n = 8) (b) Longitudinal analysis HLA-DR expression on memory CD8 and CD4 T cells from PBMC (n = 8) (c) Few mucosal CD4 T cells (y-axis) express HLA-DR at day 7 (n = 2) and 10 pi (n = 2), whereas CD3+CD4− T cells (CD8; x-axis) express high levels of HLA-DR. Analysis gates were set on CD3+ T cells. (d) Ki-67 expression of memory CD8 T cells significantly correlates with memory CD4 T cell-associated viral loads. Data from 8 animals at day 4, 7 and 10 pi each were used to derive the correlations. Memory T cells were delineated based on the expression of CD95 and CD28 with all CD95+ cells being memory T cells. Statistical analysis was performed using Mann-Whitney U test. Linear regression analysis was performed to determine line of fit, and correlations were derived using Spearman's rank test.
Interestingly, increased Ki-67 expression on memory CD8 T cells significantly correlated (r = 0.83, p < 0.0001) with SIV infection in memory CD4 T cells (Fig. 2d), suggesting that factors that drive acute immune activation likely play a role in determining the kinetics of viral infection in memory CD4 T cells. In contrast to the early activation of memory CD8 T cells, a significant increase in the proliferation of memory CD4 T cells did not appear until as late as day 14 pi (Fig. 2a), and was coincident with the massive loss of memory CD4 T cells at day 14 pi (Fig. 1c). This would suggest that the proliferation of memory CD4 T cells at day 14 pi likely represents a homeostatic response to the severe loss of memory CD4 T cells.
Next we sought to determine why memory CD8 T cells were preferentially activated as compared to CD4 T cells. We hypothesized that this might be due to the secretion of factors that preferentially targeted memory CD8 T cells. IL-15 is a proinflammatory cytokine secreted by dendritic cells, monocytes/macrophages and fibroblasts(20), and likely constitutes a part of the innate immune response to invading pathogens. Previous studies had shown that IL-15 plays a critical role in preferentially regulating homeostatic and infection-driven proliferation of memory CD8 T cells(6-8, 21). Others(9) have shown that treatment of SIV infected rhesus macaques with recombinant IL-15 during the early phase of infection led to a significant expansion of CD8 T cells. We hypothesized that the preferential expansion of memory CD8 T cells might be due to the production of IL-15 in response to early viral replication.
We observed a significant increase in IL-15 levels in the plasma of SIV infected animals at day 7 pi (Fig. 3a) that peaked at day 10 pi and declined by day 14 pi. Plasma IL-15 levels prior to day 10 pi significantly correlated with plasma (r = 0.77, p < 0.0001) and cell-associated viral loads (r = 0.78, p < 0.0001), and Ki-67 expression on memory CD8 T cells (r = 0.69, p < 0.0001) during the same period of infection (Fig. 3c to e) suggesting that increased secretion of IL-15 is a response to early viral infection. There was no significant correlation (r = 0.33, p = 0.1156) between plasma IL-15 levels and Ki-67 expression on memory CD4 T cells between day 0 and 10 pi (Suppl. Fig. 1c). The high levels of plasma IL-15 was surprising as free IL-15 is difficult to detect in the plasma, and likely reflects an early response to viral infection since by day 35 the plasma IL-15 levels had reached baseline values. Previous studies (22) have shown that IL-15 was superior to IL-2 in the generation of long-term antigen specific memory CD8 T cells. The effect of IL-15 on memory CD4 T cells is less clear. Tan et al(23) showed that unlike CD8 memory T cells, homeostatic proliferation of memory CD4 T cells was independent of both IL-7 and IL-15, and others(24-26) have demonstrated that treatment of healthy or SIV infected rhesus macaques with recombinant IL-7 increased the proliferation of both naïve and memory CD4 and CD8 T cell subsets.
Figure 3. Plasma IL-15 levels correlate with acute immune activation.
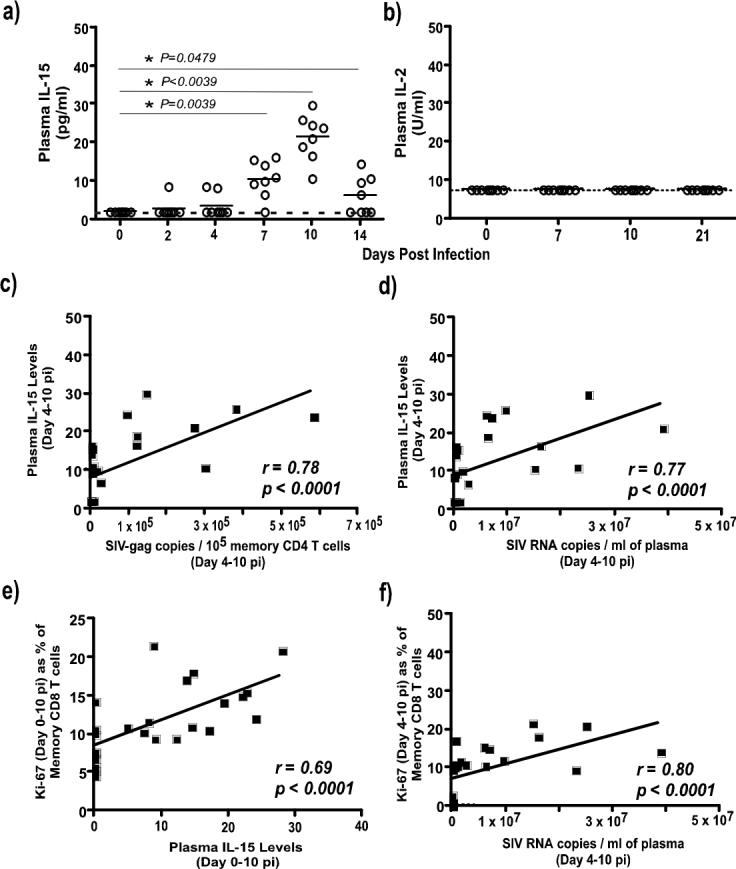
Plasma (a) IL-15 and (b) IL-2 levels were evaluated longitudinally (n = 8) during acute SIV infection. Dotted line represents limit of detection. A significant increase in plasma IL-15 but not IL-2 is seen as early as day 7 pi. Plasma IL-15 significantly correlates with (c) cell-associated viral loads in memory CD4 T cells (d) plasma viral loads and (e) Ki-67 expression on memory CD8 T cells during the early phase of acute SIV infection. Data from 8 animals at day 4, 7 and 10 pi each were used to derive the correlations. (f) Ki-67 expression on memory CD8 T cells significantly correlates with plasma viral loads. Statistical analysis was performed using Wilcoxon matched pairs test. Line of fit was determined using linear regression analysis, and correlations were derived using Spearman's rank test.
Plasma IL-2 (Fig. 3b) was below the level of detection, and no significant changes were observed in plasma IL-7 levels (Suppl. Fig. 1d) suggesting a minimal role of IL-2 and IL-7 in acute immune activation. Though IL-2 mediates its effect locally, the relative absence of IL-2 in plasma, and the demonstration of little or no CD25 expression on CD4 T cells during acute SIV infection(5) argues against a role for IL-2 in the early stages of viral infection. Though surprising, the lack of IL-2 production likely reflects the delay in the generation of SIV specific immune responses and the relatively low level of activation of CD4 T cells. Reynolds et al(27) demonstrated that SIV specific immune responses emerged much later after peak viral infection and loss of CD4 T cells. Likewise, the absence of proliferation and activation of naïve CD4 and CD8 T cells along with memory CD4 T cells during the first 10 days of infection argues against the involvement of IL-7.
CD8 T cells but not CD4 T cells express IL-2Rβ subunit of IL-15R
The absence of proliferation by memory CD4 T cells during the first 10 days of infection even in the presence of significant levels of IL-15 in plasma suggested that IL-15 likely had little effect on these cells. We hypothesized that this may be secondary to the inability of CD4 T cells to bind IL-15 due to the lack of IL-15 receptor (IL-15R) on their surface.
The IL-15R consists of the IL-15Rα subunit along with the IL-2Rα (CD122) and IL-2Rγ (CD132) subunits of the IL-2R. Interaction of the cytokine with the IL-2Rγ and IL-2Rγ on T cells is required for IL-15 induced proliferation of T cells. Studies have, however, shown that IL-15 can bind IL-2Rγ independently of the IL-15Rγ and IL-2Rγ chains(28-30). Similarly, IL-15 has been shown to bind to IL-15Rγ chain directly without the requirement of either IL-2Rγ and IL-2Rγ receptor subunits(31), but this binding does not induce proliferation or activation of T cells. IL-15Rγ is known to have a short cytoplasmic tail, and its role in cell signaling or activation is still under investigation. IL-15 is mostly presented in trans along with IL-15Rγ in vivo.
To determine if IL-15-mediated preferential proliferation of CD8 T cells was due to the differential expression of IL-15R subunits, we evaluated the expression of IL-15Rγ and IL-2Rγ on CD8 T cells and CD4 T cells. CD8 T cells expressed both receptor chains with most cells expressing high levels of IL-2Rγ (Fig. 4a & b). In contrast, CD4 T cells expressed primarily IL-15Rγ but little or very low levels of IL-2Rγ, suggesting that the low level of IL-2Rβ expression on CD4 T cells likely contributes to the low level of proliferation in these cells.
Figure 4. Kinetics of IL-15Rα and IL-2Rβ on T cells.
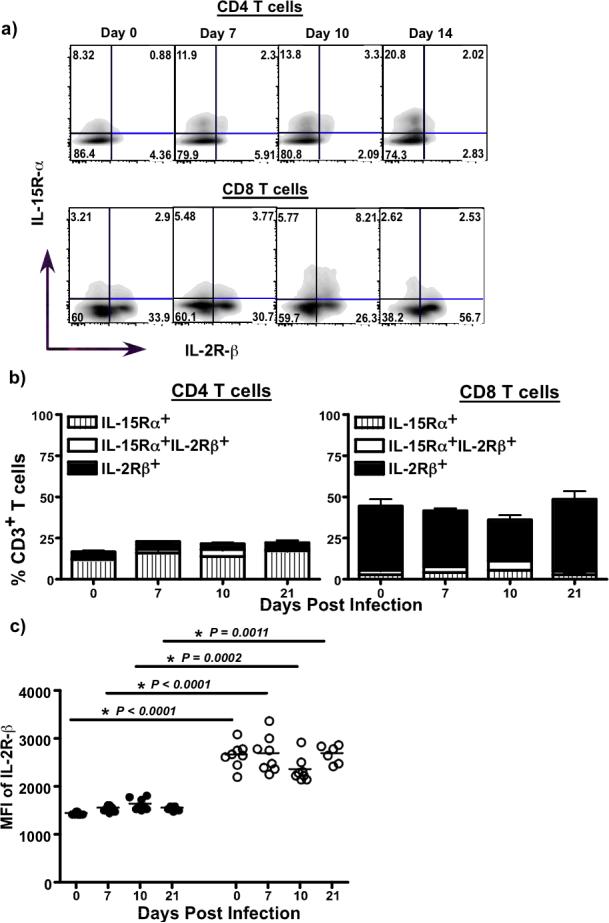
Cryopreserved PBMC samples were used to determine the expression of IL-15Rβ and IL-2Rβ using flow cytometry (a) Representative FACS plots showing the relative expression of IL-15Rβ and IL-2Rβ on total CD3+CD4+ and CD8+ T cells (b) Proportions of IL-15Rβ and IL-2Rβ expressing CD4 and CD8 T cells at day 0 (n = 8), 7 (n = 8), 10 (n = 8) and 21 pi (n = 6). CD4 T cells primarily express IL-15Rβ, whereas CD8 T cells express higher frequencies of IL-2Rβ. (c) Mean fluorescence intensity (MFI) of IL-2Rβ expression of CD3+CD4+ and CD3+CD8+ T cells. CD8 T cells express significantly higher levels of IL-2Rβ as compared to CD4+ T cells.
Density of CD4 expression is significantly upregulated on memory CD4 T cells at day 10 pi
The relative lack of activation in memory CD4 T cells between day 7 and 10 pi suggested that other factors contribute to making the memory CD4 T cells more permissive to SIV. Since SIV like HIV uses CD4 as its primary receptor and CCR5 as its coreceptor, we hypothesized that the increased permissiveness of memory CD4 T cells to SIV between day 7 and 10 pi might be due to the changes in the level of expression of either CD4 and/or CCR5 on these cells. We addressed this question by evaluating the density of CD4 and CCR5 expression on memory CD4 T cells during early SIV infection.
We observed a significant increase in the level of CD4 expression on memory CD4 T cells by day 10 pi as compared to day 7 pi (Fig. 5a & b). The increased density of CD4 expression significantly correlated (r = 0.78, p = 0.0002) with SIV infection in memory CD4 T cells (Fig. 5e). In contrast to the level of CD4 expression, CCR5 expression decreased between day 7 and 10 pi (Fig. 5c & d), and negatively correlated (r = −0.58, p = 0.0084) with SIV loads in memory CD4 T cells. These findings suggest that the high level of CD4 expression likely contributed to more efficient infection in the memory CD4 T cells. On the other hand, the negative correlation of CCR5 expression with cell-associated viral loads indicates that viral infection was associated with the down regulation of CCR5 expression on memory CD4 T cells. The lower frequency and density of CCR5 expression on memory CD4 T cells at day 10 relative to day 0 and 7 pi suggests that CCR5 was not the limiting factor in determining the level of infection in memory CD4 T cells. We have previously shown that memory CD4 T cells expressed enough CCR5 at day 3 − 10 pi to be infected with SIV(2).
Figure 5. Density of CD4 expression on memory CD4 T cells.
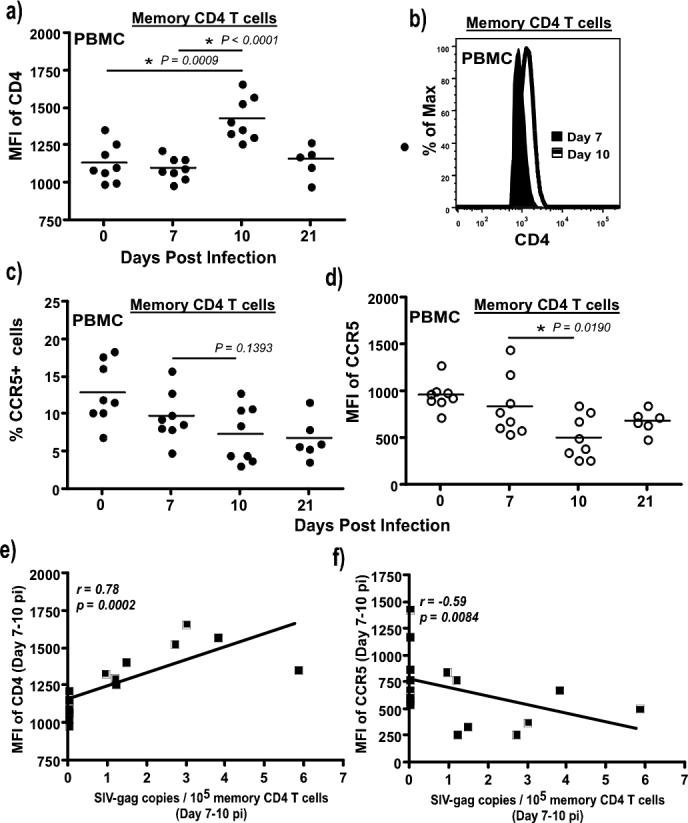
(a) CD4 expression on total memory CD4 T cells (CD95+) is significantly upregulated at day 10 pi as compared to day 0 and 7 pi. Expression of CD4 on memory CD4 T cells was determined by flow cytometry using cryopreserved PBMC from SIV infected rhesus macaques at day 7 (n = 8), 10 (n = 8) and 21 (n = 6) pi, and compared to day 0 (n = 8). All the samples were labeled and analyzed at the same time using a LSR-II instrument (b) representative histogram showing the upregulation of CD4 expression on total CD95+ memory CD4 T cells from PBMC between day 7 and 10 pi. A decrease in the (c) frequency and (d) density of CCR5 expression is observed on memory CD4 T cells between day 7 and 10 pi. (e) Significant correlation is observed between the density of CD4 expression on memory CD4 T cells at day 7 and 10 pi and memory CD4 T cell-associated viral loads, whereas (f) there is an inverse correlation between memory CD4 T cell-associated viral loads and CCR5 expression on memory CD4 T cells. Data from 8 animals at day 7 and 10 pi each were used to derive the correlations. Statistical analysis was performed using Mann-Whitney U test. Line of fit was determined using linear regression analysis, and correlations were derived using Spearman's rank test.
IL-15 upregulates CD4 expression and increases infection of CD4 T cells with SIV
To determine if IL-15 plays a role in upregulating the density of CD4 expression on memory CD4 T cells, and if this increase in the expression of CD4 coincided with a higher level of infection in memory CD4 T cells, we sorted CD4 T cells by negative selection and incubated them with or without IL-15 in the presence/absence of SIVmac251. Additionally, we cultured negatively sorted CD4 T cells in the presence of IL-15 + IL-15Rα-IgG fusion protein to block the effects of IL-15 to confirm if the increase in CD4 expression and infection in sorted cells was primarily mediated by IL-15. IL-15Rα-IgG fusion protein specifically binds and inactivates IL-15 (Fig. 6a).
Figure 6. IL-15 increases infection in CD4 T cells.
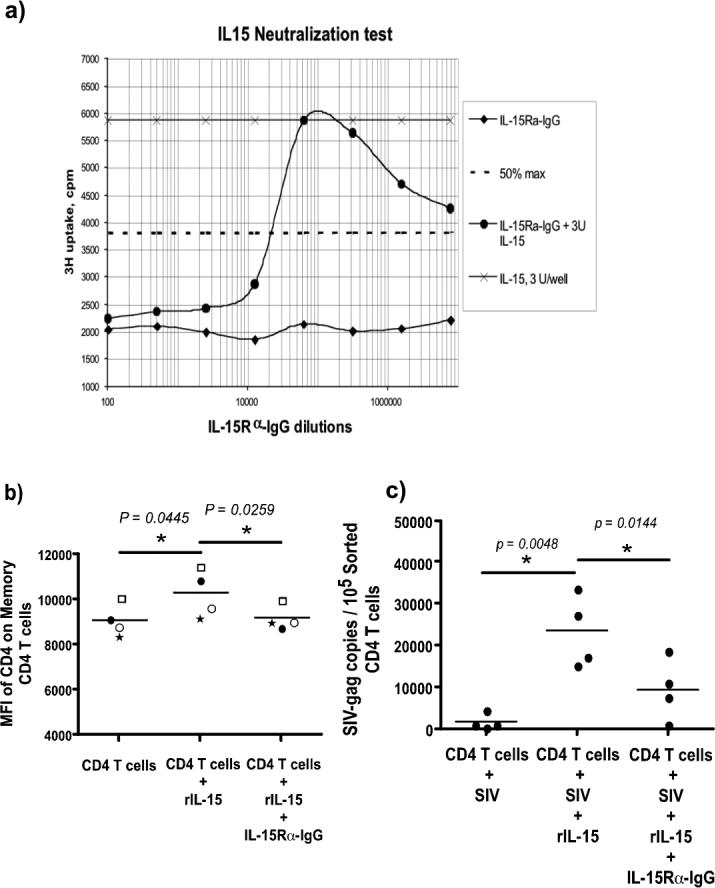
(a) IL-15Rα-IgG fusion protein blocks IL-15 mediated proliferation of HT-2 cells. Serial dilutions of rIL-15Rα-IgG (566 ug / ml) were pre-incubated with 3 Units of rMamu IL-15 for 15 minutes before addition to HT-2 cells. HT-2 cells had been starved for IL-15 for 24 hours. Proliferation was measured after 48 hours using 3H thymidine. Controls consisted of rMamuIL-15 alone, and dilutions of rMamuIL-15Rα-IgG without IL-15. Calculation of the IC50 is based on the 50% inhibition of maximal proliferation. 15Rα-IgG completely blocked IL-15 activity at concentrations as low as 0.566 ug / ml. (b) Sorted CD4 T cells (CD14−CD20−CD8−) from peripheral blood of 4 healthy rhesus macaques were equally divided and cultured in the presence or absence of IL-15 and/or rIL-15Rα-IgG or SIVmac251. The cells were not labeled with either CD3 or CD4 to avoid activating them through these molecules. Highly purified populations of CD4 T cells were used to avoid any bystander effect of cytokines released from other cells. CD4 memory T cells show significant upregulation of CD4 expression on memory CD4 T cells that was blocked by the rIL-15Rα-IgG fusion protein. (c) Sorted CD4 T cells + IL-15 + SIV had a significantly higher level of infection as compared to CD4 T cells + SIV alone or CD4 T cells + SIV + IL-15 + rIL-15Rα-IgG fusion protein. Statistical analysis was performed using Student's paired t test.
As shown in Fig. 6b, the density of expression of CD4 on the memory CD4 T cells treated with IL-15 was significantly upregulated as compared to untreated cells. IL-15Rα-IgG fusion protein blocked IL-15 mediated upregulation of CD4 expression on these cells. Increased expression of CD4 was associated with a significant increase in SIV infection (Fig 6c) in sorted CD4 T cells that was significantly blocked with 15Rα-IgG fusion protein. Taken together, the data from these in vitro culture experiments confirm our findings that IL-15-mediated upregulation of CD4 expression plays an important role in making memory CD4 T cells more susceptible to SIV infection.
Discussion
Acute HIV infection is associated with massive viral replication and loss of memory CD4 T cells in both mucosal and peripheral tissues, key aspects of viral pathogenesis that are well modeled in experimental infection of macaques with SIV. Beginning from a known time of inoculation, there is a lag, with limited viral replication for the first few days, after which infection amplifies explosively within a period of 2−3 days to reach peak by day 10 pi; this process appears to follow the same kinetics in the mucosa and periphery(1, 2). Given the highly activated mucosal tissue microenvironment, it has been generally believed that mucosal CD4 T cells were likely to be preferentially infected as compared to CD4 T cells elsewhere. However, the similarity with which infection proceeds in the mucosa and periphery suggests other factors likely play a role in this process.
Early viral infection is associated with an acute phase type response that likely induces a cascade of events leading to acute immune activation. Our results show that in experimental SIV infection, acute immune activation occurs very early, and prior to the amplification of viral infection within the memory CD4 T cell compartment. Interestingly however, the activation seen during early stages of infection was restricted to memory CD8 T cells without any apparent activation of CD4 T cells, observations in line with previous studies(5, 19). The selective activation of memory CD8 T cells but not memory CD4 T cells suggests that specific cytokines such as IL-15 play a role in this process. We found a significant correlation between the activation of CD8 T cells and plasma IL-15 levels.
It was surprising that increased levels of plasma IL-15 was not associated with increased proliferation or activation of memory CD4 T cells, but significantly correlated with the extent of viral infection in these cells. This apparent paradox was resolved by our findings that few memory CD4 T cells express IL-2Rβ (Fig. 4a-b), and those that express them do so at significantly lower levels as compared to CD8 T cells (Fig. 4c). Since IL-2Rβ signaling is critical for the activation and proliferation of T cells, these data indicate that the relative absence of increased activation and proliferation of memory CD4 T cells during the first 10 days of infection was likely due to the low levels of IL-2Rβ expression.
Burkett et al(32) found that expression of IL-15Rα on T cells was dispensable for the generation of memory CD8 T cells. In murine studies, Kamimura et al(33) showed that CD8 memory cells proliferated even after the depletion of IL-2 in vivo. These memory CD8 T cells expressed IL-2Rα, and blockade of IL-2Rα signaling completely abolished the division of memory CD8 T cells. On the other hand, our results suggest that the low levels of IL-2Rα expression on CD4 T cells likely allows IL-15 to interact with these cells. However, the relative lack of any change in Ki-67 or HLA-DR expression on CD4 T cells during the first 10 days after infection indicates that IL-15 interaction with IL-2Rα on CD4 T cells was likely not strong enough to induce activation and proliferation but was sufficient to make the CD4 T cells more susceptible to SIV infection. Previous studies(34, 35) have shown that IL-15 can enhance HIV replication in the absence of proliferation.
Kaur et al(5) reported little change in Ki-67+CD4+ T cells during the first 4 weeks of acute SIV infection. On the other hand, Picker et al(11) reported an increased proliferation of effector memory CD4 T cells after administration of IL-15 protein to rhesus macaques. We did not specifically evaluate effector memory CD4 T cells. However, it is highly likely that the high dose of exogenous IL-15 (10 ug/Kg body weight/twice a week) used by Picker et al may have contributed to the proliferation of effector memory CD4 T cells by inducing the secretion of other factors, whereas the highest level of plasma IL-15 we observed was ∼25 − 30 pg / ml at day 10 pi. Importantly, our findings indicate that increased activation or proliferation of memory CD4 T cells was not the primary cause for the increase in the level of infection in these cells at day 10 pi as compared to day 7 pi.
Though the low levels of IL-2Rβ explained the absence of any significant level of activation of memory CD4 T cells, it did not explain the highly significant correlation between plasma IL-15 levels and viral infection in memory CD4 T cells. We hypothesized that IL-15 interaction with the low levels of IL-2Rβ was sufficient to upregulate the expression of the receptors for SIV namely, CD4 and CCR5. In fact, we found a significant increase in the density of CD4 expression on memory CD4 T cells between day 7 and 10 pi that coincided with the increased level of SIV infection in these cells. SIV-gp120 binds to CD4 on CD4 T cells along with CCR5, and higher densities of CD4 expression on memory CD4 T cells significantly correlated (r = 0.78, p= 0.0002) with viral infection in these cells, suggesting that SIV may have bound to memory CD4 T cells more efficiently between day 7 and 10 pi leading to a higher a higher frequency of these cells being infected by day 10 pi.
Previous studies have shown that HIV binds weakly to cells expressing low levels of CD4(36), and that the low level of CD4 expression on macrophages played an important role in restricting the entry of T tropic SIV strains such as SIVmac239(37); this restriction could not be overcome by the overexpression of CCR5 on these cells. Kozak et al found that CD4 rather than CCR5 or CXCR4 expression determines the kinetics and pathways for gp120 binding, endocytosis, and proteolysis on cells that contain sufficient coreceptor for efficient infection(38). Platt et al showed that cells with large amounts of CD4 and low traces of CCR5 are sufficient for maximum susceptibility to HIV-1 strains(39). Finally, Pesenti et al demonstrated that susceptibility of macrophages to HIV-1 is significantly increased when they express high levels of CD4 as compared to macrophages that expressed low levels of CD4, whereas there was no significant difference in susceptibility based on CCR5 expression(40).
These studies support our observations that the low level of infection in memory CD4 T cells coincided with the lower expression of CD4 on these cells, whereas an increased level of expression was associated with near total infection of most memory CD4 T cells. It is highly unlikely that the increased viral replication is the cause for the increased density of CD4 expression. If this were true, then at day 14 pi when the plasma viral loads are 2 logs higher than at day 7 pi, all of the memory CD4 T cells present should have been infected and carry viral DNA. This is however not the case as the frequency of CD4 memory T cells in both peripheral and mucosal tissues (Fig. 1c) that carry viral DNA at day 14 pi are less than 20% whereas most of the memory CD4 T cells at day 10 pi are infected and carry viral DNA. Likewise, the plasma viral loads are quite high at day 7 pi yet the level of infection in memory CD4 T cells is much lower than what is seen at day 10 pi.
In vitro culture experiments using highly purified CD4 T cells in the presence or absence of recombinant IL-15 confirmed our hypothesis that IL-15 plays a major role in early infection. IL-15 treatment upregulated the level of CD4 expression on highly purified CD4 T cells, an effect that was accompanied by a higher frequency of sorted CD4 T cells being infected with SIV (Fig. 6). The use of a defined system comprised of purified populations of CD4 T cells, and purified recombinant IL-15 minimized potential bystander effects from other cells.
We found no significant change in the level of plasma IL-7 levels during the early phase of infection (Suppl. Fig. 1d). IL-7 is a cytokine produced by non-lymphoid cells, and plays a role in the homeostatic expansion of both naïve and memory CD4 and CD8 T cells(21, 25, 26, 41). Though we cannot completely rule out the potential role of IL-7 in acute immune activation we observed due to the small sample size, the relative lack of proliferation and activation of naïve CD4 and naïve CD8 T cells (Suppl. Fig. 2a & b), and the minimal changes in proliferation and activation of memory CD4 T cells (Fig. 2a & b) all argue against a prominent role for IL-7 in immune activation seen during the first 10 days of acute SIV infection. The exact role of IL-7 may need to be evaluated using a larger group of animals to better clarify its contribution to early infection.
In conclusion, our data shows that IL-15 plays an important role in acute immune activation and increasing the susceptibility of memory CD4 T cells to SIV infection by upregulating the expression of CD4, the primary receptor for SIV, on memory CD4 T cells. Though our studies focused on peripheral memory CD4 T cells, the similarities in the kinetics of infection between peripheral and mucosal CD4 T cells suggest that IL-15 likely plays a role in increasing the susceptibility of CD4 T cells in the mucosa to SIV infection. However, additional studies need to be performed using a larger cohort of animals to better understand the role IL-15 plays in immune activation and immunopathogenesis of SIV infection in mucosal tissues.
Supplementary Material
Supplementary Figure 1. (a) FACS plots showing purity of CD4 T cells sorted using the BD FACS Aria sorter. CD4 T cells were negatively selected using a panel of anti-CD14, CD20 and CD8. Sorted CD4 T cells was > 99% pure. (b) There was no change in the level of CCR5 expression on CD8 T cells. Cryopreserved PBMC was used to determine CCR5 expression on CD8 T cells. All the samples were labeled and analyzed at the same time using a LSR-II instrument. (c) There was no significant correlation between Ki-67 expression on memory CD4 T cells and plasma IL-15 levels. Statistical analysis was performed using Wilcoxon matched pairs test. Line of fit was determined using linear regression analysis, and correlations were derived using Spearman's rank test. (d) There is no significant change in IL-7 levels in plasma (n = 4) during the early phase of infection. Statistical analysis was performed using Mann Whitney U test
Supplementary Figure 2. Memory CD4 and CD8 T cells, but not naïve CD4 and CD8 T cells express (a) Ki-67 and (b) HLA-DR during the early phase of SIV infection. Representative FACS plots showing Ki-67 expression on naïve (CD95−) and memory (CD95+) T cells. Representative FACS plots showing HLA-DR expression on naïve (CD95−) and memory (CD95+) T cells are shown.
Supplementary Figure 3. (a) Schematic representation of rMamuIL-15Rα-IgG fusion protein. (b) Nucleotide sequence encoding mature protein with the corresponding amino acid sequence below it. Bolded nucleotides and amino acids indicate mutations and resultant amino acid substitutions L235A and P331S introduced into the IgG Fc portion of the molecule, which abolish binding to Fc receptors and to complement, respectively.
Acknowledgments
We thank Nancy Miller at the SVEU of NIAID for help with the animals; Enrico Luigi, Kaimei Song and Diane Bolton at the Vaccine Research Center; Karen Wolcott and Kateryna Lund at the Biomedical Instrumentation Center; Dr. Deborah Weiss and Jim Treece at ABL, Inc, Rockville, MD for expert assistance with the animals.
The described project was supported by Grant Number K22 AI07812 from the National Institutes of Allergy and Infectious diseases (NIAID) & Grant number R21 DE018339 from the National Institute of Dental and Craniofacial Research (NIDCR) awarded to JJM., and in part from R01 AI062437 from NIAID awarded to P.D.K. Studies were supported in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract NO1-CO-124000. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIAID or NIDCR or NCI or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 2.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 3.Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 4.Cumont MC, Diop O, Vaslin B, Elbim C, Viollet L, Monceaux V, Lay S, Silvestri G, Le Grand R, Muller-Trutwin M, Hurtrel B, Estaquier J. Early divergence in lymphoid tissue apoptosis between pathogenic and nonpathogenic simian immunodeficiency virus infections of nonhuman primates. J Virol. 2008;82:1175–1184. doi: 10.1128/JVI.00450-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaur A, Hale CL, Ramanujan S, Jain RK, Johnson RP. Differential dynamics of CD4(+) and CD8(+) T-lymphocyte proliferation and activation in acute simian immunodeficiency virus infection. J Virol. 2000;74:8413–8424. doi: 10.1128/jvi.74.18.8413-8424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmad A, Ahmad R, Iannello A, Toma E, Morisset R, Sindhu ST. IL-15 and HIV infection: lessons for immunotherapy and vaccination. Curr HIV Res. 2005;3:261–270. doi: 10.2174/1570162054368093. [DOI] [PubMed] [Google Scholar]
- 7.Alpdogan O, van den Brink MR. IL-7 and IL-15: therapeutic cytokines for immunodeficiency. Trends Immunol. 2005;26:56–64. doi: 10.1016/j.it.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 9.Mueller YM, Do DH, Altork SR, Artlett CM, Gracely EJ, Katsetos CD, Legido A, Villinger F, Altman JD, Brown CR, Lewis MG, Katsikis PD. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J Immunol. 2008;180:350–360. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller YM, Petrovas C, Do DH, Altork SR, Fischer-Smith T, Rappaport J, Altman JD, Lewis MG, Katsikis PD. Early establishment and antigen dependence of simian immunodeficiency virus-specific CD8+ T-cell defects. J Virol. 2007;81:10861–10868. doi: 10.1128/JVI.00813-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Picker LJ, Reed-Inderbitzin EF, Hagen SI, Edgar JB, Hansen SG, Legasse A, Planer S, Piatak M, Jr., Lifson JD, Maino VC, Axthelm MK, Villinger F. IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J Clin Invest. 2006;116:1514–1524. doi: 10.1172/JCI27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cline AN, Bess JW, Piatak M, Jr., Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 13.Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 14.Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 15.Lifson JD, Rossio JL, Piatak M, Jr., Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol. 2001;75:10187–10199. doi: 10.1128/JVI.75.21.10187-10199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller YM, Bojczuk PM, Halstead ES, Kim AH, Witek J, Altman JD, Katsikis PD. IL-15 enhances survival and function of HIV-specific CD8+ T cells. Blood. 2003;101:1024–1029. doi: 10.1182/blood-2002-07-1957. [DOI] [PubMed] [Google Scholar]
- 17.Onlamoon N, Rogers K, Mayne AE, Pattanapanyasat K, Mori K, Villinger F, Ansari AA. Soluble PD-1 rescues the proliferative response of simian immunodeficiency virus-specific CD4 and CD8 T cells during chronic infection. Immunology. 2008;124:277–293. doi: 10.1111/j.1365-2567.2007.02766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villinger F, Brar SS, Mayne A, Chikkala N, Ansari AA. Comparative sequence analysis of cytokine genes from human and nonhuman primates. J Immunol. 1995;155:3946–3954. [PubMed] [Google Scholar]
- 19.Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, Staprans SI, Reilly CS, Silvestri G, Haase AT. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol. 2008;180:6798–6807. doi: 10.4049/jimmunol.180.10.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weng NP, Liu K, Catalfamo M, Li Y, Henkart PA. IL-15 is a growth factor and an activator of CD8 memory T cells. Ann N Y Acad Sci. 2002;975:46–56. doi: 10.1111/j.1749-6632.2002.tb05940.x. [DOI] [PubMed] [Google Scholar]
- 21.Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17:183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Villinger F, Miller R, Mori K, Mayne AE, Bostik P, Sundstrom JB, Sugimoto C, Ansari AA. IL-15 is superior to IL-2 in the generation of long-lived antigen specific memory CD4 and CD8 T cells in rhesus macaques. Vaccine. 2004;22:3510–3521. doi: 10.1016/j.vaccine.2003.07.022. [DOI] [PubMed] [Google Scholar]
- 23.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. The Journal of experimental medicine. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beq S, Nugeyre MT, Ho Tsong Fang R, Gautier D, Legrand R, Schmitt N, Estaquier J, Barre-Sinoussi F, Hurtrel B, Cheynier R, Israel N. IL-7 induces immunological improvement in SIV-infected rhesus macaques under antiviral therapy. J Immunol. 2006;176:914–922. doi: 10.4049/jimmunol.176.2.914. [DOI] [PubMed] [Google Scholar]
- 25.Fry TJ, Moniuszko M, Creekmore S, Donohue SJ, Douek DC, Giardina S, Hecht TT, Hill BJ, Komschlies K, Tomaszewski J, Franchini G, Mackall CL. IL-7 therapy dramatically alters peripheral T-cell homeostasis in normal and SIV-infected nonhuman primates. Blood. 2003;101:2294–2299. doi: 10.1182/blood-2002-07-2297. [DOI] [PubMed] [Google Scholar]
- 26.Moniuszko M, Fry T, Tsai WP, Morre M, Assouline B, Cortez P, Lewis MG, Cairns S, Mackall C, Franchini G. Recombinant interleukin-7 induces proliferation of naive macaque CD4+ and CD8+ T cells in vivo. J Virol. 2004;78:9740–9749. doi: 10.1128/JVI.78.18.9740-9749.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds MR, Rakasz E, Skinner PJ, White C, Abel K, Ma ZM, Compton L, Napoe G, Wilson N, Miller CJ, Haase A, Watkins DI. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: too late and too little. J Virol. 2005;79:9228–9235. doi: 10.1128/JVI.79.14.9228-9235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bamford RN, Grant AJ, Burton JD, Peters C, Kurys G, Goldman CK, Brennan J, Roessler E, Waldmann TA. The interleukin (IL) 2 receptor beta chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc Natl Acad Sci U S A. 1994;91:4940–4944. doi: 10.1073/pnas.91.11.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulfone-Pau SS, Bulanova E, Pohl T, Budagian V, Durkop H, Ruckert R, Kunzendorf U, Paus R, Krause H. Death deflected: IL-15 inhibits TNF-alpha-mediated apoptosis in fibroblasts by TRAF2 recruitment to the IL-15Ralpha chain. Faseb J. 1999;13:1575–1585. doi: 10.1096/fasebj.13.12.1575. [DOI] [PubMed] [Google Scholar]
- 30.Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, Namen A, Park LS, Cosman D, Anderson D. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. Embo J. 1994;13:2822–2830. doi: 10.1002/j.1460-2075.1994.tb06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chae DW, Nosaka Y, Strom TB, Maslinski W. Distribution of IL-15 receptor alpha-chains on human peripheral blood mononuclear cells and effect of immunosuppressive drugs on receptor expression. J Immunol. 1996;157:2813–2819. [PubMed] [Google Scholar]
- 32.Burkett PR, Koka R, Chien M, Chai S, Chan F, Ma A, Boone DL. IL-15R alpha expression on CD8+ T cells is dispensable for T cell memory. Proc Natl Acad Sci U S A. 2003;100:4724–4729. doi: 10.1073/pnas.0737048100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamimura D, Ueda N, Sawa Y, Hachida S, Atsumi T, Nakagawa T, Sawa S, Jin GH, Suzuki H, Ishihara K, Murakami M, Hirano T. Evidence of a novel IL-2/15R beta-targeted cytokine involved in homeostatic proliferation of memory CD8+ T cells. J Immunol. 2004;173:6041–6049. doi: 10.4049/jimmunol.173.10.6041. [DOI] [PubMed] [Google Scholar]
- 34.Al-Harthi L, Roebuck KA, Landay A. Induction of HIV-1 replication by type 1-like cytokines, interleukin (IL)-12 and IL-15: effect on viral transcriptional activation, cellular proliferation, and endogenous cytokine production. J Clin Immunol. 1998;18:124–131. doi: 10.1023/a:1023246800353. [DOI] [PubMed] [Google Scholar]
- 35.Patki AH, Quinones-Mateu ME, Dorazio D, Yen-Lieberman B, Boom WH, Thomas EK, Lederman MM. Activation of antigen-induced lymphocyte proliferation by interleukin-15 without the mitogenic effect of interleukin-2 that may induce human immunodeficiency virus-1 expression. J Clin Invest. 1996;98:616–621. doi: 10.1172/JCI118831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozak SL, Platt EJ, Madani N, Ferro FE, Jr., Peden K, Kabat D. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J Virol. 1997;71:873–882. doi: 10.1128/jvi.71.2.873-882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bannert N, Schenten D, Craig S, Sodroski J. The level of CD4 expression limits infection of primary rhesus monkey macrophages by a T-tropic simian immunodeficiency virus and macrophagetropic human immunodeficiency viruses. J Virol. 2000;74:10984–10993. doi: 10.1128/jvi.74.23.10984-10993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozak SL, Kuhmann SE, Platt EJ, Kabat D. Roles of CD4 and coreceptors in binding, endocytosis, and proteolysis of gp120 envelope glycoproteins derived from human immunodeficiency virus type 1. The Journal of biological chemistry. 1999;274:23499–23507. doi: 10.1074/jbc.274.33.23499. [DOI] [PubMed] [Google Scholar]
- 39.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. Journal of virology. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pesenti E, Pastore C, Lillo F, Siccardi AG, Vercelli D, Lopalco L. Role of CD4 and CCR5 levels in the susceptibility of primary macrophages to infection by CCR5-dependent HIV type 1 isolates. AIDS research and human retroviruses. 1999;15:983–987. doi: 10.1089/088922299310494. [DOI] [PubMed] [Google Scholar]
- 41.Prlic M, Lefrancois L, Jameson SC. Multiple choices: regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. The Journal of experimental medicine. 2002;195:F49–52. doi: 10.1084/jem.20020767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (a) FACS plots showing purity of CD4 T cells sorted using the BD FACS Aria sorter. CD4 T cells were negatively selected using a panel of anti-CD14, CD20 and CD8. Sorted CD4 T cells was > 99% pure. (b) There was no change in the level of CCR5 expression on CD8 T cells. Cryopreserved PBMC was used to determine CCR5 expression on CD8 T cells. All the samples were labeled and analyzed at the same time using a LSR-II instrument. (c) There was no significant correlation between Ki-67 expression on memory CD4 T cells and plasma IL-15 levels. Statistical analysis was performed using Wilcoxon matched pairs test. Line of fit was determined using linear regression analysis, and correlations were derived using Spearman's rank test. (d) There is no significant change in IL-7 levels in plasma (n = 4) during the early phase of infection. Statistical analysis was performed using Mann Whitney U test
Supplementary Figure 2. Memory CD4 and CD8 T cells, but not naïve CD4 and CD8 T cells express (a) Ki-67 and (b) HLA-DR during the early phase of SIV infection. Representative FACS plots showing Ki-67 expression on naïve (CD95−) and memory (CD95+) T cells. Representative FACS plots showing HLA-DR expression on naïve (CD95−) and memory (CD95+) T cells are shown.
Supplementary Figure 3. (a) Schematic representation of rMamuIL-15Rα-IgG fusion protein. (b) Nucleotide sequence encoding mature protein with the corresponding amino acid sequence below it. Bolded nucleotides and amino acids indicate mutations and resultant amino acid substitutions L235A and P331S introduced into the IgG Fc portion of the molecule, which abolish binding to Fc receptors and to complement, respectively.