

Abstract
Current literature suggests that chronic nitric oxide synthase (NOS) inhibition has differential effects on endothelium-dependent dilation (EDD) of conduit arteries vs. arterioles. Therefore, we hypothesized that chronic inhibition of NOS would impair EDD of porcine left anterior descending (LAD) coronary arteries but not coronary arterioles. Thirty-nine female Yucatan miniature swine were included in the study. Animals drank either tap water or water with NG-nitro-L-arginine methyl ester (L-NAME; 100 mg/l), resulting in control and chronic NOS inhibition (CNI) groups, respectively. Treatment was continued for 1–3 mo (8.3 ± 0.6 mg · kg−1 · day−1). In vitro EDD of coronary LADs and arterioles was assessed via responses to ADP (LADs only) and bradykinin (BK), and endothelium-independent function was assessed via responses to sodium nitroprusside (SNP). Chronic NOS inhibition diminished coronary artery EDD to ADP and BK. Incubating LAD rings with L-NAME decreased relaxation responses of LADs from control pigs but not from CNI pigs such that between-group differences were abolished. Neither indomethacin (Indo) nor sulfaphenazole incubation significantly affected relaxation responses of LAD rings to ADP or BK. Coronary arteries from CNI pigs showed enhanced relaxation responses to SNP. In contrast to coronary arteries, coronary arterioles from CNI pigs demonstrated preserved EDD to BK and no increase in dilation responses to SNP. L-NAME, Indo, and L-NAME + Indo incubation did not result in significant between-group differences in arteriole dilation responses to BK. These results suggest that although chronic NOS inhibition diminishes EDD of LAD rings, most likely via a NOS-dependent mechanism, it does not affect EDD of coronary arterioles.
Keywords: endothelium-dependent dilation, NG-nitro-L-arginine methyl ester
A healthy vascular endothelium promotes arterial homeostasis; in contrast, an injured endothelium portends arterial dysfunction (28). Although the mechanisms by which endothelial dysfunction exacts vascular damage remain to be fully elucidated, it is thought that reduced nitric oxide (NO) production and/or scavenging play a crucial role (27).
Chronically decreased NO production by nitric oxide synthase (NOS) inhibition results in significant hemodynamic alterations. Systemically, NOS inhibition via NG-nitro-L-argi-nine methyl ester (L-NAME) or other NOS inhibitor administration increases blood pressure and peripheral resistance while decreasing heart rate and cardiac output (4, 11, 18, 21, 24, 29). The fact that chronic NOS inhibition causes increased mean arterial pressure in vivo could be evidence that chronic NOS inhibition has relatively greater effects on arterioles/resistance arteries. That is, if chronic NOS inhibition causes increased blood pressure because of a greater effect of this treatment on arterioles, one might expect that chronic NOS inhibition would have greater effects on intrinsic endothelium-dependent dilation of arterioles than of conduit arteries.
In contrast to this line of reasoning that suggests chronic NOS inhibition has greater effects on endothelial function of arterioles than of conduit arteries, current literature indicates that chronic NOS inhibition has greater effects on the endothelium of conduit arteries than arterioles. For example, chronic administration of NOS inhibitors has been shown to decrease endothelium-dependent dilation of the coronary (24), femoral (24), aorta (18), mesenteric (11), and basilar (21) arteries. Likewise, blunted endothelial function has been demonstrated in the carotid artery (6) and aorta (2, 16, 17) of endothelial NOS (eNOS) gene-disrupted mice. In contrast, gracilis (25), coronary (9), and pial arterioles (20) demonstrated preserved endothelial function in eNOS knockouts, and chronic NOS inhibition had no effect on gracilis arterioles (29). Indeed, upregulation of alternative pathways within the endothelium have been shown to compensate for a lack of NO in arterioles (9, 25, 29). Although the literature summarized suggests greater effects of chronic NOS inhibition on endothelium-dependent dilation of conduit arteries than arterioles, this hypothesis has not been previously tested. Therefore, the purpose of this study was to test the hypothesis that chronic L-NAME treatment would impair endothelium-dependent function of conduit arteries more than arterioles in the coronary arterial tree. We tested this hypothesis by chronically treating pigs with L-NAME and then comparing endothelium-dependent dilation of porcine left anterior descending (LAD) coronary arteries and left ventricular (LV) coronary arterioles in normal and treated pigs.
METHODS
Experimental Design
Thirty-nine female Yucatan miniature swine were included in the study. Animals drank either tap water or water with L-NAME (100 mg/l), resulting in control (n = 21) and chronic NOS inhibition (CNI; n = 18) groups, respectively. Treatment was continued for 1–3 mo (8.3 ± 0.6 mg · kg−1 · day−1) and was efficacious, as indicated by significantly different mean arterial blood pressures between control (84 ± 4 mmHg, n = 8) and CNI (118 ± 7 mmHg, n = 8) groups. In addition, plasma NO metabolite levels were significantly reduced in the CNI group (12.2 ± 1.9 μM, n = 19 vs. 6.8 ± 0.7 μM, n = 14).
In Vitro Assessment of Coronary Vasomotor Function
Isolation of coronary arteries and arterioles
Under anesthesia, hearts were removed and placed in cold Krebs-bicarbonate buffer (4°C). The LAD was dissected from the same location in all pigs and subsequently trimmed of connective tissue and fat. The LAD was segmented into rings, and their outer diameter, inner diameter, and axial length were measured with an Olympus microscope and NIH ImageJ software. As previously described (26), rings were mounted on myographs and diameter was adjusted to elicit a maximal response to KCl (Lmax). In addition, myocardial arterioles (75–150 μm in diameter) were isolated from the LAD field within the LV apex. As previously described (10), arterioles were cannulated with micropipettes and pressurized to 60 cmH2O. With an inverted microscope, arterioles were visualized on a monitor; video micrometers were utilized to measure diameter.
Coronary artery protocol
Mounted rings were preconstricted with prostaglandin F2α (PGF2α; 30 μM), and in vitro endothelial function was assessed via isometric relaxation responses to ADP (10−9 to 10−4 M) and bradykinin (BK; 10−11 to 10−6 M). Endothelium-independent function was then assessed via relaxation responses to sodium nitroprusside (SNP; 10−10 to 10−4 M). For each pig, eight LAD rings were studied: ring 1 was left untreated, ring 2 was incubated with L-NAME (300 μM), ring 3 was incubated with indomethacin (Indo; 5 μM), ring 4 was incubated with sulfaphenazole (Sulf; 10 μM), ring 5 was incubated with L-NAME + Indo, ring 6 was incubated with L-NAME + Sulf, ring 7 was incubated with L-NAME + Indo +Sulf, and ring 8 was incubated with L-NAME +Indo + Sulf + KCl (20 μM). L-NAME incubation inhibited NOS, Indo inhibited cyclooxygenase (COX), Sulf inhibited cytochrome P-450 epoxygenase (8), and KCl inhibited the action(s) of endothelium-derived hyperpolarizing factor (EDHF); hence, the roles of NO, prostacyclin (PGI2), and EDHF in vasorelaxation could be elucidated. In addition, a subset of rings from control pigs had their endothelium denuded. Following generation of each dose-response curve, Krebs-bicarbonate buffer solution was replaced and rings were allowed to stabilize at resting tension.
Coronary arteriole protocol
In vitro endothelial function of coronary arterioles was assessed via dilatory responses to BK (3 × 10−15 to 10−8 M), and endothelium-independent function was assessed via dilation responses to SNP (10−9 to 10−4 M). In addition, the contributions of NO, PGI2, and NO + PGI2 to endothelium-dependent dilation were evaluated by incubating arterioles with L-NAME (300 μM), Indo (5 μM), or L-NAME + Indo before generating dose-response curves, respectively. Repeatability of BK dose-response curves (i.e., time control) was verified in a subset of untreated arterioles.
Analysis of eNOS Gene Expression
Isolation of coronary arteries
Expression of eNOS (protein content in LAD and arterioles, eNOS mRNA in arterioles) was examined in arteries isolated from five control and 4 CNI pigs. Pigs were anesthetized, and hearts were removed and maintained at 0–4°C in Krebs solution during vessel isolation as described above. Segments of LAD and LV arterioles were carefully dissected from LV myocardium, fat, and connective tissue. Arterioles were isolated from myocardium (LV free wall) by dissecting along the length of small arteries to the smallest branches. Single arterioles 1–2 mm in length and similar in diameter were isolated, and five arterioles were pooled into one sample to provide sufficient total protein in each sample. All samples were placed in microcentrifuge tubes, immediately frozen, and stored at −70°C.
Immunoblot procedures
All samples of arteries and arterioles were solubilized in Laemmli buffer (62.5 mM Tris, pH 6.8, 6 M urea, 160 mM 1,4-dithiothreitol, 2% SDS, and 0.0001% bromphenol blue), boiled, vortexed, and sonicated. Cell lysates were analyzed for total protein content using Nano-Orange protein assay (Invitrogen, Molecular Probes). Each well of the gel was loaded with 10 μg of total protein and subjected to SDS-PAGE under reducing conditions. Proteins were transferred to polyvinylidene difluoride membrane (Hybond-ECL; Amersham) and blocked (1 h at 25°C) with 5% nonfat milk in TBST (20 mM Tris · HCl, 137 mM NaCl, and 0.1% Tween 20). Blots were incubated overnight (25°C) with primary antibody against eNOS (1:1,000; Transduction Laboratories), followed by incubation for 1 h with secondary antibody (1:2,500 horseradish peroxidase-conjugated anti-mouse). For comparisons between control and CNI samples, equal numbers of samples from both groups were loaded in the same gel. Blots were subsequently reprobed overnight for superoxide dismutase (SOD)-1 (1:5,000; Stressgen) and SOD-3 (1:1,000; Stressgen), both with 1:2,500 anti-rabbit secondary antibody. Specific protein bands were detected with chemiluminescence (Pierce SuperSignal West) and quantified with a Kodak 4000R Imager and Molecular Imaging software.
Measurement of eNOS mRNA in coronary arterioles
eNOS mRNA measurement was performed as described previously (19). Briefly, isolated arterioles were lysed with 100 mM Tris·HCl, 500 mM LiCl, 10 mM EDTA, 1.0 ml/100 ml lithium disulfide, and 5 mM dithiothreitol, pH 7.5. Using magnetic beads with oligo(dT) and reverse transcriptase (200 U/μl), mRNA was isolated and first-strand synthesis was performed. Polymerase chain reaction (PCR) was used to amplify the eNOS cDNA with the primers 5′-AGG CAT CAC CAG GAA GAA GA-3′ (forward) and 5′-GGC CAG TCT CAG AGC CAT AC-3′ (reverse) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA with primers 5′-ACT CTA CCC ACG GCA AGT TC-3′ (forward) and 5′-TAC TCA GCA CCA GCA TCA CC-3′ (reverse). The (40×) PCR cycles were 30 s at 95°C, 60 s at 60°C, and 60 s at 75°C. The reaction mixture consisted of a master mixture (dNTPs, DNA polymerase, SYBR green), MgCl2 (2.5 mM), primers (100 nM), and either 5.0 μl of eNOS cDNA or 2.0 μl of GAPDH cDNA. eNOS expression was normalized to GAPDH expression.
Solutions and Drugs
Krebs-bicarbonate buffer solution contained (in mM) 131.5 NaCl, 5.0 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 11.2 glucose, 20.8 NaHCO3, 0.003 propranolol, and 0.025 EDTA. Solutions were aerated with 95% O2-5% CO2 (pH 7.4) and maintained at 37°C. All drugs and chemicals were purchased from Sigma Chemical.
Statistical Analysis
Values are expressed as means (SD) or ± SE. Percent relaxation of conduits was calculated as 100 × (PGF2α-induced tension − dose tension)/(PGF2α-induced tension − resting tension). Percent possible dilation of arterioles was calculated as 100 × (response diameter − tone diameter)/(maximum diameter −tone diameter), where response diameter is the diameter after drug administration, tone diameter is the diameter at baseline with spontaneous tone, and maximum diameter is the Ca2+-free diameter (determined at the end of the experiment). Contributions of NO and prostaglandins in both artery and arteriole dilation were calculated by subtracting the dose-response in the presence of inhibitor (L-NAME or Indo) from that in the absence of the inhibitor. Half-maximal effective dose (ID50) values were calculated using GraphPad Prism. Differences between groups with respect to ring characteristics, maximal responses, and ID50 (expressed as negative log of molar concentration) were determined via unpaired t-test. Dose-response curves of both conduits and arterioles were analyzed via a general linear model with (dose) repeated measures, and individual curves were compared with Fisher’s protected least significant difference post hoc analysis. Significance was set at P < 0.05. All data were analyzed in SuperANOVA.
RESULTS
Coronary Artery Characteristics
There were no significant differences between control and CNI LAD length, outer diameter, inner diameter, or wall thickness (Table 1). Likewise, there were no significant differences in resting tension or percent stretch to Lmax. We also examined tensions of LAD rings following incubation with L-NAME before dose-response curves, and we found that L-NAME incubation significantly increased resting tension within the control (change = 1.51 ± 0.48 g, P = 0.002) but not the CNI group (change = 0.31 ± 0.45 g, P = 0.487).
Table 1.
Coronary artery characteristics
| Variable | Control | CNI |
|---|---|---|
| n | 14 | 14 |
| Outer diameter, mm | 1.86 (0.31) | 1.84 (0.26) |
| Inner diameter, mm | 1.18 (0.26) | 1.16 (0.17) |
| Wall thickness, mm | 0.34 (0.08) | 0.34 (0.06) |
| Axial length, mm | 3.43 (0.60) | 3.37 (0.53) |
| Resting tension at Lmax, g | 5.97 (1.54) | 5.77 (1.37) |
| Percent stretch to Lmax, % | 168 (9) | 166 (9) |
Values are means (SD); n, no. of pigs. CNI, chronic nitric oxide synthase inhibition. Independent t-tests revealed no significant differences between groups.
Coronary Artery In Vitro Function
LAD ADP responses
Increasing concentrations of ADP elicited increasing relaxation responses in LADs of both control and CNI groups (Fig. 1 and Table 2). LADs from CNI pigs showed significantly diminished responses to ADP compared with control rings, as well as lower ID50 values, but not lower maximal responses (P = 0.116). Incubating rings with L-NAME alone significantly decreased relaxation responses of LADs from control pigs but not from CNI pigs (Fig. 1B), and between-group differences were abolished with respect to dose-response curves and ID50 values. Indo incubation did not affect dose-response curves of either group (Fig. 1C). L-NAME + Indo incubation had the same effect as L-NAME incubation, abolishing between-group differences (Fig. 1D). Sulf treatment alone tended to shift the dose-response curves for ADP-induced relaxation in both groups to the left (P = 0.065 in control, P = 0.051 in CNI). Although Sulf incubation did not affect between-group differences, L-NAME + Sulf, L-NAME + Indo + Sulf, and L-NAME + Indo + Sulf + KCl incubation abolished between-group differences (data not shown). The contribution of NO produced by NOS to ADP-induced relaxation (reflected in the amount of relaxation inhibited by L-NAME) was greater in control than in CNI LADs (Fig. 2A).
Fig. 1.

ADP-induced vasorelaxation responses of preconstricted coronary artery rings. Values are means ± SE in control and chronic nitric oxide synthase (NOS) inhibition (CNI) groups. A: untreated CNI rings displayed decreased relaxation responses compared with untreated control rings. B: NG-nitro-L-arginine methyl ester (L-NAME) incubation decreased control but not CNI responses such that between-group differences were abolished. C: indomethacin (Indo) incubation did not affect relaxation responses. D: L-NAME + Indo incubation had the same effects as L-NAME incubation. [1]Significantly different from control. [2]Significantly different from control + inhibitor.
Table 2.
Agonist-induced maximal relaxation and ID50 values of coronary arteries
| ADP |
BK |
SNP |
||||
|---|---|---|---|---|---|---|
| Incubation | Control | CNI | Control | CNI | Control | CNI |
| n | 8 | 9 | 8 | 9 | 8 | 9 |
| Untreated | ||||||
| Maximal, % | 98.35 (8.48) | 81.64 (18.26) | 110.61 (13.16) | 90.26 (36.68) | 111.30 (10.85) | 122.23 (10.41)* |
| ID50, -log M | 6.12 (0.44) | 5.50 (0.50)* | 9.18 (0.54) | 8.59 (0.22)* | 6.56 (0.44) | 6.94 (0.27)* |
| L-NAME | ||||||
| Maximal, % | 70.74 (26.88) | 65.48 (24.47) | 95.23 (22.39) | 65.90 (42.39)* | 115.86 (16.32) | 138.24 (26.51)* |
| ID50, -log M | 5.45 (0.28) | 5.10 (0.37) | 8.68 (0.44) | 8.59 (0.42) | 6.96 (0.49) | 7.23 (0.37) |
| Indo | ||||||
| Maximal, % | 101.83 (3.97) | 83.07 (22.83) | 103.91 (4.67) | 75.28 (23.85)* | 108.13 (10.80) | 111.31 (7.60) |
| ID50, -log M | 6.31 (0.35) | 5.44 (0.42)* | 9.37 (0.42) | 8.80 (0.32)* | 6.70 (0.51) | 7.23 (0.31)* |
| L-NAME + Indo | ||||||
| Maximal, % | 75.81 (23.63) | 73.30 (16.48) | 71.64 (29.90) | 58.47 (28.64) | 109.78 (8.50) | 113.23 (8.14) |
| ID50, -log M | 5.35 (0.37) | 5.15 (0.20) | 8.66 (0.44) | 8.75 (0.41) | 7.09 (0.46) | 7.44 (0.36) |
Values are means (SD); n, no. of pigs. (For untreated rings, n = 14.) BK, bradykinin; SNP, sodium nitroprusside; L-NAME, NG-nitro-L-arginine methyl ester; Indo, indomethacin; ID50, half-maximal effective dose.
P < 0.05, significantly different from control.
Fig. 2.
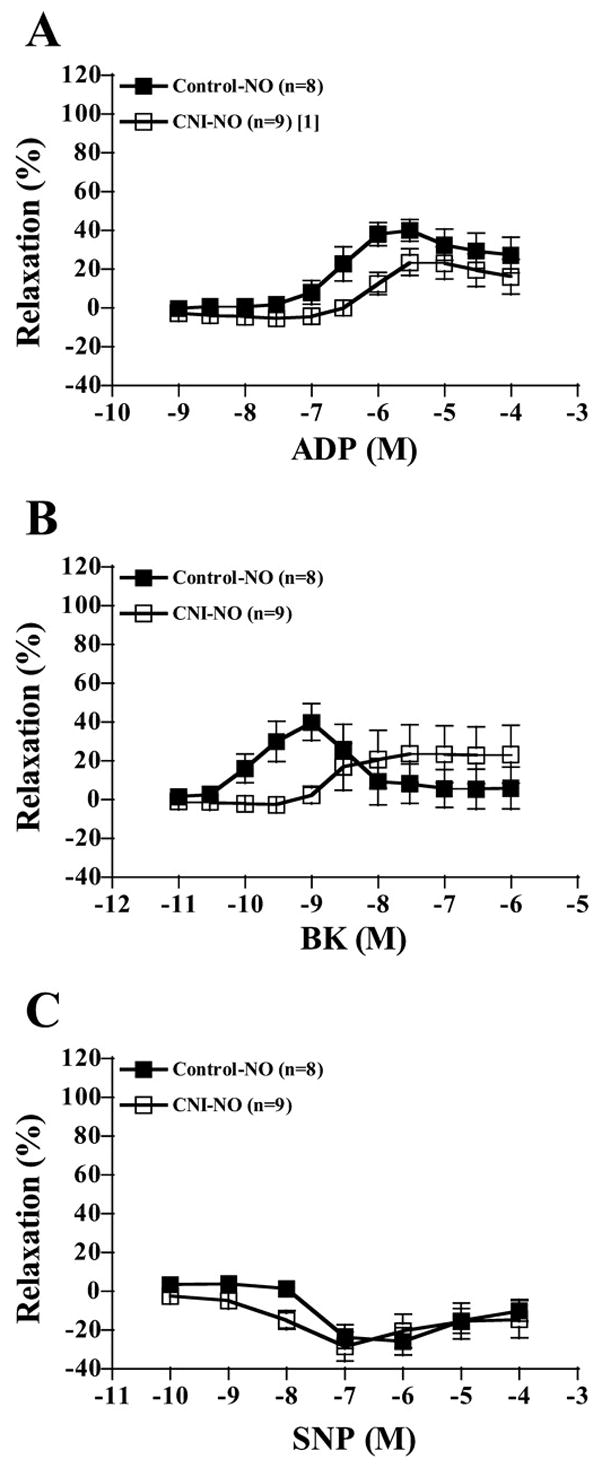
Contribution of NO to ADP-, bradykinin (BK)-, and sodium nitroprusside (SNP)-induced vasorelaxation responses of preconstricted coronary artery rings. Values are means ± SE. A: control rings displayed significantly more NO-mediated relaxation to ADP compared with CNI rings. B: control rings displayed significantly (in the 10−10 to 10−9 range) more NO-mediated relaxation to BK compared with CNI rings. C: groups did not differ in the amount of NO-mediated relaxation to SNP. [1]Significantly different from control.
LAD BK responses
Increasing concentrations of BK elicited increasing relaxation responses in LADs of both control and CNI groups (Fig. 3 and Table 2). LADs from CNI pigs showed significantly diminished responses to BK compared with control rings, as well as lower ID50 values, but not maximal responses (P = 0.115). Incubating rings with L-NAME alone significantly decreased relaxation responses of LADs from control pigs but not from CNI pigs such that between-group differences were abolished (Fig. 3B) with respect to dose-response curves and ID50 values. Indo incubation did not affect dose-response curves of either group (Fig. 3C). L-NAME + Indo incubation had the same effect as L-NAME incubation in that it abolished between-group differences (Fig. 3D). As was true for ADP-induced relaxation, the contribution of NO, produced by NOS, to BK-induced relaxation was greater (in the 10−10 to 10−9 M range) in control than in CNI LADs (Fig. 2B).
Fig. 3.

BK-induced vasorelaxation responses of preconstricted coronary artery rings. Values are means ± SE. A: untreated CNI rings displayed decreased relaxation responses compared with untreated control rings. B: L-NAME incubation decreased control but not CNI responses such that between-group differences were abolished. C: Indo incubation did not affect relaxation responses. D: L-NAME + Indo incubation had the same effects as L-NAME incubation, except that CNI + L-NAME + Indo (CNI + Both) was significantly decreased compared with untreated CNI. [1]Significantly different from control. [2]Significantly different from control + inhibitor. [3]Significantly different from CNI.
Sulf treatment alone tended to shift the dose-response curves for BK-induced relaxation in LADs to the left in both groups, but this effect was not significant in either control (P = 0.450) or CNI pigs (P = 0.389, Figs. 4A and 5A). However, in the presence of L-NAME, Sulf treatment decreased BK-induced relaxation of control LADs from 80% (Fig. 3B) with L-NAME alone to 60% with L-NAME + Sulf (Fig. 4B). In contrast, Sulf tended to increase BK-induced relaxation of CNI LADs in the presence of L-NAME treatment so that BK-induced relaxation was no longer different from untreated CNI (P = 0.290, Fig. 5B). Sulf treatment in the presence of L-NAME + Indo did not have an effect on BK-induced relaxation, since L-NAME + Indo treatment resulted in 60% relaxation in both groups (Fig. 3D) and L-NAME + Indo + Sulf treatment also resulted in 60% relaxation in both groups (Figs. 4C and 5C). Finally, denuding the endothelium resulted in complete abolishment of BK responses (data not shown).
Fig. 4.

BK-induced vasorelaxation responses of preconstricted control coronary artery rings. Values are means ± SE. A: sulfaphenazole (Sulf) treatment alone tended to shift the dose-response curves to the left, but this effect was not significant (P = 0.450). B: however, in the presence of L-NAME, Sulf treatment decreased BK-induced relaxation of control LADs to 60%. C: Sulf treatment in the presence of L-NAME + Indo did not have an effect on BK-induced relaxation, since L-NAME + Indo + Sulf treatment resulted in 60% relaxation in both groups. D: relaxation in the presence of L-NAME + Indo + Sulf + KCl. *Significantly different from control.
Fig. 5.

BK-induced vasorelaxation responses of preconstricted CNI coronary artery rings. Values are means ± SE. A: Sulf treatment alone tended to shift the dose-response curves to the left, but this effect was not significant (P = 0.389). B: Sulf tended to increase BK-induced relaxation in the presence of L-NAME treatment so that BK-induced relaxation was no longer different from untreated CNI (P = 0.290). C: Sulf treatment in the presence of L-NAME + Indo did not have an effect on BK-induced relaxation, since L-NAME + Indo + Sulf treatment resulted in 60% relaxation in both groups. D: relaxation in the presence of L-NAME + Indo + Sulf + KCl. *Significantly different from CNI.
LAD SNP responses
Increasing concentrations of SNP elicited increasing relaxation responses in LADs of both control and CNI groups (Fig. 6 and Table 2). LADs from CNI pigs showed significantly enhanced responses to SNP compared with control rings, as well as higher maximal responses and ID50 values. Incubating rings with L-NAME alone potentiated SNP-induced relaxation of LADs from CNI pigs and tended to do the same in control pigs (Fig. 6B). Hence, control + L-NAME was significantly lower compared with CNI + L-NAME with respect to dose-response curves. Indo incubation did not affect LAD relaxation responses in either control or CNI groups (Fig. 6C), but Indo incubation did abolish between-group differences with respect to dose-response curves and maximal relaxation but not ID50 values. Curiously, L-NAME + Indo incubation abolished between-group differences with respect to dose-response curves, maximal relaxation, and ID50 values (Fig. 6D). In addition, although Sulf incubation did not affect between-group differences (except maximal relaxation, P = 0.072), L-NAME + Sulf, L-NAME + Indo + Sulf, and L-NAME + Indo + Sulf + KCl incubation, as well as denuding the endothelium, abolished between-group differences (data not shown). Neither inhibition of NOS nor COX activity resulted in significantly different SNP dose-response relaxation in control rings compared with CNI rings (Fig. 2C).
Fig. 6.

SNP-induced vasorelaxation responses of preconstricted coronary artery rings. Values are means ± SE. A: untreated CNI rings displayed enhanced relaxation responses compared with untreated control rings. B: L-NAME incubation increased CNI but not control responses such that between-group differences remained. C: Indo incubation did not affect relaxation responses, although between-group differences were no longer significant. D: L-NAME + Indo incubation had the same effects as Indo incubation. [1]Significantly different from control. [2]Significantly different from control + inhibitor. [3]Significantly different from CNI. [4]Significantly different from CNI + inhibitor.
Coronary Arteriole Characteristics
There were no significant differences between control and CNI arteriole maximal diameters (111 ± 3 μm, n = 14 vs. 110 ± 4 μm, n = 13, P = 0.887), amount of spontaneous tone before BK curves (38 ± 4 μm, n = 15 vs. 46 ± 4 μm, n = 13, P = 0.427), or amount of spontaneous tone before SNP curves (46 ± 5 μm, n = 15 vs. 52 ± 6 μm, n = 11, P = 0.603). Incubation with L-NAME, Indo, or both did not significantly affect arteriole tone (P = 0.394).
Coronary Arteriole In Vitro Function
Arteriole BK responses
Increasing concentrations of BK elicited increasing dilatory responses in arterioles of both control and CNI groups (Fig. 7). There were no significant between-group differences in percent possible dilation responses to BK, regardless of whether arterioles were left untreated or incubated with L-NAME, Indo, or L-NAME + Indo (Fig. 7). In addition, although L-NAME incubation decreased dilatory responses in arterioles from control pigs, differences were only significant at a P value of 0.078 (Fig. 7B). Indo incubation did not affect responses of either group (Fig. 7C). Furthermore, the effect of L-NAME + Indo incubation on dilatory responses did not reach significance in either group (control, P = 0.063; CNI, P = 0.085). Calculated NO contribution to percent relaxation tended to be greater in control compared with CNI arterioles (Fig. 8A, P = 0.075), but calculated PGI2 and combined NO + PGI2 contributions did not differ between groups (Fig. 8, B and C).
Fig. 7.

BK-induced vasodilation responses of coronary arterioles. Values are means ± SE. A: untreated control arterioles did not differ in dilatory responses from untreated CNI arterioles. B: L-NAME incubation significantly decreased dilation of control but not CNI arterioles, and control + L-NAME did not significantly differ from CNI + L-NAME. C: Indo incubation did not affect dilatory responses of either group. D: L-NAME + Indo incubation did not significantly alter dilatory responses of either group, although CNI + Both was significantly decreased compared with untreated control. [1]Significantly different from control. [2]Significantly different from CNI.
Fig. 8.
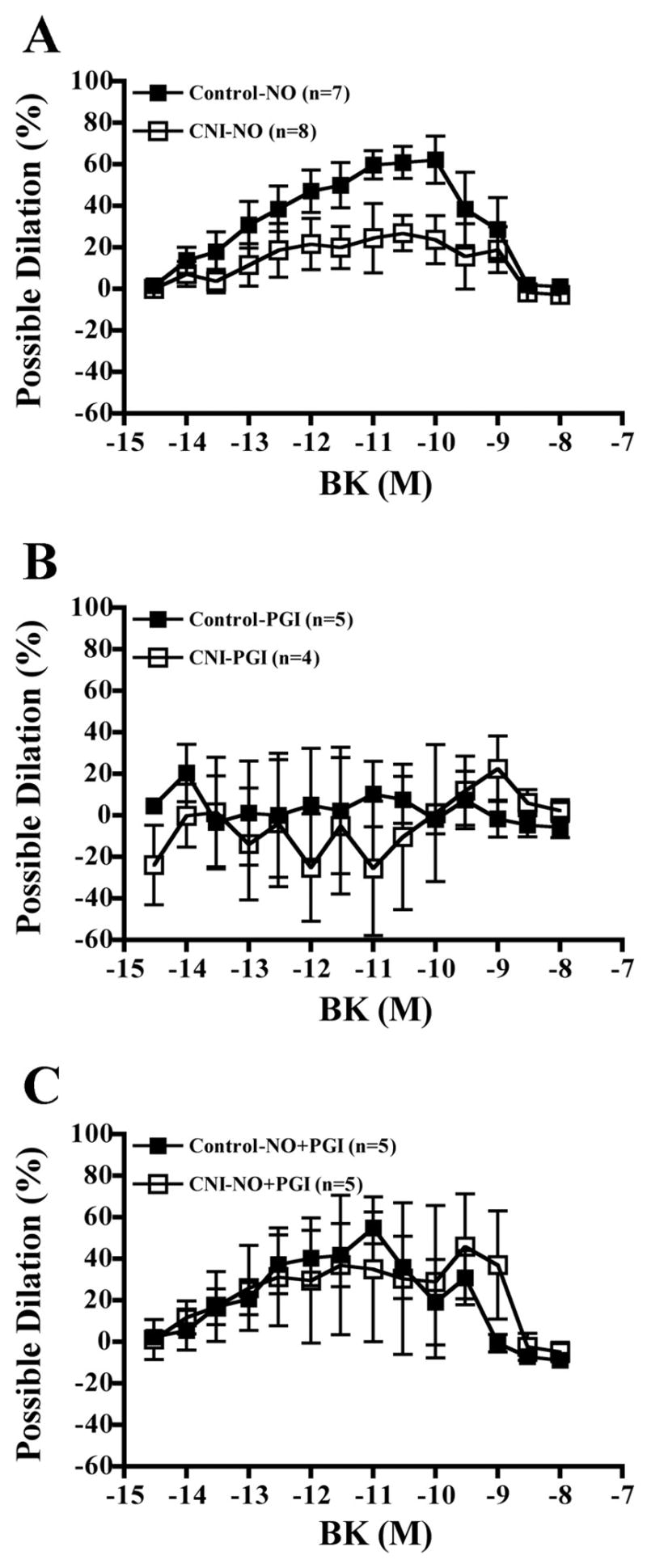
Contribution of NO, prostaglandins (PGI2), and NO + PGI2 to BK-induced vasodilation responses of coronary arterioles. Values are means ± SE. A: between-group differences in contribution of NO to vasodilation did not reach significance (P = 0.07), although individual doses did differ significantly. B: groups did not differ in contribution of PGI2 to BK-induced vasodilation. C: groups did not differ in contribution of NO + PGI2 to BK-induced vasodilation.
Arteriole SNP responses
Increasing concentrations of SNP elicited increasing dilatory responses in arterioles of both control and CNI groups (Fig. 9). There were no significant differences in dilator responses to SNP of arterioles from control and CNI groups (Fig. 9A). Likewise, incubation with L-NAME or L-NAME + Indo did not significantly affect SNP-induced dilation in either group (Fig. 9, B and C).
Fig. 9.
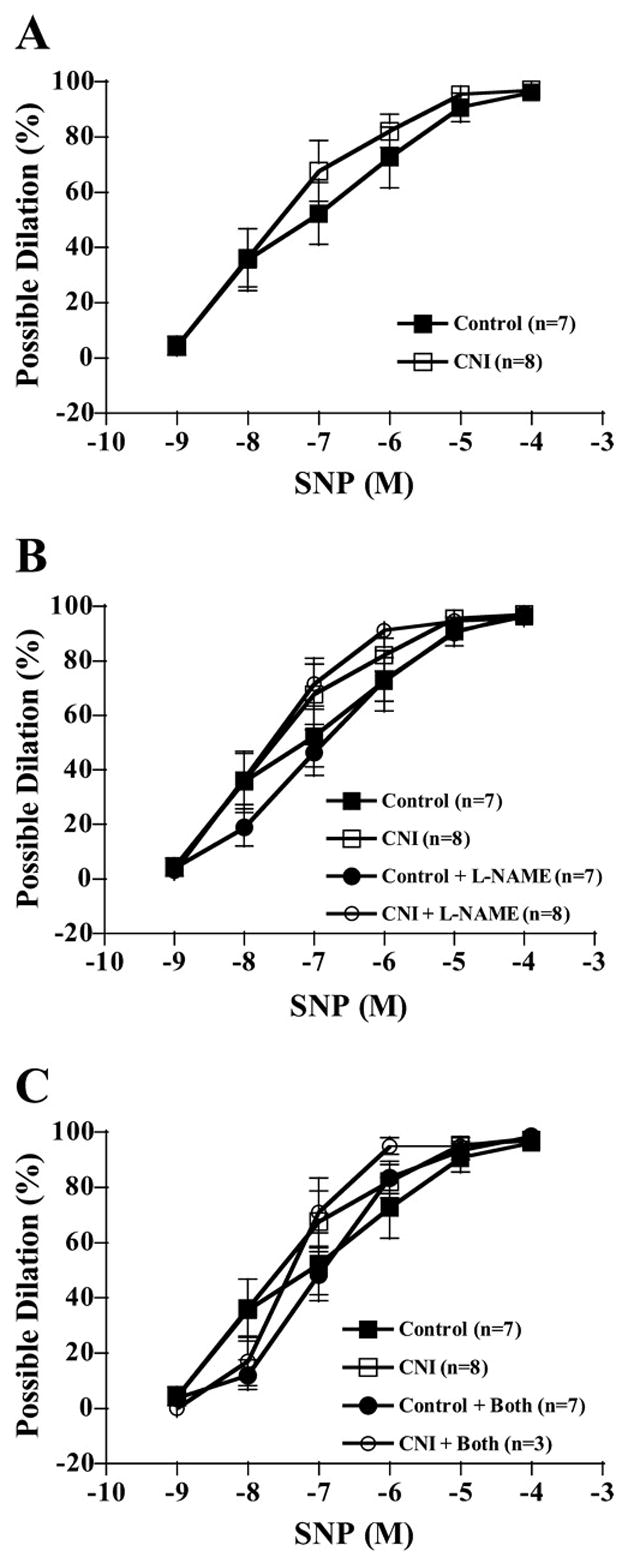
SNP-induced vasodilation responses of coronary arterioles. Values are means ± SE. A: untreated control arterioles did not differ in dilatory responses from untreated CNI arterioles. B: L-NAME incubation did not affect relaxation responses between groups. C: L-NAME + Indo did not affect relaxation responses. No significant differences were detected between groups or incubations.
eNOS and SOD expression in LADs and arterioles from L-NAME-treated pigs
As shown in Fig. 10, L-NAME treatment tended to increase eNOS protein content of whole LAD and of LAD endothelial cells scraped from the LADs. However, there were no significant differences in eNOS protein content between groups. In addition, there was no significant difference in eNOS protein content of coronary arterioles isolated from control and CNI pigs. Finally, RT-PCR results from coronary arterioles also indicated no difference in eNOS mRNA of arterioles from control (25.44 ± 0.26 cycles, 1.05 ± 0.02 normalized, n = 4) and CNI pigs (26.06 ± 1.40 cycles, 1.06 ± 0.05 normalized, n = 3).
Fig. 10.

Endothelial NOS (eNOS) expression in LADs and arterioles from L-NAME-treated pigs. Values are means + SE. A: L-NAME treatment did not significantly affect whole vessel eNOS protein content of coronary arteries or arterioles. B: sample eNOS blot.
SOD protein content was measured in arterial samples from control and CNI pigs. Protein content is expressed relative to the amount of protein in control samples on each blot as described in METHODS. Extracellular SOD did not differ between groups in whole vessel conduits (1.00 ± 0.22, n = 5 vs. 0.90 ± 0.15, n = 4; P = 0.718), conduit endothelial cell scrapes (1.00 ± 0.48, n = 5 vs. 1.13 ± 0.70, n = 4; P = 0.883), or arterioles (1.00 ± 0.24, n = 5 vs. 1.32 ± 0.31, n = 4; P = 0.447). Cytoplasmic SOD (SOD-1) did not differ between groups in whole vessel conduits (1.00 ± 0.17, n = 5 vs. 1.01 ± 0.24, n = 4; P = 0.971), conduit endothelial cell scrapes (1.00 ± 0.08, n = 5 vs. 1.10 ± 0.09, n = 4; P = 0.449), or arterioles (1.00 ± 0.15, n = 5 vs. 1.06 ± 0.07, n = 4; P = 0.695). Finally, although mitochondrial SOD (SOD-2) did not differ between groups in conduit endothelial cell scrapes (1.00 ± 0.08, n = 5 vs. 1.23 ± 0.29, n = 4; P = 0.503), it was significantly lower in arterioles from CNI pigs compared with control pigs (1.00 ± 0.13, n = 5 vs. 0.56 ± 0.06, n = 4; P = 0.024).
DISCUSSION
A strength of this study is that we examined the effects of chronic NOS inhibition in conduit arteries and arterioles of the coronary arterial tree of the same animals. Our main findings are that chronic NOS inhibition 1) diminished conduit artery (LAD) endothelium-dependent function via an eNOS mechanism and that it 2) enhanced endothelium-independent function in the LAD; in contrast, 3) endothelium-dependent function of coronary arterioles was preserved, and 4) endothelium-independent function of arterioles was unaffected. Although previous studies have reported that chronic NOS inhibition diminishes conduit artery endothelial function (11, 18, 21, 24), and separate studies have demonstrated preserved endothelial function of arterioles (29), this is the first study that directly tested the hypothesis that chronic L-NAME treatment would decrease endothelium-dependent function of the conduit LAD coronary artery but not decrease endothelium-dependent function of resistance coronary arterioles. Demonstration of this pattern in the coronary vasculature is of particular importance because of the clinical significance of the coronary arterial tree and the fact that previous studies have suggested divergent results in this vascular bed (24).
Acute Effects of NOS Inhibition Do Not Predict Effects of Chronic NOS Inhibition
Acute inhibition of NOS in vivo has been reported to produce greater constriction of large-bore resistance vessels (>25 μm in diameter) than in small arterioles (<25 μm in diameter) in feline skeletal muscle (5). Ekelund and Mellander (5) noted that the constriction produced by NOS inhibition may have produced “secondary metabolic/myogenic inhibition of tone opposing the response to NOS inhibition” in the small arterioles, masking the true effect of treatment. The secondary metabolic dilation would result from decreased tissue blood flow caused by constriction of the arteries, whereas secondary myogenic dilation would result from the decreased microvascular pressures caused by the NOS inhibition-induced constriction. To avoid these secondary contributions, we determined effects of acute NOS inhibition on intrinsic endothelium-dependent dilator responses in isolated arteries and arterioles from control pigs. It is important when interpreting our results to keep in mind that the methods we used in these experiments measure responses of conduit arteries as changes in force developed, whereas responses of arterioles were measured as changes in diameter. Our results from control arteries and arterioles demonstrate that acute NOS inhibition in vitro produced greater inhibition of BK-induced dilation in coronary arterioles (Fig. 6, ~60%) than of BK-induced relaxation of conduit coronary arteries (Fig. 2, ~40%). This is interesting given that the results of this study also show that chronic NOS inhibition produced greater effects on endothelial function in conduit arteries than in arterioles, as discussed below.
The results summarized above suggest that the increased blood pressure observed in CNI pigs might be the result of decreased arterial compliance produced by NOS inhibition, in addition to increased arterial resistance. Indeed, both acute and chronic NOS inhibitions have been reported to decrease large artery compliance in rats (12, 13) independently of changes in mean arterial pressure; similar effects of acute inhibition of NOS have been reported in humans (14, 15). These previous findings suggest that the blunted conduit endothelial function observed in the present study might be due to an L-NAME-induced decrease in compliance.
Divergent Effects of Chronic NOS Inhibition on Coronary Artery and Arteriole Endothelium-Dependent Function
LAD coronary arteries from CNI pigs had significantly diminished relaxation responses to the endothelium-dependent agonists ADP and BK (Figs. 1A and 3A). These findings are in agreement with previous studies that demonstrated attenuated endothelium-dependent relaxation of conduit arteries in the face of chronic NOS inhibition (11, 18, 21, 24). Although neither Indo nor Sulf incubation affected this finding, L-NAME incubation abolished between-group differences (Figs. 1B and 3B). These results suggest that the observed between-group differences are a result of differences in NO generated by NOS, but not COX or non-NOS, non-COX, pathways. Because of these findings, we were surprised that our results indicated that eNOS protein content and eNOS mRNA were not different from control in LAD or coronary arterioles of CNI pigs (Fig. 10).
A previous study reported that conduit coronary arteries from dogs treated with Nω-nitro-L-arginine (L-NNA) for 7 days (24) demonstrated an increased contribution of the COX pathway to endothelium-dependent relaxation. Our results indicate no role of the COX pathway in endothelium-dependent relaxation of either the control or CNI groups, given that Indo incubation had no effect on ADP or BK dose-response curves (Figs. 1C and 3C). Likewise, a previous study reported no role of the COX pathway in endothelium-dependent relaxation of the porcine LAD coronary artery (26). Differences in results among these studies may be attributed to the relatively short duration of the canine experiment, between-species differences (canine vs. porcine), or between-vessel differences (circumflex vs. LAD).
Sulfaphenazole is a selective inhibitor of cytochrome P-450 (CYP2C9) epoxygenase, which has been reported to attenuate EDHF-mediated responses in porcine coronary arteries (8). Our results indicate that Sulf did not significantly influence ADP- or BK-induced relaxation in untreated LADs from control or CNI pigs (Figs. 4A and 5A). Comparison of the effects of Sulf in control and CNI LADs treated with L-NAME reveals that Sulf attenuated BK-induced relaxation in control LADs (Figs. 3B and 4B) but that Sulf did not have this effect on BK-induced relaxation of CNI LADs in the presence of L-NAME (Figs. 3B and 5B). Sulf treatment did not alter BK-induced relaxation in control or CNI LADs in the presence of L-NAME + Indo (Figs. 4C and 5C). Fichtlscherer et al. (7) and Passauer et al. (22, 23) reported similar results in normal human forearm, where Sulf treatment did not have a significant effect on BK-induced dilation. In patients with atherosclerotic disease, Sulf treatment enhanced endothelium-dependent dilation in the forearm (7). Our results indicate that the CYP2C9 P450 pathway has a minor contribution to production of EDHF in normal or CNI Yucatan coronary arteries. Results also suggest an interaction between the NO pathway and Sulf-sensitive pathway in normal Yucatan LADs, but this interaction is no longer apparent following chronic L-NAME treatment (CNI).
In contrast to the coronary conduit arteries, coronary arterioles from CNI pigs demonstrated preserved endothelium-dependent dilation (Fig. 7A). These results are consistent with previous reports of preserved arteriolar endothelial function following chronic NOS inhibition (29). In addition, eNOS gene-disrupted mice have demonstrated preserved arteriolar endothelial function (9, 20, 25). Arterioles from CNI pigs showed a trend for decreased contribution of NO to endothelium-dependent dilation (Fig. 8A), suggesting an increased role of PGI2 and/or EDHF. Indeed, previous experiments have shown that NO inhibits PGI2 and EDHF synthesis (1, 3). By corollary, chronic inhibition of NOS may cause an increased synthesis of PGI2 and EDHF. In support of this notion, endothelium-dependent vasodilation in male rats chronically treated with L-NAME has been shown to be solely prostaglandin mediated, and vasodilation in female L-NAME treated rats was mediated solely by EDHF (29). On the basis of these results (29), we expected that the female pigs in the present study would demonstrate preserved endothelium-dependent dilation via a more active EDHF pathway. Our results are not consistent with this hypothesis, however, because there were no between-group differences in dilatory responses to BK when arterioles were incubated with L-NAME + Indo (hence, the state in which the NO and COX, but not EDHF, pathways were inhibited). Moreover, the existence of no between-group differences in PGI2 contribution to vasodilation (Fig. 8B) and no effect of Indo incubation on vasodilatory responses of arterioles from either group (Fig. 7C) suggests a minimal role of the COX pathway in coronary arteriolar vasodilation. Nonetheless, our results indicate that chronic NOS inhibition does not decrease endothelium-dependent vasodilator properties of coronary arterioles.
Divergent Effects of Chronic NOS Inhibition on Coronary Artery and Arteriole Endothelium-Independent Function
In the present study, chronic NOS inhibition potentiated LAD artery endothelium-independent relaxation to SNP (Fig. 6A). This finding is in agreement with previous studies in which chronic inhibition of NOS in mice resulted in an increased sensitivity to SNP in the aorta (18). Furthermore, both aorta and carotid arteries of eNOS gene-disrupted mice demonstrated greater sensitivity to SNP compared with controls (2, 6, 16). In a study in which dogs were treated with L-NNA for 7 days, however, relaxation responses to Sin-1 (an NO donor) were similar in the coronary and femoral arteries of control and treatment groups (24). In addition, the aortas of eNOS gene-disrupted mice had significantly decreased relaxation responses to SNP compared with controls (17). Although results of the present study offer no explanation for the observed potentiation of coronary artery endothelium-independent relaxation, previous studies (2) suggest that chronic NOS inhibition may increase arterial sensitivity to nitrovasodilators via increased soluble guanylyl cyclase pathway activity. Interestingly, the fact that between-group differences were abolished in the present study when the endothelium was denuded suggests that the mechanism may be endothelium dependent. Arterioles from CNI pigs demonstrated similar relaxation responses to SNP compared with controls (Fig. 9). These results are consistent with a study using NOS gene-disrupted mice that showed no significant differences in pial arteriole responses to SNP (20).
Effects of Chronic NOS Inhibition on eNOS Expression in Coronary Arteries and Arterioles
Western blot analysis revealed that chronic NOS inhibition did not alter eNOS expression in either LADs or coronary arterioles. These results suggest that the decreased contribution of NOS-generated NO release in relaxation of conduit coronary arteries in CNI pigs is the result of decreased eNOS activity and/or decreased NO bioavailability, not decreased eNOS protein expression.
In summary, this study established that chronic NOS inhibition differentially affected function of coronary arteries and arterioles measured in vitro. Coronary arteries demonstrated diminished endothelium-dependent relaxation and enhanced SNP-induced relaxation, whereas coronary arterioles showed unaltered endothelium-dependent and -independent dilation. The decrease in endothelium-dependent relaxation of conduit coronary arteries appears to be the result of a decreased NO release by a NOS-dependent mechanism. It is not clear how the CNI arterioles sustained normal intrinsic endothelium-dependent dilator sensitivity.
Acknowledgments
We thank Pamela K. Thorne for work on microvessels and eNOS protein data, Robert Johnson for work on conduits, and Miles Tanner for help with eNOS mRNA data.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants RR-18276, HL-36088, and HL-52490.
References
- 1.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 2.Brandes RP, Kim D, Schmitz-Winnenthal FH, Amidi M, Godecke A, Mulsch A, Busse R. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- 3.Doni MG, Whittle BJ, Palmer RM, Moncada S. Actions of nitric oxide on the release of prostacyclin from bovine endothelial cells in culture. Eur J Pharmacol. 1988;151:19–25. doi: 10.1016/0014-2999(88)90687-5. [DOI] [PubMed] [Google Scholar]
- 4.Dowell FJ, Henrion D, Duriez M, Michel JB. Vascular reactivity in mesenteric resistance arteries following chronic nitric oxide synthase inhibition in Wistar rats. Br J Pharmacol. 1996;117:341–346. doi: 10.1111/j.1476-5381.1996.tb15196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekelund U, Mellander S. Role of endothelium-derived nitric oxide in the regulation of tonus in large-bore arterial resistance vessels, arterioles and veins in cat skeletal muscle. Acta Physiol Scand. 1990;140:301–309. doi: 10.1111/j.1748-1716.1990.tb09004.x. [DOI] [PubMed] [Google Scholar]
- 6.Faraci FM, Sigmund CD, Shesely EG, Maeda N, Heistad DD. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am J Physiol Heart Circ Physiol. 1998;274:H564–H570. doi: 10.1152/ajpheart.1998.274.2.H564. [DOI] [PubMed] [Google Scholar]
- 7.Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher AM, Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109:178–183. doi: 10.1161/01.CIR.0000105763.51286.7F. [DOI] [PubMed] [Google Scholar]
- 8.Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 9.Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 10.Henderson KK, Turk JR, Rush JW, Laughlin MH. Endothelial function in coronary arterioles from pigs with early-stage coronary disease induced by high-fat, high-cholesterol diet: effect of exercise. J Appl Physiol. 2004;97:1159–1168. doi: 10.1152/japplphysiol.00261.2004. [DOI] [PubMed] [Google Scholar]
- 11.Henrion D, Dechaux E, Dowell FJ, Maclour J, Samuel JL, Levy BI, Michel JB. Alteration of flow-induced dilatation in mesenteric resistance arteries of L-NAME treated rats and its partial association with induction of cyclo-oxygenase-2. Br J Pharmacol. 1997;121:83–90. doi: 10.1038/sj.bjp.0701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu CT, Chang HR, Hsu YH, Liu CJ, Chen HI. Ventricular hypertrophy and arterial hemodynamics following deprivation of nitric oxide in rats. Life Sci. 2005;78:164–173. doi: 10.1016/j.lfs.2005.04.061. [DOI] [PubMed] [Google Scholar]
- 13.Hu CT, Chang KC, Wu CY, Chen HI. Acute effects of nitric oxide blockade with L-NAME on arterial haemodynamics in the rat. Br J Pharmacol. 1997;122:1237–1243. doi: 10.1038/sj.bjp.0701496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kielstein JT, Donnerstag F, Gasper S, Menne J, Kielstein A, Martens-Lobenhoffer J, Scalera F, Cooke JP, Fliser D, Bode-Boger SM. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke. 2006;37:2024–2029. doi: 10.1161/01.STR.0000231640.32543.11. [DOI] [PubMed] [Google Scholar]
- 15.Kinlay S, Creager MA, Fukumoto M, Hikita H, Fang JC, Selwyn AP, Ganz P. Endothelium-derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053. doi: 10.1161/hy1101.095329. [DOI] [PubMed] [Google Scholar]
- 16.Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc Res. 1999;42:206–213. doi: 10.1016/s0008-6363(98)00315-0. [DOI] [PubMed] [Google Scholar]
- 17.Lake-Bruse KD, Faraci FM, Shesely EG, Maeda N, Sigmund CD, Heistad DD. Gene transfer of endothelial nitric oxide synthase (eNOS) in eNOS-deficient mice. Am J Physiol Heart Circ Physiol. 1999;277:H770–H776. doi: 10.1152/ajpheart.1999.277.2.H770. [DOI] [PubMed] [Google Scholar]
- 18.Linder AE, Weber DS, Whitesall SE, D’Alecy LG, Webb RC. Altered vascular reactivity in mice made hypertensive by nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46:438–444. doi: 10.1097/01.fjc.0000175879.14994.63. [DOI] [PubMed] [Google Scholar]
- 19.McAllister RM, Albarracin I, Jasperse JL, Price EM. Thyroid status and endothelium-dependent vasodilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2005;288:R284–R291. doi: 10.1152/ajpregu.00061.2003. [DOI] [PubMed] [Google Scholar]
- 20.Meng W, Ma J, Ayata C, Hara H, Huang PL, Fishman MC, Moskowitz MA. ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am J Physiol Heart Circ Physiol. 1996;271:H1145–H1150. doi: 10.1152/ajpheart.1996.271.3.H1145. [DOI] [PubMed] [Google Scholar]
- 21.Moreau P, Takase H, Kung CF, van Rooijen MM, Schaffner T, Luscher TF. Structure and function of the rat basilar artery during chronic nitric oxide synthase inhibition. Stroke. 1995;26:1922–1928. doi: 10.1161/01.str.26.10.1922. [DOI] [PubMed] [Google Scholar]
- 22.Passauer J, Bussemaker E, Lassig G, Pistrosch F, Fauler J, Gross P, Fleming I. Baseline blood flow and bradykinin-induced vasodilator responses in the human forearm are insensitive to the cytochrome P450 2C9 (CYP2C9) inhibitor sulphaphenazole. Clin Sci (Lond) 2003;105:513–518. doi: 10.1042/CS20030118. [DOI] [PubMed] [Google Scholar]
- 23.Passauer J, Pistrosch F, Lassig G, Herbrig K, Bussemaker E, Gross P, Fleming I. Nitric oxide- and EDHF-mediated arteriolar tone in uremia is unaffected by selective inhibition of vascular cytochrome P450 2C9. Kidney Int. 2005;67:1907–1912. doi: 10.1111/j.1523-1755.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 24.Puybasset L, Bea ML, Ghaleh B, Giudicelli JF, Berdeaux A. Coronary and systemic hemodynamic effects of sustained inhibition of nitric oxide synthesis in conscious dogs. Evidence for cross talk between nitric oxide and cyclooxygenase in coronary vessels. Circ Res. 1996;79:343–357. doi: 10.1161/01.res.79.2.343. [DOI] [PubMed] [Google Scholar]
- 25.Sun D, Huang A, Smith CJ, Stackpole CJ, Connetta JA, Shesely EG, Koller A, Kaley G. Enhanced release of prostaglandins contributes to flow-induced arteriolar dilation in eNOS knockout mice. Circ Res. 1999;85:288–293. doi: 10.1161/01.res.85.3.288. [DOI] [PubMed] [Google Scholar]
- 26.Thompson MA, Henderson KK, Woodman CR, Turk JR, Rush JW, Price E, Laughlin MH. Exercise preserves endothelium-dependent relaxation in coronary arteries of hypercholesterolemic male pigs. J Appl Physiol. 2004;96:1114–1126. doi: 10.1152/japplphysiol.00768.2003. [DOI] [PubMed] [Google Scholar]
- 27.Vita JA. Endothelial function and clinical outcome. Heart. 2005;91:1278–1279. doi: 10.1136/hrt.2005.061333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vita JA, Keaney JF., Jr Endothelial function: a barometer for cardiovascular risk? Circulation. 2002;106:640–642. doi: 10.1161/01.cir.0000028581.07992.56. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2456–H2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]