

Abstract
BACKGROUND
Inflammation is pivotal in all phases of atherosclerosis. Among the numerous inflammatory biomarkers, the largest amount of published data supports a role for C-reactive protein (CRP) as a robust and independent risk marker in the prediction of primary and secondary adverse cardiovascular events. In addition to being a risk marker, there is much evidence indicating that CRP may indeed participate in atherogenesis.
CONTENT
In this review, we focus on the role of CRP in promoting atherothrombosis by discussing its effects on endothelial cells, endothelial progenitor cells, monocyte-macrophages, and smooth muscle cells.
CONCLUSIONS
CRP is clearly a risk marker for cardiovascular disease and is recommended for use in primary prevention. In addition, CRP appears also to contribute to atherogenesis. However, much further research is needed, especially in appropriate animal models, to confirm the possible role of CRP in promoting atherothrombosis.
Inflammation and Atherosclerosis
A large amount of evidence supports a pivotal role for inflammation in all phases of atherosclerosis, from initiation of the fatty streak to final culmination in acute coronary syndromes (1, 2). The earliest event in atherogenesis appears to be endothelial cell dysfunction precipitated by various noxious insults including obesity, hypertension, diabetes, smoking, and dyslipidemia. Endothelial cell dysfunction manifests primarily as deficiency of nitric oxide (NO)2 and an increase in moieties such as endothelin 1 (ET-1), angiotensin II (Ang II), plasminogen activator inhibitor 1 (PAI-1), cellular adhesion molecules, and cytokines/chemokines. With onset of endothelial cell dysfunction, mononuclear cells such as monocytes and T lymphocytes tether and roll along the endothelium, initially loosely; thereafter, they adhere firmly to the endothelium and transmigrate into the subendothelial space. The rolling and tethering of leukocytes on the endothelium is orchestrated by adhesion molecules such as selectins (E-selectin, P-selectin), cell adhesion molecules [intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1)], and integrins. Chemotaxis and entry of monocytes into the subendothelial space is promoted by monocyte chemoattractant protein 1 (MCP-1), interleukin (IL)-8, and fractal-kine. Thereafter, macrophage colony-stimulating factor (M-CSF) promotes the differentiation of monocytes into macrophages. Macrophages incorporate lipids from retained LDL that can undergo oxidative or enzymatic modification (OxLDL or E-LDL) and are taken up via the scavenger receptor (SR) pathway [CD36, SR-A, CD68, lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), SR-B1], becoming foam cells, the hallmark of the early fatty streak lesion. After the fatty streak lesion, smooth muscle cells migrate into the intima, proliferate, and form the fibrous cap. It is currently believed that lipid-laden macrophages, during the process of cytokine stimulation, CD40 ligation, necrosis, and apoptosis, release matrix metalloproteinases (MMPs), which cause a thinning of the endothelial layer. Because the lipid-laden macrophage is enriched in tissue factor, this is released from the macrophage and activates factor VII, initiating the coagulation cascade, resulting in thrombus formation and acute coronary syndromes (unstable angina and myocardial infarction). Macrophages also interact with T cells and other cells via activation of the CD40–CD40 ligand pathway, which contributes to a more atheromatous and less fibrous lesion that is prone to plaque rupture. Various gene knockouts and transgenic experiments have underscored the importance of the various cytokines, chemokines, and adhesion molecules in atherogenesis, emphasizing the pivotal role of inflammation in atherosclerosis (1, 2).
Numerous inflammatory biomarkers have been shown in various studies to predict cardiovascular events. These include cell adhesion molecules, cytokines, chemokines, and acute-phase reactants such as fibrinogen, serum amyloid A (SAA), and C-reactive protein (CRP). In this review, we discuss the inflammatory marker CRP, since the largest amount of published data supports a role for CRP as a robust and independent risk marker for cardiovascular disease, as has been reviewed (3, 4). In addition to being a risk marker, there is much evidence suggesting that CRP may indeed be a culprit in atherogenesis, and this potential role of CRP is the focus of this review.
C-Reactive Protein
C-reactive protein is the prototypic marker of inflammation in humans and a member of a highly conserved family of proteins called the pentraxins. It comprises 5 noncovalently associated protomers arranged symmetrically around a central pore and has a molecular weight of 118 000 Da (5). It is a nonglycosylated protein in humans, and its gene has been mapped to chromosome 1. To date, in phagocytes, it has been shown to bind Fcγ receptor (FcγR) I and II, and its function appears to clear apoptotic and necrotic cells (opsonophagocytosis).
It has been proposed that distinct forms of CRP are made during inflammation. Conformationally rearranged CRP, which expresses many epitopes not seen in native CRP, is referred to as modified or monomeric CRP (mCRP). mCRP antigens have been reported to be detected in the wall of normal human blood vessels (6). Although several in vitro studies have described the proatherogenic effects of mCRP (7, 8), the presence of mCRP in serum and atheroma has not been reported, and its existence is questionable since the calcium concentration is >2 mmol/L in both serum and lesions (CRP remains in the pentameric form in presence of calcium). Furthermore, 2 recent studies have shown that mCRP may indeed be antiatherogenic (9, 10). Treatment with native pentameric CRP for 8 weeks (2.5 mg/kg subcutaneously, weekly) resulted in a 4-fold–higher mean aortic plaque area in 14-week-old female ApoE−/− mice compared with the saline controls. In contrast, mean plaque size was decreased by approximately 50% in mCRP-treated ApoE−/− mice (2.5 mg/kg subcutaneously, weekly). In a subsequent study, female ApoE−/− mice fed a Western-type diet (42% fat) were treated with either human native CRP or mCRP (2.5 mg/kg subcutaneously, weekly) or saline for 8 weeks. Endothelium-dependent relaxation was impaired in native CRP-treated but not in mCRP-treated ApoE−/− mice. These findings, coupled with the lack of evidence of mCRP in atheroma or in the circulation, argue against a role for monomeric/modified CRP in atherothrombosis.
CRP is predominantly synthesized in hepatocytes as an acute-phase reactant and is transcriptionally driven by IL-6, with synergistic enhancement by IL-1. Some recent data challenge the dogma that CRP is exclusively produced by the liver, however, and suggest that it is produced in the atherosclerotic lesion (especially by smooth muscle cells and macrophages), the kidney, neurons, and alveolar macrophages (11–14). mRNA and protein for CRP is expressed in arterial plaque tissue, and both CRP mRNA and protein levels are 10-fold higher in plaque compared with the normal artery (13). Also, we have shown that human aortic endothelial cells (HAECs) synthesize and secrete CRP (15). The most potent agonist for CRP production from HAECs is the combination of IL-1 and IL-6. Thus, synthesis and secretion of CRP by cells in the atherosclerotic lesion by paracrine/autocrine loops could result in local concentrations of CRP far in excess of plasma concentrations and could contribute to proinflammatory, proatherogenic effects. Ouchi et al. (16) have demonstrated CRP mRNA in human adipose tissue. Our group has reported recently (17) that adiponectin, an adipocytokine, significantly decreases CRP mRNA and protein levels, whereas leptin has been shown to enhance CRP synthesis in endothelial cells incubated with IL-1 and IL-6 (18).
CRP and Atherogenesis
CRP AND ENDOTHELIAL CELLS
Several studies have shown an inverse relationship between CRP levels and endothelial function (19–21). CRP induces increased expression of ICAM, VCAM, E-selectin, and the chemokine MCP-1, resulting in increased adhesion of U937 cells (monocytic cell line) to human umbilical vein endothelial cells (22, 23). A critical enzyme in endothelial cells is endothelial nitric oxide synthase (eNOS). In HAECs, we found that CRP resulted in significant reductions in mRNA and protein for eNOS (24) and eNOS activity [i.e., conversion of L-arginine to L-citrulline and cyclic guanosine monophosphate (GMP) release]. Recently, we have delineated the mechanisms by which CRP causes decreased eNOS. CRP uncouples eNOS, resulting in increased superoxide production, decreased NO production, and altered eNOS phosphorylation (25). These effects appear to be mediated via the Fcγ receptors CD32 and CD64. Treatment of porcine coronary arterioles with CRP significantly attenuated NO release and vasodilatation and also increased NADPH oxidase activity and superoxide production in the arterial endothelium (26). Recently, Nagaoka et al. (27) showed that CRP inhibits endothelium-dependent NO-mediated dilation in retinal arterioles by producing superoxide from NADPH oxidase; this appeared to be downstream of RhoA (Ras homolog family member A) and p38 mitogen-activated protein kinase (MAPK) activation. These data further support a role for CRP in mediating endothelial dysfunction. Also, lectin-like OxLDL receptor-1 (LOX-1), a newly identified endothelial receptor for OxLDL, plays a pivotal role in OxLDL-induced endothelial dysfunction. LOX-1 mRNA and protein are induced by CRP, resulting in increased human monocyte adhesion to endothelial cells and Ox-LDL uptake by these cells (28). By virtue of inhibiting eNOS expression and NO release, CRP blocks NO-dependent processes. Furthermore, several groups have shown that CRP in vivo impairs endothelial vasoreactivity (29–31). CRP-transgenic (CRPtg) mice have been shown to have endothelial dysfunction (29). Aortic segments from turpentine-induced CRP-overexpressing CRPtg mice demonstrated impaired endothelium-dependent vasodilation compared with vehicle-treated CRPtg controls (P <0.05, n = 6). NO release as well as phosphorylated eNOS protein expression from isolated aortic segments of CRPtg mice overexpressing CRP were markedly reduced compared to that from vehicle-treated controls. Schwartz et al. (30) have reported that CRP downregulates eNOS and attenuates reendothelialization in vivo in mice. We have recently shown that CRP-treated rats compared with human serum albumin (HuSA)-treated rats exhibit impaired endothelium-dependent vasorelaxation; this was associated with increased aortic superoxide, decreased aortic tetrahydrobiopterin (BH4), and uncoupling of eNOS as evidenced by decreased eNOS dimer: monomer ratio (Fig. 1). Bisoendial et al. (31) also demonstrated that CRP infusion in patients with hypercholesterolemia resulted in marked deterioration of endothelial vasoreactivity.
Fig. 1. CRP promotes endothelial dysfunction in rats in vivo by uncoupling of eNOS.
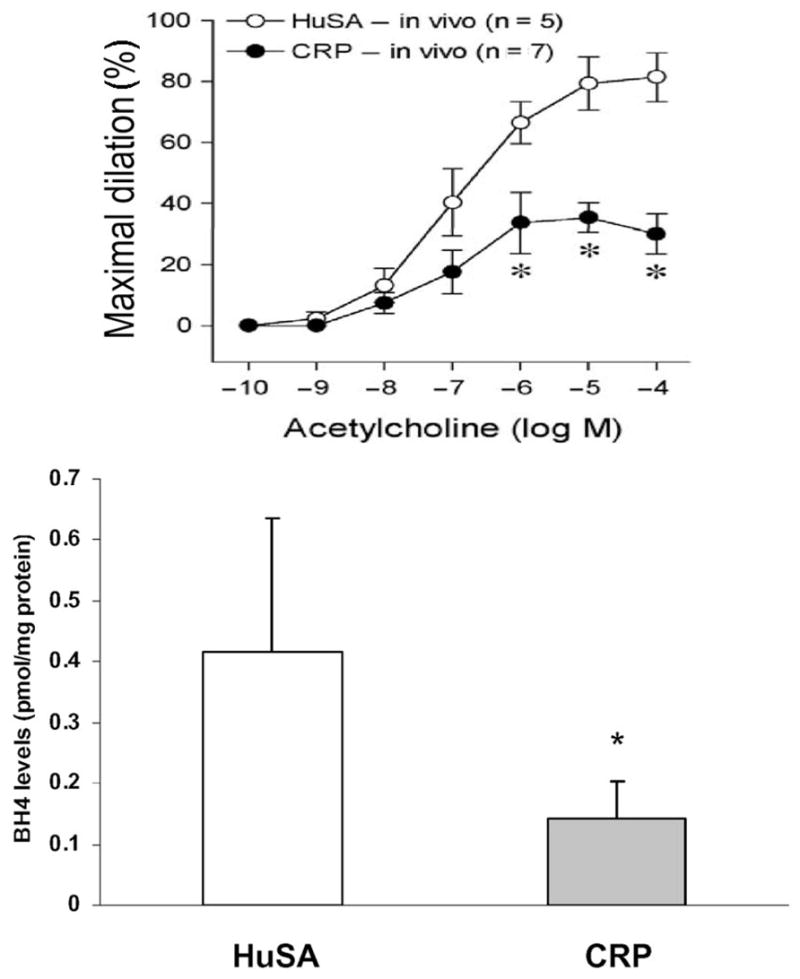
Rats were injected with human CRP (n = 7) or HuSA as control (n = 5) intraperitoneally (10 mg/kg) for 3 consecutive days. Endothelium-dependent vasodilation was significantly impaired in mesenteric arterioles of CRP-injected compared with HuSA-injected rats. Aortic levels of BH4 were significantly decreased, indicating uncoupling of the enzyme. *P < 0.05 vs HuSA. (Unpublished data, December 2008, I. Jialal.)
Another important product of endothelial cells (ECs) is prostacyclin, a potent vasodilator, inhibitor of platelet aggregation, and inhibitor of smooth muscle cell proliferation. In our studies, we showed that CRP caused a decrease in the release of the stable metabolite of prostacyclin, prostaglandin F-1α (PGF-1α), in both HAECs and human coronary artery ECs (32) via increased nitration of the enzyme prostaglandin synthase.
PAI-1, a marker of impaired fibrinolysis and atherothrombosis, is increased in patients with coronary artery disease. Increased PAI-1 gene expression is present in human atherosclerotic arteries and correlates with the degree of atherosclerosis, and PAI-1 deficiency protects against atherosclerotic progression in the mouse carotid artery. CRPtg mice develop coronary artery thrombosis (33). We have shown that CRP induces PAI-1 mRNA, antigen, and activity in HAECs (34). CRP-induced PAI-1 appears to occur via activation of Rho kinase and the nuclear factor (NF)-κB pathway (35). CRP inhibits tissue plasminogen activator (tPA) activity via generation of proinflammatory cytokines [IL-1β and tumor necrosis factor-α (TNF-α)] (36). These studies provide additional evidence that CRP may be a procoagulant, which has implications for atherothrombosis.
In venous endothelium, CRP has been shown to promote the release of the potent endothelial-derived contracting factor ET-1 (37). ET-1 is not only a potent vasoconstrictor, but it also appears to be a mediator of CRP-induced upregulation of adhesion molecules and MCP-1 in venous ECs.
An important chemokine is IL-8, a powerful trigger of adhesion of monocytes to endothelium. The mouse homolog of IL-8 triggers arrest of monocytes in carotid arteries of ApoE−/− mice. Knockout of the homolog of IL-8 receptor, CXCR2, decreases intimal accumulation of macrophages and decreases progression of atherosclerotic lesions (38). CRP induces IL-8 expression in HAECs and human coronary artery endothelial cells (HCAECs) via activation of NF-κB (39). This was confirmed by Wang et al. (40), who showed that IL-8 release was induced by CRP via upregulation of MEK (mitogen-activated protein kinase or extracellular signal-regulated kinase) and extracellular signal-regulated kinase (ERK) 1/2. Also, the CD40–CD40 ligand (CD40L) pathway plays an important role in plaque stability, and CRP has been shown to upregulate surface expression of CD40 and CD40L (41).
The Du Clos laboratory and others have shown that CRP binds mainly to FcγR2 (CD32) on leukocytes (42). We showed for the first time that CRP binds to both CD32 and CD64 on HAECs (43) but not CD16, and that CRP mediates its biological effects in EC via these 2 receptors. Studies have also shown that CRP binds to LOX-1 and may exert proatherogenic effects by binding to the receptors for advanced glycation end products (28). In macrophages, we have also shown that CRP-mediated OxLDL uptake can be inhibited by blocking both CD32 and CD64, and also the scavenger receptors CD36 and SR-A (44).
CRP AND ENDOTHELIAL PROGENITOR CELLS
There is tremendous interest in a special subtype of progenitor cells isolated from peripheral blood and bone marrow of adults, endothelial progenitor cells (EPCs), which have the capacity to circulate, proliferate, and differentiate into mature endothelial cells (45, 46). Importantly, the measurement of circulating EPCs is a surrogate marker of endothelial dysfunction and future cardiovascular events. Clinical studies suggest that traditional risk factors for coronary atherosclerosis are associated with lower levels of circulating EPCs (45, 46). Importantly, a negative correlation between systemic CRP levels and circulating EPC number has been reported (47) in patients with unstable angina. Also, low levels of EPCs are associated with adverse cardiovascular events (48). Additionally, pioglitazone treatment in type 2 diabetic patients revealed that the decrease in CRP level was the only parameter associated with increase in EPC number in multivariate analyses (49). In vitro studies report that CRP at concentrations ≥15 mg/L significantly reduced EPC number, inhibited the expression of the endothelial cell–specific markers tunica internal endothelial cell, kinase-2 (Tie-2), EC lectin, and vascular endothelial cadherin, significantly increased EPC apoptosis, and impaired EPC-induced angiogenesis (50). A recent article suggests that in mature ECs, CRP appears to stimulate angiogenesis and may be a mediator of neovessel formation in the intima of vulnerable plaques, thus suggesting different biological effects on EPCs and mature ECs (51). EPC-induced angiogenesis was dependent on the presence of NO, and CRP treatment caused a decrease in eNOS mRNA expression by EPCs. Also, subsequently, CRP was demonstrated to increase oxidative stress and induce apoptosis in these EPCs. Thus, CRP may negatively affect EPCs by quenching antioxidant defenses and promoting telomerase inactivation.
Thus, a large body of evidence from in vitro studies points to a proatherogenic, prothrombotic role for CRP in ECs. These effects of CRP may result in endothelial dysfunction and impairment of EPC survival and differentiation. These studies, especially the effects on EPCs, need to be confirmed in vivo.
CRP AND MONOCYTE-MACROPHAGES
Cermak et al. (52) were one of the first groups to report on the effects of CRP on monocytes. These investigators showed that CRP induced monocyte tissue factor (TF) secretion. In that study, they showed that CRP induced TF antigen and procoagulant activity. However, no studies were undertaken to elucidate the mechanism. Later studies have shown that peripheral blood mononuclear cells also respond in a similar fashion to CRP (53). Cirillo et al. (54) demonstrated that incubation of ECs and smooth muscle cells (SMCs) with CRP resulted in a dose-dependent activation of cell proliferation, which was mediated by activation of the ERK1/2 pathway. In addition, CRP also induced TF expression in both cell types in a dose-dependent fashion, exerting its effect at the transcriptional level, as demonstrated by semiquantitative and real-time PCR. Activation of the transcription factor NF-κB by CRP was demonstrated by electrophoretic mobility shift assay, and suppression of TF expression by the NF-κB inhibitor pyrrolidine-dithio-carbamate ammonium was also demonstrated. Recently, we demonstrated that intraperitoneal administration of CRP (20 mg/kg) compared with HuSA increased superoxide anion release and TF activity from peritoneal macrophages in vivo (P < 0.01) (55). This was confirmed using intra-pouch administration of CRP (25 μg/mL) compared with HuSA. In rat pouch macrophages in vivo, we showed that CRP induced TF antigen and activity was inhibited by inhibitors to NADPH oxidase. Furthermore, we demonstrated that inhibition of NF-κB activity with caffeic acid phenethyl ester resulted in significant inhibition of CRP-induced TF activity, and blockade of CD32 and CD64 reduced CRP-induced TF.
Several investigators have demonstrated that CRP induces oxidative stress in vitro. Prasad (56) showed that in neutrophils, CRP significantly and dose-dependently induces superoxide anion release in vitro; however, no mechanistic studies were undertaken. Exposure of rat mesangial cells to CRP for 60 min led to a dose-dependent increase in superoxide anion release that was nearly 3-fold greater than in control conditions (P < 0.0001) (57). Trachtman et al. also showed that coincubation with diphenylene iodinium (10 μmol/L), an inhibitor of NADPH oxidase, prevented the increase in superoxide production by rat mesangial cells induced by CRP (100 μg/mL). Whereas they used high concentrations of CRP in their in vitro studies, we demonstrated that lower concentrations of CRP (25 μg/mL) in vivo induce superoxide anion release and that this is reversed by inhibition of protein kinase C (PKC) and NADPH oxidase using apocynin. In that article, in rat pouch macrophages, we also demonstrated that the effects of CRP on superoxide anion release were inhibited via inhibition of ERK and c-Jun N-terminal kinase (JNK) but not p38 MAPK (55). These effects are abrogated by blocking Fcγ receptors CD32 and CD64. Another important in vivo demonstration we made recently is the induction of myeloperoxidase activity in macrophages in vivo by human CRP administration (58).
CRP also induces proinflammatory cytokine release from human monocytes. Ballou and Lozanski (59) conducted a study in which they incubated human monocytes with CRP at different doses for 16 h and were able to demonstrate significantly increased levels of IL-1β, TNF-α, and IL-6 at concentrations of CRP >5 mg/L. This induction of cytokine release was unaffected by polymixin B but was completely abrogated by boiling of CRP, confirming that this effect of CRP was not due to lipopolysaccharide (LPS) contamination. Subsequently, we have also shown that CRP inhibits antiinflammatory cytokine activity. Incubation of human monocyte-derived macrophages with CRP significantly decreased LPS-induced second-wave IL-10 mRNA and intracellular and secreted IL-10 protein and destabilized IL-10 mRNA (60). Also, CRP alone increased secretion of IL-8, IL-6, and TNF from human monocyte-derived macrophages. FcγRI antibodies significantly reversed CRP-mediated IL-10 inhibition. CRP significantly decreased intracellular cAMP, phospho-cAMP response element binding protein, and adenyl cyclase activity. cAMP agonists reversed CRP- mediated IL-10 inhibition. Furthermore, administration of human CRP to rats significantly decreased IL-10 levels.
A single report has shown increased CD11b expression on monocytes incubated with CRP, accompanied by increased adhesion of these monocytes to LPS-activated human umbilical vein endothelial cells (61). We have shown that CRP increases monocyte-endothelial cell adhesion under shear stress and static conditions via increased NF-κB and increased ICAM-1 and VCAM-1 expression. These effects were abrogated by blockade of CD32 and CD64 (62). We have also recently shown that CRP promotes M-CSF release from human macrophages and induces macrophage proliferation; this effect appears to be mediated via NF-κB activation and through the Fcγ receptors CD32 and CD64 (63).
CRP and oxidized LDL uptake
There has been a report that CRP promotes uptake of native LDL (64). However, this has been brought into question by the Witztum group, who showed in an elegant study that CRP promotes the uptake of oxidized but not native LDL due to certain unexposed phosphocholine epitopes on OxLDL (65). Recently, CRP has been shown to significantly reduce cholesterol efflux from THP-1 (human myelogenous leukemia cell line) and peripheral blood mononuclear cells to apoA-I or HDL. CRP significantly decreased the expression of ATP-binding membrane cassette transporter A1 (ABCA1) and ABCG1, whereas it increased superoxide anion production. Furthermore, CRP substantially activated ERK1/2 in THP-1–derived foamlike cells (66). Reducing superoxide anion by antioxidant seleno-L-methionine or superoxide dismutase mimetic [manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP)] effectively abolished the CRP-induced decrease in cholesterol efflux and the expression of ABCA1 and ABCG1. Inhibiting ERK1/2 activation by its specific inhibitor PD98059 or by a dominant negative mutant of ERK2 could also block CRP action on THP-1 cells. We have recently demonstrated that in vivo administration of CRP promotes uptake of oxidized LDL and intracellular cholesterol ester accumulation in rat macrophages in vivo (44). The increased uptake of OxLDL by CRP was inhibited by pretreatment with antibodies to CD32, CD64, CD36, and fucoidin, suggesting uptake by both scavenger receptors and Fcγ receptor.
CRP and MMP
Williams et al., in an article that further supports the role of CRP in later stages of atherosclerosis (67), demonstrated that CRP stimulated MMP-1 mRNA, protein, and collagenase activity in human monocyte/macrophages. This activity appeared to be orchestrated via FcγRII, and the signal pathway appeared to be via ERK. CRP had no effect on tissue inhibitor of metalloproteinase 1 (TIMP-1). Furthermore, CRP treatment increased MMP-9 activity in macrophages compared with HuSA, which was abrogated by inhibitors to p38 MAPK, ERK, and NF-κB but not JNK before human CRP treatment.
In human monocytes, CRP promotes MCP-1–mediated chemotaxis by upregulating CC-chemokine receptor 2 (CCR2) expression (68). CCR2 is the most dominant chemotaxis receptor in monocytes, and mediates chemotactic movement of monocytes in response to MCP-1. CCR2 plays a pivotal role in atherosclerotic plaque formation, and genetic disruption of CCR2 in atherosclerosis-prone mice markedly decreased atherosclerosis. CRP-induced upregulation of CCR2 expression involved binding of CRP to the FcγR, most notably FcγRI, and phospholipase D1 activation.
High mobility group box-1 (HMGB1), a primarily nuclear protein, is released into the extracellular milieu by necrotic/damaged cells and is actively secreted by monocyte-macrophages. CRP induces active release of HMGB1 from macrophages via an FcγR/p38 MAPK signaling pathway (69).
Thus, it is clear that CRP is proatherogenic in monocyte-macrophages since it increases tissue factor expression, promotes monocyte chemotaxis and adhesion to ECs, reactive oxygen species release, MMP-1, CCR2, cytokines, and M-CSF, and promotes OxLDL uptake, thus leading to increased foam cell formation. Furthermore, CRP is present in foam cells in the atherosclerotic lesion and activates complement (12).
CRP AND SMOOTH MUSCLE CELLS
The angiotensin type I receptor (AT1-R) is a key atherosclerotic switch facilitating Ang II–induced ROS production and vascular smooth muscle cell (VSMC) migration, proliferation, and remodeling. Given thecentral importance of AT1-R in the development and clinical course of atherosclerosis (70), the demonstration by the Verma laboratory that CRP upregulates AT1-R mRNA and protein in VSMCs and increases the number of AT1-R binding sites in VSMCs could have major implications with regard to atherogenesis (71). CRP also augmented Ang II–induced VSMC migration and proliferation, further supporting a functional relationship between CRP and Ang II in mediating VSMC pathology. In an in vivo model of carotid balloon angioplasty, CRP exposure facilitated AT1-R expression, with resulting increases in neointimal formation, VSMC migration, and proliferation and promoted production of collagen and elastin, key matrix proteins in the vessel wall. These effects were attenuated by angiotensin receptor blockade. Therefore, CRP exerts direct proatherosclerotic effects at the level of VSMCs. Also, in VSMCs, CRP has been shown to upregulate inducible NO synthase (iNOS) and certain cell signal transduction pathways including MAPK pathway and NF-κB (72). Although accumulation of VSMCs in the intima is a key event in the development of arterial lesions, apoptosis of VSMCs also plays an important role in progression of atherosclerotic lesions and contributes to increased plaque vulnerability. CRP promotes apoptosis of VSMCs and thus may contribute to plaque instability (73). Given existing data that CRP promotes VSMC proliferation in vitro and induces apoptosis in vitro, the role of CRP in VSMCs and primary atherosclerosis remains to be delineated.
Kobayashi et al. (13) showed that the incubation of cultured coronary artery smooth muscle cells with CRP resulted in enhanced p22phox protein expression and the generation of intracellular reactive oxygen species (ROS). In cultured VSMCs obtained from human coronary arteries, Ryu et al. (74) showed CRP-induced ROS generation by VSMCs, which requires functional activation of FcγRIIa and NADPH oxidase 4, orchestrates proinflammatory activities of VSMCs, and may eventually promote atherogenesis and plaque rupture. Also, Ang II has been shown to upregulate CRP synthesis in VSMCs via upregulation of AT1-Rs and ERK (75).
Recently, Wu et al. (76) reported that TF mRNA, protein, and activity levels were significantly higher in VSMCs isolated from CRPtg mice than from wild-type (WT) mice. Tissue factor pathway inhibitor (TFPI) expression was significantly downregulated in CRPtg vs WT VSMCs. Transfection of human VSMCs with CRP expression plasmid significantly increased TF expression and decreased TFPI expression. Gene silencing of FcγRIIIa blocked the effect of CRP on VSMC TF expression. CRP activated p44/42 MAPK, and the effect of CRP on TF expression was blocked by pharmacological inhibitor of p44/42 MAPK. ROS scavengers blocked CRP-induced upregulation of VSMC TF expression. In vivo analyses revealed significant increases in TF expression and decreases in TFPI expression in carotid arteries of CRPtg mice vs WT mice. Thus, CRP increases TF and decreases TFPI expression by VSMCs in vitro and in vivo. Induction of TF expression by CRP is mediated by FcγRIIIa, p44/42 MAPK, and ROS generation.
IN VIVO EVIDENCE THAT CRP PROMOTES ATHEROTHROMBOSIS
In the CRPtg mice, Danenberg et al. (33) showed that compared with WT mice, in which CRP levels are undetectable, in CRPtg mice, CRP increased to 18.6 mg/L. Following transluminal wire injury to the femoral artery, there was complete thrombotic occlusion in the femoral artery in 75% of the human CRPtg mice compared with 17% of the WT mice at 28 days (P < 0.05). Furthermore, arterial photochemical injury to the carotids shortened clot formation time in the human CRPtg mice compared with WT. Whereas the human CRP transgene resulted in increased atherosclerotic lesions on an apolipoprotein E–knockout background, it should be pointed out that the effect was seen only in male mice and was modest, raising questions regarding the validity of the mouse model. The model of transgenic CRP-overexpressing mice to study atherogenesis is also fraught with problems and has resulted in conflicting observations, as reviewed previously (4). Although Paul et al. (77) demonstrated increased atherosclerosis in CRPtg apolipoprotein E–deficient mice, Hirschfield et al. (78) were unable to uncover a similar phenotype. The rat model is preferred over the mouse model based on the following reasons: in mice the main acute-phase reactant is serum amyloid P component and not CRP, human CRP is antiinflammatory in mice, rat CRP shares 79% identity with human CRP, and injection of human CRP into rats increased infarct size by 40%. Human CRP activates complement in rats. Human CRP has been shown to be proinflammatory in the rat and increases iNOS expression in cytokine-stimulated rat cardiac myocytes as well as iNOS in rat macrophages and stimulates NF-κB in rat vascular smooth muscle cells. These attributes make the rat, and not the mouse, a suitable model with which to test the proinflammatory effect of human CRP in vivo. Griselli et al. (79) showed that parenteral injection of human CRP in experimental acute myocardial infarction produced by coronary artery ligation reproducibly enhanced infarct size by approximately 40%. Gill et al. (80) also showed that adult rats subjected to middle cerebral artery occlusion and then treated with human CRP similarly developed significantly larger cerebral infarcts compared with control subjects receiving human serum albumin.
Furthermore, in 7 male volunteers, Bisoendial et al. (31) showed that an infusion of 1.25 mg/kg recombinant human CRP (rhCRP) on 2 occasions resulted in an increase in plasma CRP (from 1.9 to 23.9 mg/L), and subsequently both biomarkers of inflammation and coagulation were activated, as evidenced by increased serum IL-6, IL-8, SAA, prothrombin 1 and 2, and D-dimer.
We have provided several lines of evidence with our in vivo studies of human CRP in the rat model and have demonstrated that CRP induces superoxide anion release and tissue factor activity in vivo and promotes OxLDL uptake, MMP release, and myeloperoxidase from macrophages.
Recently, Pepys et al. (81) reported the design, synthesis, and efficacy of 1,6-bis(phosphocholine)-hexane as a specific small-molecule inhibitor of CRP. Five molecules of this palindromic compound are bound by 2 pentameric CRP molecules, cross-linking and occluding the ligand-binding B-face of CRP and blocking its functions. Administration of 1,6-bis(phosphocholine)-hexane to rats undergoing acute myocardial infarction significantly abrogated the increase in infarct size and cardiac dysfunction produced by injection of human CRP.
The field of CRP has been plagued by reports of artifacts such as contamination of CRP with endotoxin and azide. It is very important to emphasize that some of the studies with CRP have used recombinant CRP, and there is the possibility that endotoxin and azide may have explained the effects seen with CRP in some of the studies. However, studies from the Jialal laboratory (15, 17, 24, 32, 34, 36, 39, 56, 58, 60, 62, 63) and the Stroes laboratory (31) have used CRP purified from human ascites fluid, purified of endotoxin and dialyzed free of azide. Several other lines of evidence also suggest that the observed effects are due to CRP and not to artifacts: (1) for the in vitro experiments, boiled and trypsinized CRP have been used as controls; (2) addition of polymixin B fails to abrogate the effects of CRP; (3) whereas LPS activates eNOS, CRP decreases eNOS activity; (4) the most convincing argument for the proatherogenic effects of CRP per se comes from studies in which Toll-like receptor 4 (TLR4) small interfering RNA (siRNA) (which is responsible for the proinflammatory actions of LPS) was knocked out, and CRP still exerted its proatherogenic effects in these cells (Fig. 2); (5) the biological effects of CRP appear to be mediated via the Fcγ receptors CD32 and CD64. These data strongly support that the effects observed are due to CRP per se and not other artifacts.
Fig. 2. TLR4 knockdown fails to abrogate proatherogenic effects of CRP from endothelial cells.

HAECs (n = 5) were transfected with scrambled siRNA (Sc siRNA) or TLR-4 siRNA and then incubated with either CRP (25 μg/mL) or LPS (1 μg/mL), and release of IL-1β, IL-8, PAI-1, and eNOS activity (arginine to citrulline release, pmol/min/mg protein) were assessed. As shown in the figure, TLR4 knockdown failed to abrogate effects of CRP and only decreased effects seen with LPS (TLR4 ligand). *P < 0.05 vs control; †P < 0.05 vs CRP. Adapted from Dasu et al. (82).
Conclusion
In summary, CRP is clearly a risk marker for cardiovascular disease and is recommended for use in primary prevention. In addition, CRP appears also to contribute to atherogenesis as shown in Table 1. Recent results from Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER), especially in those subjects with hsCRP > mg/L but no other risk factors (n = 6000), showed a significant reduction in cardiovascular events with rosuvastatin. This study provides further support in human subjects that CRP appears to be an active participant in atherothrombosis and that specifically targeting CRP in the future may also prove beneficial.
Table 1.
Proatherogenic effects of CRP.
| Endothelial cells | Monocyte-macrophages | Smooth muscle cells |
|---|---|---|
| Increased VCAM, ICAM-1, E-selectin, MCP-1, monocyte adhesion | Increased tissue factor | Increased AT-1 and VSMC migration and proliferation |
| Increased PAI-1, IL-8, CD40/CD40L, MMP-1, ET-1, M-CSF | Increased superoxide and myeloperoxidase | Increased neointimal formation in vivo |
| Decreased tPA | Increased proinflammatory cytokines and decreased IL-10 | Increased iNOS |
| Decreased prostacyclin | Increased CD11b, CCR2 | Increased ROS |
| Increased superoxide, iNOS | Promoted OxLDL uptake and decreases cholesterol efflux | Increased tissue factor |
| Decreased eNOS (uncoupling) | Increased MMPs, HMGB1 | |
| Promoted endothelial dysfunction in vivo | Increased M-CSF and proliferation | |
| Impaired EPC number and function in vitro |
Acknowledgments
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Nonstandard abbreviations: NO, nitric oxide; ET-1, endothelin 1; Ang II, angiotensin II; PAI-1, plasminogen activator inhibitor 1; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; MCP-1, monocyte chemoattractant protein 1; IL, interleukin; M-CSF, macrophage colony-stimulating factor; OxLDL, oxidatively modified LDL; E-LDL, enzymatically modified LDL; SR, scavenger receptor; LOX-1, lectin-like oxidized low-density lipoprotein receptor 1; MMP, matrix metalloproteinase; SAA, serum amyloid A; CRP, C-reactive protein; FcγR, Fcγ receptor; mCRP, monomeric CRP; HAEC, human aortic endothelial cell; eNOS, endothelial nitric oxide synthase; GMP, guanosine monophosphate; MAPK, mitogen-activated protein kinase; CRPtg, CRP-transgenic; HuSA, human serum albumin; BH4, tetrahydrobiopterin; EC, endothelial cell; PGF-1μ, prostaglandin F-1α; NF, nuclear factor; tPA, tissue plasminogen activator; TNF-α, tumor necrosis factor-α; CXCR2, mouse homolog of IL-8 receptor; HCAEC, human coronary artery endothelial cell; MEK, mitogen-activated protein kinase or extracellular signal-regulated kinase; ERK, extracellular signal-regulated kinase; CD40L, CD40 ligand; EPC, endothelial progenitor cell; Tie-2, tunica internal endothelial cell, kinase-2; TF, tissue factor; SMC, smooth muscle cell; PKC, protein kinase C; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; ABC, ATP-binding membrane cassette transporter; MnTBAP, manganese (III) tetrakis (4-benzoic acid) porphyrin; TIMP-1, tissue inhibitor of metalloproteinase 1; CCR2, CC-chemokine receptor 2; HMGB1, high mobility group box 1; AT1-R, angiotensin type I receptor; VSMC, vascular smooth muscle cell; iNOS, inducible NO synthase; ROS, reactive oxygen species; WT, wild-type; TFPI, tissue factor pathway inhibitor; rhCRP, recombinant human CRP; TLR4, Toll-like receptor 4; siRNA, small interfering RNA.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures of Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Expert Testimony: None declared.
Research Funding: I. Jialal, NIH (K24AT00596, RO1HL74360).
References
- 1.Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 3.Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat Clin Pract Cardiovasc Med. 2005;2:29–36. doi: 10.1038/ncpcardio0074. [DOI] [PubMed] [Google Scholar]
- 4.Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. 2006;113:2135–50. [PubMed] [Google Scholar]
- 5.Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7:169–77. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 6.Khreiss T, József L, Potempa LA, Filep JG. Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ Res. 2005;97:690–7. doi: 10.1161/01.RES.0000183881.11739.CB. [DOI] [PubMed] [Google Scholar]
- 7.Khreiss T, József L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation. 2004;109:2016–22. doi: 10.1161/01.CIR.0000125527.41598.68. [DOI] [PubMed] [Google Scholar]
- 8.Khreiss T, József L, Potempa LA, Filep JG. Opposing effects of C-reactive protein isoforms on shear-induced neutrophil-platelet adhesion and neutrophil aggregation in whole blood. Circulation. 2004;110:2713–20. doi: 10.1161/01.CIR.0000146846.00816.DD. [DOI] [PubMed] [Google Scholar]
- 9.Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner C, Potempa, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2005;112:1016–23. doi: 10.1161/CIRCULATIONAHA.105.556530. [DOI] [PubMed] [Google Scholar]
- 10.Schwedler SB, Kuhlencordt PJ, Ponnuswamy PP, Hatiboglu G, Quaschning T, Widder J, et al. Native C-reactive protein induces endothelial dysfunction in ApoE−/− mice: implications for iNOS and reactive oxygen species. Atherosclerosis. 2007;195:e76– 84. doi: 10.1016/j.atherosclerosis.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Calabro P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 2003;108:1930–2. doi: 10.1161/01.CIR.0000096055.62724.C5. [DOI] [PubMed] [Google Scholar]
- 12.Yasojima K, Schwab C, McGeer EG, McGeer PL. Generation of C-reactive protein and complement components in atherosclerotic plaques. Am J Pathol. 2001;158:1039–51. doi: 10.1016/S0002-9440(10)64051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi S, Inoue N, Ohashi Y, Terashima M, Matsui K, Mori T, et al. Interaction of oxidative stress and inflammatory response in coronary plaque instability: important role of C-reactive protein. Arterioscler Thromb Vasc Biol. 2003;23:1398– 404. doi: 10.1161/01.ATV.0000081637.36475.BC. [DOI] [PubMed] [Google Scholar]
- 14.Dong Q, Wright JR. Expression of C-reactive protein by alveolar macrophages. J Immunol. 1996;156:4815–20. [PubMed] [Google Scholar]
- 15.Venugopal SK, Devaraj S, Jialal I. Macrophage conditioned medium induces the expression of C-reactive protein in human aortic endothelial cells: potential for paracrine/autocrine effects. Am J Pathol. 2005;166:1265–71. doi: 10.1016/S0002-9440(10)62345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouchi N, Kihara S, Funahashi T, Nakamura T, Nishida M, Kumada M, et al. Reciprocal association of C-reactive protein with adiponectin in blood stream and adipose tissue. Circulation. 2003;107:671– 4. doi: 10.1161/01.cir.0000055188.83694.b3. [DOI] [PubMed] [Google Scholar]
- 17.Devaraj S, Torok N, Dasu MR, Samols D, Jialal I. Adiponectin decreases C-reactive protein synthesis and secretion from endothelial cells: evidence for an adipose tissue-vascular loop. Arterioscler Thromb Vasc Biol. 2008;28:1368–74. doi: 10.1161/ATVBAHA.108.163303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh P, Hoffmann M, Wolk R, Shamsuzzaman AS, Somers VK. Leptin induces C-reactive protein expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:e302–7. doi: 10.1161/ATVBAHA.107.148353. [DOI] [PubMed] [Google Scholar]
- 19.Fichtlscherer S, Rosenberger G, Walter DH, Breuer S, Dimmeler S, Zeiher AM. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. 2000;102:1000– 6. doi: 10.1161/01.cir.102.9.1000. [DOI] [PubMed] [Google Scholar]
- 20.Cleland SJ, Sattar N, Petrie JR, Forouhi NG, Elliott HL, Connell JM. Endothelial dysfunction as a possible link between C-reactive protein levels and cardiovascular disease. Clin Sci (Lond) 2000;98:531–5. [PubMed] [Google Scholar]
- 21.Tomai F, Crea F, Gaspardone A, Versaci F, Ghini AS, Chiariello L, Gioffre PA. Unstable angina and elevated C-reactive protein levels predict enhanced vasoreactivity of the culprit lesion. Circulation. 2001;104:1471– 6. doi: 10.1161/hc3801.096354. [DOI] [PubMed] [Google Scholar]
- 22.Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165– 8. doi: 10.1161/01.cir.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 23.Pasceri V, Cheng JS, Willerson JT, Yeh ET, Chang J. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation. 2001;103:2531– 4. doi: 10.1161/01.cir.103.21.2531. [DOI] [PubMed] [Google Scholar]
- 24.Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439– 41. doi: 10.1161/01.cir.0000033116.22237.f9. [DOI] [PubMed] [Google Scholar]
- 25.Singh U, Devaraj S, Vasquez-Vivar J, Jialal I. C-reactive protein decreases endothelial nitric oxide synthase activity via uncoupling. J Mol Cell Cardiol. 2007;43:780–91. doi: 10.1016/j.yjmcc.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25:995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- 27.Nagaoka T, Kuo L, Ren Y, Yoshida A, Hein TW. C-reactive protein inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Invest Ophthalmol Vis Sci. 2008;49:2053– 60. doi: 10.1167/iovs.07-1387. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Roumeliotis N, Sawamura T, Renier G. C-reactive protein enhances LOX-1 expression in human aortic endothelial cells: relevance of LOX-1 to C-reactive protein-induced endothelial dysfunction. Circ Res. 2004;95:877– 83. doi: 10.1161/01.RES.0000147309.54227.42. [DOI] [PubMed] [Google Scholar]
- 29.Teoh H, Quan A, Lovren F, Wang G, Tirgari S, Szmitko PE, et al. Impaired endothelial function in C-reactive protein overexpressing mice. Atherosclerosis. 2008;201:318–25. doi: 10.1016/j.atherosclerosis.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz R, Osborne-Lawrence S, Hahner L, Gibson LL, Gormley AK, Vongpatanasin W, et al. C-reactive protein downregulates endothelial NO synthase and attenuates reendothelialization in vivo in mice. Circ Res. 2007;100:1452–9. doi: 10.1161/01.RES.0000267745.03488.47. [DOI] [PubMed] [Google Scholar]
- 31.Bisoendial RJ, Kastelein JJ, Peters SL, Levels JH, Birjmohun R, Rotmans JI, et al. Effects of CRP infusion on endothelial function and coagulation in normocholesterolemic and hypercholesterolemic subjects. J Lipid Res. 2007;48:952– 60. doi: 10.1194/jlr.P600014-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Venugopal SK, Devaraj S, Jialal I. C-reactive protein decreases prostacyclin release from human aortic endothelial cells. Circulation. 2003;108:1676– 8. doi: 10.1161/01.CIR.0000094736.10595.A1. [DOI] [PubMed] [Google Scholar]
- 33.Danenberg HD, Szalai AJ, Swaminathan RV, Peng L, Chen Z, Seifert P, et al. Increased thrombosis after arterial injury in human C-reactive protein-transgenic mice. Circulation. 2003;108:512–5. doi: 10.1161/01.CIR.0000085568.13915.1E. [DOI] [PubMed] [Google Scholar]
- 34.Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. 2003;107:398– 404. doi: 10.1161/01.cir.0000052617.91920.fd. [DOI] [PubMed] [Google Scholar]
- 35.Nakakuki T, Ito M, Iwasaki H, Kureishi Y, Okamoto R, Moriki N, et al. Rho/Rho-kinase pathway contributes to C-reactive protein-induced plasminogen activator inhibitor-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:2088–93. doi: 10.1161/01.ATV.0000183607.50230.9f. [DOI] [PubMed] [Google Scholar]
- 36.Singh U, Devaraj S, Jialal I. C-reactive protein decreases tissue plasminogen activator activity in human aortic endothelial cells: evidence that C-reactive protein is a procoagulant. Arterioscler Thromb Vasc Biol. 2005;25:2216–21. doi: 10.1161/01.ATV.0000183718.62409.ea. [DOI] [PubMed] [Google Scholar]
- 37.Verma S, Li SH, Badiwala MV, Weisel RD, Fedak PW, Li RK, et al. Endothelin antagonism and interleukin-6 inhibition attenuate the proatherogenic effects of C-reactive protein. Circulation. 2002;105:1890– 6. doi: 10.1161/01.cir.0000015126.83143.b4. [DOI] [PubMed] [Google Scholar]
- 38.Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest. 1998;101:353– 63. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devaraj S, Kumaresan PR, Jialal I. Effect of C-reactive protein on chemokine expression in human aortic endothelial cells. J Mol Cell Cardiol. 2004;36:405–10. doi: 10.1016/j.yjmcc.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Zhu X, Xu Q, Ding X, Chen YE, Song Q. Effect of C-reactive protein on gene expression in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2005;288:H1539– 45. doi: 10.1152/ajpheart.00963.2004. [DOI] [PubMed] [Google Scholar]
- 41.Lin R, Liu J, Gan W, Yang G. C-reactive protein-induced expression of CD40-CD40L and the effect of lovastatin and fenofibrate on it in human vascular endothelial cells. Biol Pharm Bull. 2004;27:1537– 43. doi: 10.1248/bpb.27.1537. [DOI] [PubMed] [Google Scholar]
- 42.Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leukocytes is Fcgamma receptor II. J Exp Med. 1999;190:585–90. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359– 63. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- 44.Singh U, Dasu MR, Yancey PG, Afify A, Devaraj S, Jialal I. Human C-reactive protein promotes oxidized low density lipoprotein uptake and matrix metalloproteinase-9 release in Wistar rats. J Lipid Res. 2008;49:1015–23. doi: 10.1194/jlr.M700535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fadini GP, Agostini C, Sartore S, Avogaro A. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194:46–54. doi: 10.1016/j.atherosclerosis.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 46.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593– 600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 47.George J, Goldstein E, Abashidze S, Deutsch V, Shmilovich H, Finkelstein A, et al. Circulating endothelial progenitor cells in patients with unstable angina: association with systemic inflammation. Eur Heart J. 2004;25:1003– 8. doi: 10.1016/j.ehj.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 48.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 49.Wang CH, Ting MK, Verma S, Kuo LT, Yang NI, Hsieh IC, Wang SY, Hung A, Cherng WJ. Pioglitazone increases the numbers and improves the functional capacity of endothelial progenitor cells in patients with diabetes mellitus. Am Heart J. 2006;152:1051.e1–8. doi: 10.1016/j.ahj.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 50.Fujii H, Li SH, Szmitko PE, Fedak PW, Verma S. C-reactive protein alters antioxidant defenses and promotes apoptosis in endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2006;26:2476– 82. doi: 10.1161/01.ATV.0000242794.65541.02. [DOI] [PubMed] [Google Scholar]
- 51.Turu MM, Slevin M, Matou S, West D, Rodríguez C, Luque A, et al. C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signalling pathways and gene expression. BMC Cell Biol. 2008;9:47–53. doi: 10.1186/1471-2121-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood. 1993;82:513–20. [PubMed] [Google Scholar]
- 53.Paffen E, Vos HL, Bertina RM. C-reactive protein does not directly induce tissue factor in human monocytes. Arterioscler Thromb Vasc Biol. 2004;24:975– 81. doi: 10.1161/01.ATV.0000126681.16619.69. [DOI] [PubMed] [Google Scholar]
- 54.Cirillo P, Golino P, Calabrò P, Calì G, Ragni M, De Rosa S, et al. C-reactive protein induces tissue factor expression and promotes smooth muscle and endothelial cell proliferation. Cardiovasc Res. 2005;68:47–55. doi: 10.1016/j.cardiores.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Devaraj S, Dasu MR, Singh U, Rao LV, Jialal I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. doi: 10.1016/j.atherosclerosis.2008.05.060. [Epub ahead of print 2008 Jun 13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prasad K. C-reactive protein increases oxygen radical generation by neutrophils. J Cardiovasc Pharmacol Ther. 2004;9:203–9. doi: 10.1177/107424840400900308. [DOI] [PubMed] [Google Scholar]
- 57.Trachtman H, Futterweit S, Arzberger C, Bod J, Goldschmiedt J, Gorman H, et al. Nitric oxide and superoxide in rat mesangial cells: modulation by C-reactive protein. Pediatr Nephrol. 2006;21:619–26. doi: 10.1007/s00467-006-0066-x. [DOI] [PubMed] [Google Scholar]
- 58.Singh U, Devaraj S, Jialal I. C-reactive protein stimulates myeloperoxidase release from polymorphonuclear cells and monocytes: implications for acute coronary syndromes. Clin Chem. 2009;55:361– 4. doi: 10.1373/clinchem.2008.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine. 1992;4:361– 8. doi: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 60.Singh U, Devaraj S, Dasu MR, Ciobanu D, Reusch J, Jialal I. C-reactive protein decreases interleukin-10 secretion in activated human monocyte-derived macrophages via inhibition of cyclic AMP production. Arterioscler Thromb Vasc Biol. 2006;26:2469–75. doi: 10.1161/01.ATV.0000241572.05292.fb. [DOI] [PubMed] [Google Scholar]
- 61.Woollard KJ, Phillips DC, Griffiths HR. Direct modulatory effect of C-reactive protein on primary human monocyte adhesion to human endothelial cells. Clin Exp Immunol. 2002;130:256– 62. doi: 10.1046/j.1365-2249.2002.01978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devaraj S, Davis B, Simon SI, Jialal I. CRP promotes monocyte-endothelial cell adhesion via Fc-gamma receptors in human aortic endothelial cells under static and shear flow conditions. Am J Physiol Heart Circ Physiol. 2006;291:H1170– 6. doi: 10.1152/ajpheart.00150.2006. [DOI] [PubMed] [Google Scholar]
- 63.Devaraj S, Yun JM, Duncan-Staley C. C-reactive protein promotes MCSF release and macrophage proliferation. J Leukoc Biol. doi: 10.1189/jlb.0808458. [Epub ahead of print 2008 Nov 13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation. 2001;103:1194–7. doi: 10.1161/01.cir.103.9.1194. [DOI] [PubMed] [Google Scholar]
- 65.Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci U S A. 2002;99:13043– 8. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Liao D, Bharadwaj U, Li M, Yao Q, Chen C. C-reactive protein inhibits cholesterol efflux from human macrophage-derived foam cells. Arterioscler Thromb Vasc Biol. 2008;28:519–26. doi: 10.1161/ATVBAHA.107.159467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams TN, Zhang CX, Game BA, He L, Huang Y. C-reactive protein stimulates MMP-1 expression in U937 histiocytes through FcgammaRII and extracellular signal-regulated kinase pathway: an implication of CRP involvement in plaque destabilization. Arterioscler Thromb Vasc Biol. 2004;24:61– 6. doi: 10.1161/01.ATV.0000104014.24367.16. [DOI] [PubMed] [Google Scholar]
- 68.Han KH, Hong KH, Park JH, Ko J, Kang DH, Choi KJ, et al. C-reactive protein promotes monocyte chemoattractant protein-1-mediated chemotaxis through upregulating CC chemokine receptor 2 expression in human monocytes. Circulation. 2004;109:2566–71. doi: 10.1161/01.CIR.0000131160.94926.6E. [DOI] [PubMed] [Google Scholar]
- 69.Kawahara K, Biswas KK, Unoshima M, Ito T, Kikuchi K, Morimoto Y, et al. C-reactive protein induces high-mobility group box-1 protein release through activation of p38MAPK in macrophage RAW264.7 cells. Cardiovasc Pathol. 2008;17:129–318. doi: 10.1016/j.carpath.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 70.Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation. 2002;105:393– 6. doi: 10.1161/hc0302.102618. [DOI] [PubMed] [Google Scholar]
- 71.Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, Szmitko P, et al. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation. 2003;107:1783–90. doi: 10.1161/01.CIR.0000061916.95736.E5. [DOI] [PubMed] [Google Scholar]
- 72.Hattori Y, Matsumura M, Kasai K. Vascular smooth muscle cell activation by C-reactive protein. Cardiovasc Res. 2003;58:186–95. doi: 10.1016/s0008-6363(02)00855-6. [DOI] [PubMed] [Google Scholar]
- 73.Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, et al. C-reactive protein induces apoptosis in human coronary vascular smooth muscle cells. Circulation. 2004;110:579– 87. doi: 10.1161/01.CIR.0000136999.77584.A2. [DOI] [PubMed] [Google Scholar]
- 74.Ryu J, Lee CW, Shin JA, Park CS, Kim JJ, Park SJ, Han KH. FcgammaRIIa mediates C-reactive protein-induced inflammatory responses of human vascular smooth muscle cells by activating NADPH oxidase 4. Cardiovasc Res. 2007;75:555–65. doi: 10.1016/j.cardiores.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 75.Peng N, Liu JT, Gao DF, Lin R, Li R. Angiotensin II-induced C-reactive protein generation: inflammatory role of vascular smooth muscle cells in atherosclerosis. Atherosclerosis. 2007;193:292– 8. doi: 10.1016/j.atherosclerosis.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 76.Wu J, Stevenson MJ, Brown JM, Grunz EA, Strawn TL, Fay WP. C-reactive protein enhances tissue factor expression by vascular smooth muscle cells: mechanisms and in vivo significance. Arterioscler Thromb Vasc Biol. 2008;28:698–704. doi: 10.1161/ATVBAHA.107.160903. [DOI] [PubMed] [Google Scholar]
- 77.Paul A, Ko KW, Li L, Yechoor V, McCrory MA, Szalai AJ, Chan L. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–55. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 78.Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM, et al. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2005;102:8309–14. doi: 10.1073/pnas.0503202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Griselli M, Herbert J, Hutchinson WL, Taylor KM, Sohail M, Krausz T, Pepys MB. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. 1999;190:1733– 40. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab. 2004;24:1214– 8. doi: 10.1097/01.WCB.0000136517.61642.99. [DOI] [PubMed] [Google Scholar]
- 81.Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature (Lond) 2006;440:1217–21. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 82.Dasu MR, Devaraj S, Du Clos TW, Jialal I. The biological effects of CRP are not attributable to endotoxin contamination: evidence from TLR4 knockdown human aortic endothelial cells. J Lipid Res. 2007;48:509–12. doi: 10.1194/jlr.C600020-JLR200. [DOI] [PubMed] [Google Scholar]