

Abstract
Rationale: Bronchopulmonary dysplasia (BPD) continues to be a major morbidity in preterm infants. The lung pathology in BPD is characterized by impaired alveolar and capillary development. An imbalance between proteases and protease inhibitors in association with changes in lung elastic fibers has been implicated in the pathogenesis of BPD.
Objective: To investigate the expression and activity levels of papain-like lysosomal cysteine proteases, cathepsins B, H, K, L, S, and their inhibitors, cystatins B and C, in a baboon model of BPD.
Methods: Real-time reverse transcriptase–polymerase chain reaction, immunohistochemistry, immunoblotting, active site labeling of cysteine proteases, and in situ hybridization were performed.
Measurements and Main Results: The steady-state mRNA and protein levels of all cathepsins were significantly increased in the lung tissue of baboons with BPD. In contrast, the steady-state mRNA and protein levels of two major cysteine protease inhibitors, cystatin B and C, were unchanged. Correlating with these alterations, the activity of cysteine proteases in lung tissue homogenates and bronchoalveolar lavage fluid was significantly higher in the BPD group. The levels of cathepsin B, H, and S increased and cathepsin K decreased with advancing gestation. All cathepsins, except for cat K, were immunolocalized to macrophages in BPD. In addition, cathepsin H and cystatin B were colocalized in type 2 alveolar epithelial cells. Cathepsin L was detected in some bronchial epithelial, endothelial, and interstitial cells. Cathepsin K was localized to some perivascular cells by in situ hybridization.
Conclusions: Cumulatively, these findings demonstrate an imbalance between cysteine proteases and their inhibitors in BPD.
Keywords: baboon, bronchopulmonary dysplasia, cathepsin, macrophage, protease
Bronchopulmonary dysplasia (BPD) was originally described in 1967 as a chronic pulmonary disease that developed in premature infants who were treated with prolonged ventilation and 80 to 100% O2 (1). The lung pathology in classic BPD was characterized by widespread bronchial and bronchiolar disease with fibroproliferative changes, smooth muscle hyperplasia, and alternating areas of emphysema with atelectasis (1, 2). Since the original description of BPD, major advances in neonatology have significantly improved the care and survival rates of very low birth weight infants (birth weight < 1,500 g). Despite these advances, a milder form of the classic BPD, “new BPD,” continues to be a significant morbidity of premature infants (3, 4). Lung pathology in new BPD is markedly different than in the old BPD and is primarily characterized by impaired alveolar and capillary development (5). Approximately 25 to 30% of very low birth weight infants develop the new BPD (6, 7). Subsequently, these infants are more likely to have impaired growth, abnormal neurodevelopment, reactive airway disease, and respiratory infections requiring hospitalization (8–10).
The pathogenesis of BPD is multifactorial and incompletely understood. Lung immaturity, barotrauma/volutrauma, oxygen toxicity, and inflammation are well-recognized risk factors (3, 11, 12). An imbalance between proteases and protease inhibitors also has been implicated in the pathogenesis of BPD (12). Numerous studies in human infants with the classic BPD have shown increased elastase and decreased elastase inhibitory activity in the airway secretions (13–16). Elastin, the target molecule of elastolytic enzymes, is an important structural component of the lung. In elastin-deficient mice, impaired terminal airway branching is accompanied by fewer and dilated distal air sacs (17). Abnormal patterns of elastin expression and accumulation are observed in baboon and lamb models of BPD as well as in autopsy specimens of infants who died of BPD (16, 18–21).
Thus far, studies that investigated protease–antiprotease imbalance in BPD have focused on two groups of elastolytic proteases: serine proteases, such as neutrophil elastase (NE) and trypsin (15, 22), and metalloproteinases (MMPs), such as MMP-2 and MMP-9 (23, 24). Recently, a third group of proteases, the papain family of cysteine proteases, has been implicated in the pathogenesis of lung disorders (25). These proteases, cathepsins (cat) B, H, K, L, and S, play important roles in diverse processes, some of which are relevant to the pathophysiology of BPD, such as fibrosis (26), apoptosis (27), and extracellular matrix remodeling (28, 29). We therefore hypothesized that cysteine proteases have a role in the development of BPD. As the first step toward exploring this hypothesis, we characterized the mRNA and protein expression and cellular distribution of cat B, H, K, L, and S, and two of their major inhibitors, cystatin B and C, and determined the activity of cathepsins in a well-characterized baboon model of BPD (30). The ontogeny of cathepsins and cystatins in the baboon lung between 125 and 185 d gestation was also characterized. Our findings demonstrate an imbalance between cysteine proteases and their inhibitors in BPD. Some of the results of this study have been reported in the form of an abstract (31).
METHODS
Animal Model
Frozen and paraffin-embedded baboon lung tissues and necropsy bronchoalveolar lavage (BAL) samples were provided by the Southwest Foundation for Biomedical Research (San Antonio, TX). All procedures were approved by the Institutional Animal Care and Use Committee at the Southwest Foundation for Biomedical Research. Details of the model have been described previously (30). Briefly, baboons that were delivered by hysterotomy at 125 d developed characteristic features of the new BPD after surfactant (Survanta; Ross, Columbus, OH) replacement, mechanical ventilation, and “as needed” oxygen treatment for 14 d (BPD group). Baboons that were delivered at 125 or 140 d and killed immediately served as the gestational control (GC) animals (125- or 140-d groups). Another control group consisted of baboons that were born via natural delivery (ND) at full-term gestation (∼ 185 d) and killed 2 to 3 d later.
Isolation of Total RNA and Reverse Transcription
Total RNA was isolated from frozen baboon lung tissues or BAL cell pellets using TRIzol reagent (Gibco BRL, Rockville, MD). First-strand cDNA was synthesized from 0.5 μg of RNA using the Superscript First-Strand Synthesis System (Invitrogen, Carlsbad, CA) following the manufacturer's instructions.
Real-Time Polymerase Chain Reaction Analysis
Real-time polymerase chain reaction (PCR) analysis was performed using Mx4000 Multiplex Quantitative PCR System (Stratagene, Inc., La Jolla, CA) and the brilliant SYBR Green QPCR Master Mix (Stratagene). Oligonucleotide primer sequences are provided in Table E1 in the online supplement. A standard curve was plotted for each primer set with the critical threshold cycle (Ct) values obtained from amplification of 10-fold dilutions of cDNA from normal baboon lung tissue. GAPDH was used as an internal reference to normalize the target transcripts, and the relative differences were calculated.
Antibodies
The sources and dilutions of antibodies are provided in the online supplement.
Immunoblotting
Frozen lung tissue samples were homogenized in 10 volumes of phosphate buffer, pH 6.8, supplemented with a protease inhibitor cocktail (1 tablet/100 ml; Roche Applied Science, Indianapolis, IN). Immunoblotting was performed under reducing conditions as previously described (32). The protein bands were visualized by chemiluminescence (ECL Western Blotting Analysis System; Amersham Biosciences, Piscataway, NJ), and quantitated by densitometry with NIH-Image 163.
Active Site Labeling of Cysteine Proteases and Immunoprecipitation
Active site labeling of cysteine proteases and immunoprecipitation was performed as previously described (33). Lung lysates or BAL fluid (BALF) was normalized to total protein and incubated with a biotin-labeled E-64 analog, JPM565 (gift of H. Ploegh, Boston, MA). Samples were immunoprecipitated with cat B, H, K, L, or S antibodies, resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and visualized by chemiluminescence.
Immunohistochemistry
Immunohistochemistry was performed as previously reported (34). Antigen retrieval with trypsin–ethylenediamenetetraacetic acid (Invitrogen) digestion for 10 min at 37°C was used for cat B and CD68 immunostaining. Isotype-matched antibodies and omission of the primary antibody served as negative controls.
In Situ Hybridization
A 35S-labeled antisense riboprobe specific for cat K was generated from a reverse transcriptase–PCR product subcloned into pBluescript (Stratagene) and in situ hybridization was performed as previously described (35).
Statistical Analysis
Group data were analyzed with a Mann-Whitney test using SAS version 9.1 (SAS Institute, Cary, NC). Values are expressed as mean ± SEM. A p value of less than 0.05 was considered significant.
RESULTS
Steady-State mRNA Levels of Cysteine Proteases and Cystatins in BPD
Steady-state mRNA levels of cat B, H, K, L, and S, and cystatin B and C were analyzed by real-time PCR after reverse transcription of total RNA isolated from fresh frozen baboon lung tissues (Figure 1). In BPD, the steady-state mRNA levels of cat B, H, K, L, and S were increased about 1.5, 2.5, 9, 2.5, and 3 times, respectively, in comparison to the 140-d GC group (p < 0.05). Cat K, L, and S mRNA levels in BPD were also significantly higher than in the ND group (66-, 2.8-, and 1.9-fold, respectively; p < 0.05). The steady-state mRNA levels of cat B, H, and S were significantly higher in the full-term lungs (ND group) than in the 125-d GC group, whereas the cat S mRNA level was significantly lower in the ND group than in the 125-d GC group. In contrast to the alterations observed in cysteine proteases, the steady-state mRNA levels of cystatin B and C were similar in all GC groups as well as in the BPD group (Figure 1).
Figure 1.

Cathepsin mRNA levels during lung development and in BPD. Relative steady-state mRNA levels of cathepsin (cat) B, cat H, cat K, cat L, cat S, cystatin (cys) B, and cys C were determined by quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) using RNA obtained from 125-d, 140-d, natural delivery (ND), and bronchopulmonary dysplasia (BPD) group baboons. n = 6–8 animals/group. Data are expressed as mean ± SEM from two experiments performed in triplicates. *p < 0.05 versus 140 d; **p < 0.05 versus 125 d; ¶p < 0.05 versus ND.
Cysteine Protease and Cystatin Protein Expression in BPD
To characterize the expression pattern of cat B, H, K, L, and S, and cystatin B and C at the protein level, baboon lung lysates were analyzed by immunoblotting and densitometry (Figure 2). In BPD, cat B, H, K, L, and S proteins were increased about 2, 25, 2.5, 2, and 18 times, respectively, in comparison to the 140-d GC group (p < 0.05). Parallel with the alterations observed at the mRNA level, cat K, L, and S protein levels were also significantly higher in the BPD samples in comparison to the ND group (15, 2.2, and 3.4 times, respectively; p < 0.05). During lung development between 125-d and full-term gestation, the expression level of cat B, H, and S proteins increased and cat K decreased significantly, whereas that of cat L was similar in all GC groups. The protein levels of cystatin B and C, a major intracellular and a secreted inhibitor of cystein proteases, respectively, were similar in all groups. Similar results were obtained when samples were normalized to either total protein (i.e., same amount of total protein was loaded per lane on each gel) or β-actin (data not shown). Proenzyme forms of cathepsin B, L, and S were detected in some samples, but there was no statistically significant difference among the study groups. Cat H was detected in both single- and heavy-chain forms in ND and BPD groups. In all samples, densitometry was performed using only the mature single-chain form of the enzymes.
Figure 2.

Cathepsin (cat) proteins during lung development and in BPD. Immunoblot and densitometry analysis for cat B, H, K, L, and S, and cystatin (cys) B and C proteins were performed in total lung homogenates obtained from 125-d, 140-d, ND, and BPD group baboons. Results are representative of three independent experiments using a total of 6–10 different study animals/group. The marker indicates 28 and 15 kD for cathepsin and cystatin immunoblots, respectively. Mean ± SEM values for all proteins are normalized to values in the 125-d gestation lungs. The total amount of protein loaded per lane is 4 μg for cat L, 5 μg for cys B, 10 μg for cat B, H, and K, and 15 μg for cat S immunoblots. *p < 0.05 versus 140 d; **p < 0.05 versus 125 d; ¶p < 0.05 versus ND.
Cysteine Protease Activity in BPD
To determine if the activity of cathepsins showed a parallel increase to the levels of these proteins in the lung tissues and BALF, active site labeling of cysteine proteases was performed in 140-d, ND, and BPD group samples (Figure 3). Both in lung tissues (Figure 3A) and BALF (Figure 3B), there was increased activity of cysteine proteases in BPD relative to both the 140-d and ND controls. The individual cathepsins that contributed to the total cysteine protease activity in BPD samples were determined by immunoprecipitation of active site–labeled lung homogenates using their specific antibodies (Figure 3C).
Figure 3.

Increased activity of cathepsins in BPD. Lung lysates (A) and bronchoalveolar lavage fluid (BALF; B) obtained from 140-d, ND, and BPD group baboons were normalized to total protein, incubated with a biotin-labeled E-64 analog (JPM-565), and resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and chemiluminescence. Data are representative of three independent experiments using a total of 6–10 different study animals/group. (C) To identify the relative molecular masses of active site–labeled cathepsins in the baboon lung, active site–labeled and pooled lung homogenates from three baboons in the BPD group were immunoprecipitated using cat B, H, K, L, and S antibodies. All immunoprecipitated cathepsins were detected with horseradish peroxidase–streptavidin and chemiluminescence.
Detection of Cysteine Proteases and Cystatins in BALF
To determine if active forms of cysteine proteases can be detected in BALF samples of baboons with BPD and to determine their relative levels, BALF samples were analyzed by immunoblotting and densitometry (Figure 4). Single-chain active forms of cat B and L enzymes were detected in BALF of 140-d and ND samples as well as the BPD samples. Cat B was also detected as a proenzyme in some ND and BPD samples. In BPD, the cat B level was similar to that in the ND group, but significantly higher than in the 140-d GC group, whereas the cat L level was significantly increased in comparison to both the 140-d and ND groups (p < 0.05). Interestingly, cat S was present only as a proenzyme in the 140-d group and as a single-chain enzyme (the only mature form of cat S) in the ND and BPD groups. By densitometry, the BALF cat S level was about three times higher in the BPD group than in the ND group. In overexposed immunoblots, small amounts of pro-cat H was detected in the 140-d samples, whereas ND and BPD groups had single-chain cat H (data not shown). Cat K was not detectable in BALF samples of either the BPD group or the GC groups. Both cystatins B and C were detectable in BALF in all groups tested in similar levels.
Figure 4.

Detection of cathepsins (cat) in BALF. Immunoblot analysis for cat B, L, and S, and cystatin (cys) B and C were performed on baboon necropsy lavage fluids of 140-d, ND, and BPD group baboons. Results are representative of at least two experiments using samples from a total of 6–10 different study animals/group. Black and white arrows indicate procathepsins and mature cathepsins, respectively. The total amount of protein loaded per lane is 4 μg for cat B and cys B and C, and 7 μg for cat L and S immunoblots. *p < 0.05 versus 140 d; ¶p < 0.05 versus ND.
Macrophages in the 125-d Baboon Model of BPD
Macrophages are the primary sources of several cathepsins. To determine if increased cathepsin levels were associated with an increased number of macrophages in the 125-d baboon model of BPD, we analyzed the mRNA expression of the macrophage marker CD68 in the lung tissue and BALF cell pellets by quantitative reverse transcriptase–PCR. In addition, BALF total and differential cell counts were performed on cytospin preparations. In the lung tissue, the steady-state mRNA level of CD-68 was significantly higher in the BPD group in comparison to 140-d GCs (Figure 5A; p < 0.05). The ND group was used as the control group for CD68 expression in BALF cells because the 140-d group had very few cells and RNA isolation was not successful. The BPD group had an approximate 3.5-fold increase in CD68 mRNA expression in comparison to the ND group (Figure 5B; p < 0.05). Absolute macrophage counts in BAL were increased about 10 and 2 times, respectively, in comparison to the 140-d and ND groups (Figure 5C; p < 0.05). The expression of CD68 at the protein level was determined semiquantitatively by immunohistochemistry using an anti-CD68 antibody in paraffin-embedded lung tissue sections of 140-d (Figure 5D), ND (Figure 5E), and BPD (Figure 5F) groups.
Figure 5.

Macrophages in baboon lung tissues and BALF in BPD. Relative steady-state mRNA levels of the activated macrophage marker CD68 were determined by quantitative RT-PCR using RNA obtained from total baboon lung homogenates (A) or BAL cells (B); n = 4–5 animals/group. Data are expressed as mean ± SEM from two experiments performed in triplicate. Absolute macrophage numbers were determined in BAL cell pellets of 140-d, ND, and BPD group animals (C); n = 10–12 animals/group. Immunohistochemistry was performed on paraffin-embedded lung tissue sections from 140-d (D), ND (E), and BPD (F) groups using a CD68 antibody. Methyl green was used as the counterstain. Arrowheads point to CD68-positive macrophages in representative tissue sections. Scale bar, 100 μm. *p < 0.05 versus 140 d; ¶p < 0.05 versus ND.
Immunolocalization of Cathepsins and Cystatin B in BPD
To characterize the cellular distribution of cathepsins and the intracellular cysteine protease inhibitor cystatin B in BPD, immunohistochemistry was performed on paraffin-embedded baboon lung tissue sections using specific antibodies (Figure 6). Pro-surfactant protein C (pro-SPC) and CD68 were used as markers for type 2 alveolar epithelial cells and macrophages, respectively. Cat B was detected in macrophages and some CD68-negative interstitial cells (Figures 6A–6C). Some of these interstitial cells were identified as fibroblasts on the basis of their morphology (Figures 6B and 6C). Cat H protein was detected in type 2 alveolar epithelial cells in addition to macrophages (Figures 6D–6F). Type 2 cells were identified on the basis of their reactivity with pro-SPC antibody in serial sections (shown in the online supplement). Cat L was detected in CD68-positive cells (Figure 6G) as well as in some CD68-negative interstitial cells, some of which were fibroblasts (Figure 6J). In the ciliated, pseudostratified bronchial epithelium, cat L was detected in a supranuclear, apical localization (Figure 6H). Cat L immunoreactivity was also detected in some vascular and lymphoid endothelial cells in a subset of BPD samples (Figures 6I and 6J). Cat S expression was restricted to CD68-positive cells (Figures 6K and 6L). Cystatin B immunoreactivity was detected in macrophages, type 2 alveolar epithelial cells, as well as large airway epithelial cells (Figures 6M and 6N). Mouse (Figure 6O) and rabbit (not shown) primary antibody isotype controls did not show any immunoreactivity with the baboon lung tissues.
Figure 6.

Immunolocalization of cathepsin and cystatin B proteins in BPD. Immunohistochemistry was performed on formalin-fixed, paraffin-embedded lung tissue sections of baboons with BPD using specific antibodies for cat B, H, L, and S, and cystatin B. Methyl green was used as the counterstain. Photomicrographs are representative of the results obtained in a minimum of four experiments using samples from four to five study animals. Scale bar, 100 μm. Shown are cat B immunoreactivity in macrophages (A; black arrow), interstitial cells (B; white arrows), and fibroblasts (C; white arrows); cat H staining in type 2 alveolar epithelial cells (D, E, and F; arrowheads), macrophages (D, E, and F; black arrows), and rare interstitial cells (E; white arrows); cat L immunoreactivity in macrophages (G; black arrows), bronchial epithelial cells in supranuclear localization (H; black arrowhead), vascular (I; black arrow) and lymphatic endothelial cells (J; black arrow), and a fibroblast (J; white arrowhead); cat S staining in macrophages (K and L; black arrows); cystatin B immunoreactivity in type 2 alveolar epithelial cells (M; arrowhead), macrophages (M; black arrow), and bronchial epithelial cells (N) and a bronchial gland (N; black arrowhead); negative control stained with mouse primary antibody isotype control showing no staining in macrophages (O; black arrows).
Cat K mRNA Detection in BPD by In Situ Hybridization
The lung tissue distribution of cat K protein could not be reliably determined by immunohistochemistry due to poor reactivity of the antibodies with lung tissue sections of the BPD group baboons. To determine the mRNA distribution of cat K in baboon lung tissues in BPD, in situ hybridization was performed. Cathepsin mRNA was localized to some cells in the perivascular areas (Figure 7).
Figure 7.
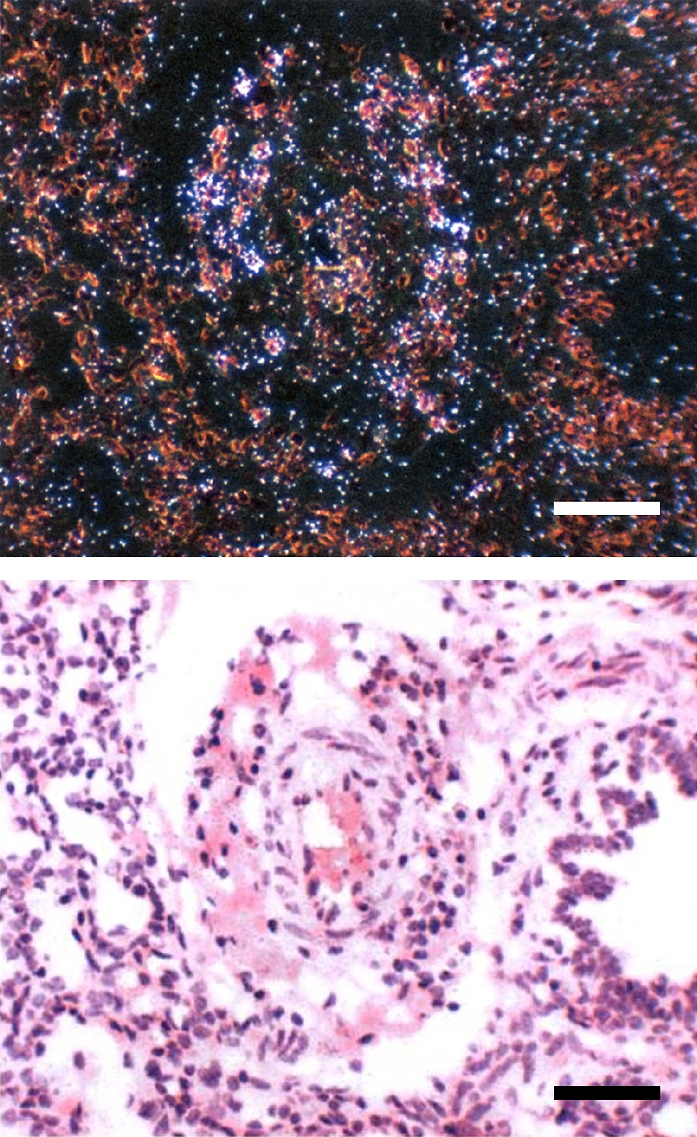
Cat K mRNA expression in BPD. In situ hybridization using a 35S-labeled antisense riboprobe specific for cat K was performed. Shown are paired bright and dark-field views of lung sections hybridized in situ for cat K mRNA. Positive signal is visible as white silver grains in the dark-field view. Note individual positive cells localizing to perivascular areas. Scale bar, 100 μm.
DISCUSSION
This study has shown the following in a baboon model of new BPD: (1) the cysteine proteases cat B, H, K, L, and S were up-regulated at the mRNA and protein levels in the lung; (2) cat B, H, L, and S protein levels were increased in the necropsy BALF samples; (3) the levels of two major inhibitors of cysteine proteases, cystatin B and C, in the lung and BALF were similar in BPD and control tissues; (4) increased activity of cat B, H, K, L, and S proteins were detected in lung tissue and BALF by active site labeling; (5) CD68-positive cells were increased both in the lung tissue and the airways; (6) cat B, H, L, and S, and cystatin B were expressed in CD68-positive cells in BPD. In addition, cat H and cystatin B were colocalized in type 2 alveolar epithelial cells, whereas cat L was detected in some bronchial epithelial and endothelial cells. Cat B and L immunoreactivity was detected in some CD68-negative interstitial cells, some of which were fibroblasts. Cat K mRNA was expressed in some cells that were localized to perivascular areas. Cumulatively, these findings demonstrate an imbalance between cysteine proteases and their inhibitors that results in an overall increase in cysteine protease activity in BPD.
A second goal of this study was to examine the developmental changes in expression levels of cysteine proteases and cystatins. The expression levels of cat H and S were very low in the immature 125-d baboon lung and increased with advancing gestational age. Cat B levels also were higher in full-term animals than in the 125- or 140-d groups, whereas cat L, cystatin B, and cystatin C levels did not show any significant alterations between 125 and 185 d gestation. Interestingly, cat K levels decreased with advancing gestational age and were almost undetectable in the ND group. Consistent with a study by Buhling and coworkers (36), we did not detect any cat K immunoreactivity in either interstitial or alveolar macrophages. Instead, cat K was detected in mast cells in 125- and 140-d lung tissues by immunohistochemistry (S.C., unpublished data) and in some interstitial cells in perivascular locations, also likely mast cells, in BPD by in situ hybridization. Mast cell hyperplasia is a well-known feature of BPD (37, 38) and, taken together with our findings, this phenomenon can explain the source of increased cat K levels in BPD. Although cat K expression was previously reported in human bronchial and alveolar epithelial cells in one study (39), we could not confirm these findings in baboon lung tissues.
Cat B, H, K, L, and S are members of the papain family of cysteine proteases (reviewed in References 25, 40). These proteases are synthesized in the endoplasmic reticulum as pre-proproteins and activated in the acidic environment of the lysosomes. Initially, the function of cathepsins was believed to be restricted to nonspecific degradation of proteins in the lysosomes. However, studies over the last decade have shown that, despite having some functional overlaps, cathepsins differ significantly in their substrate specificities, tissue distributions, and intracellular functions. For example, cat L and S play key roles in processing major histocompatibility complex class II–associated invariant chain in antigen-presenting cells (41, 42). Cat K, the major cysteine protease expressed in osteoclasts, has potent collagenolytic activity and plays a pivotal role in bone remodeling (43, 44). Cat H, a protease with both amino- and endoprotease activities, is involved in processing of SPB and SPC in type 2 alveolar epithelial cells (45, 46).
BPD is a unique disease of the immature lung that results in “disrupted lung development.” However, several pathologic features, such as airway enlargement, inflammation, and fibrosis, are common findings in some adult lung diseases and BPD. Therefore, they may also share similar underlying mechanisms of lung injury. Although this is the first report to demonstrate an association between cysteine proteases and BPD, recent studies in genetically modified mice have suggested a role for cathepsins in the development of pathologic processes in the lung, such as fibrosis and emphysema. For example, overexpression of interleukin 13 or IFN-γ in the adult murine lung causes emphysema and inflammation that are accompanied by induction of several cathepsins in addition to MMPs (28, 29). In another study, cat K–deficient mice deposited significantly more extracellular matrix in response to bleomycin than did wild-type control mice (26). This finding suggests that cat K has a protective role to counter the excessive deposition of collagen matrix in this model of lung injury.
This study showed that the expression levels and activities of cat B, H, K, L, and S were up-regulated in BPD. This finding does not necessarily imply that these cathepsins collectively function as mediators or protective molecules in BPD. For example, cat K, as indicated above, could have a protective role in the lung through its antifibrotic activity. In contrast, cat L is a potential candidate to function as a mediator of BPD. In addition to being a potent elastolytic protease, cat L can cleave and inactivate protective molecules in the lung, such as α1-antitrypsin, secretory leukocyte protease inhibitor, and defensins (47–49). Similarly, cat B, a known inducer of apoptosis (50, 51), is more likely to be a mediator than a protective molecule in BPD.
Although NE is regarded as the major proteinase responsible from tissue damage in old BPD, a paucity of literature on NE and its inhibitors in new BPD together with emerging data on other classes of proteases, such as cathepsins and MMPs, raise questions about the relative role of NE in new BPD. Sequential tracheal aspirate fluids of baboons with new BPD showed a higher number of macrophages than neutrophils at each time point tested, although this difference did not reach statistical significance because of the variability (30). We noted significantly increased number of macrophages in necropsy BALF samples in the same baboon model of BPD. On the basis of these findings, we propose that, in the surfactant era, the predominant inflammatory cell type in BPD has become the macrophage instead of the neutrophil. If our speculation is supported with data from human infants, then neutrophil to macrophage switch could account for the changing profile of proteases in new BPD. Another protease implicated in new BPD is MMP-9, which is secreted by both neutrophils and macrophages. However, it is intriguing that, although the imbalance between cysteine proteases and inhibitors in BPD is a consequence of increased levels of cysteine proteases, MMP-9–tissue inhibitor of metalloproteinase (TIMP) 1 imbalance is a consequence of decreased levels of the inhibitor TIMP-1 (24).
With the exception of cat S, cathepsins have an acidic pH optimum and to maintain their activity extracellularly, they must be released into an acidic environment (25, 40, 52). An interesting finding of this study was the detection of mature and active forms of cat B, H, L, and S in BALF in BPD. Both pro and mature forms of cathepsins can be released from different cell types, including activated macrophages (cat B, L, S, K) (53, 54), smooth muscle cells (cat S) (55), and fibroblasts (cat B) (56). Furthermore, Punturieri and colleagues have shown that macrophages can acidify their pericellular space by using a vacuolar type H+-ATPase before releasing cathepsins (54). This important observation explains how extracellular cathepsins in BALF could maintain their activity in the airways, provided that they are released from certain cells, such as macrophages. Alternatively, detection of cathepsins in BALF may be due to release from detached and necrotic airway cells.
Our study has some limitations that need to be considered. First, the sample size of the study groups is relatively small for statistical analysis. Second, cysteine protease activity levels are measured by active site labeling and could be affected by potential contribution of other cysteine proteases in addition to cat B, H, K, L, and S. Finally, cysteine protease and cystatin measurements in BALF were performed by normalizing the samples to total protein in BALF. This method could give rise to inaccurate results in the presence of capillary leakage, such as that occurs in BPD. Nevertheless, the magnitude of differences observed in BALF cathepsins were unlikely to be affected by any biases introduced by normalization methods and were also consistent with the tissue levels.
In summary, we have shown significant increases in cat B, H, K, L, and S mRNA and protein levels in association with increased activities of these enzymes in the lung tissues of baboons with BPD. The increased levels of cathepsins accompany an influx of macrophages in the lung tissue and airways in BPD. In addition to macrophages, cathepsins are expressed in several different cell types in BPD. Cumulatively, these results suggest that cysteine proteases may play a role in the development of BPD. Considering the diverse functions of these proteases, it is possible that they could also have differential roles in the pathogenesis of BPD. Further studies are required to define the specific roles of cysteine proteases in the lung and to determine whether inhibition of any cathepsin(s) could ameliorate pathologic findings associated with BPD.
Supplementary Material
Acknowledgments
The authors thank all the personnel at the Bronchopulmonary Dysplasia Resource Center in San Antonio for their work related to the baboon model. They also thank Dr. Steve D. Shapiro for critical review of the manuscript and helpful suggestions; Drs. Gary A. Silverman, Thomas Mariani, Mark Exley, Helen Christou, Don McCurnin, and Harry Kozakewich for valuable discussions; and Rita Medek for excellent technical support.
Supported by National Institutes of Health grants HL075904 (S.C.), HL04403 (S.C.), HL63387 (R.A.P.), and P51 RR13986 for facility support; Survanta was donated by Ross Laboratories, Columbus, Ohio, and ventilators were donated by InfantStar (Infrasonics, San Diego, CA).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.200503-425OC on September 15, 2005
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Northway WJ, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N Engl J Med 1967;276:357–368. [DOI] [PubMed] [Google Scholar]
- 2.Bonikos DS, Bensch KG. Pathogenesis of bronchopulmonary dysplasia. In: Merritt TA, Northway WH, editors. Bronchopulmonary dysplasia. Boston, MA: Blackwell; 1988. pp. 33–58.
- 3.Bancalari E, Claure N, Sosenko IR. Bronchopulmonary dysplasia: changes in pathogenesis, epidemiology and definition. Semin Neonatol 2003;8:63–71. [DOI] [PubMed] [Google Scholar]
- 4.Tanswell AK, Jankov RP. Bronchopulmonary dysplasia: one disease or two? Am J Respir Crit Care Med 2003;167:1–2. [DOI] [PubMed] [Google Scholar]
- 5.Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol 2003;8:73–81. [DOI] [PubMed] [Google Scholar]
- 6.Lemons JA, Bauer CR, Oh W, Korones SB, Papile LA, Stoll BJ, Verter J, Temprosa M, Wright LL, Ehrenkranz RA, et al. Very low birth weight outcomes of the National Institute of Child Health and Human Development Neonatal Research Network, January 1995 through December 1996. Pediatrics 2001;107:E1. [DOI] [PubMed] [Google Scholar]
- 7.Walsh MC, Yao Q, Gettner P, Hale E, Collins M, Hensman A, Everette R, Peters N, Miller N, Muran G, et al. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics 2004;114:1305–1311. [DOI] [PubMed] [Google Scholar]
- 8.Gross SJ, Iannuzzi DM, Kveselis DA, Anbar RD. Effect of preterm birth on pulmonary function at school age: a prospective controlled study. J Pediatr 1998;133:188–192. [DOI] [PubMed] [Google Scholar]
- 9.Halvorsen T, Skadberg BT, Eide GE, Roksund OD, Carlsen KH, Bakke P. Pulmonary outcome in adolescents of extreme preterm birth: a regional cohort study. Acta Paediatr 2004;93:1294–1300. [PubMed] [Google Scholar]
- 10.Short EJ, Klein NK, Lewis BA, Fulton S, Eisengart S, Kercsmar C, Baley J, Singer LT. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics 2003;112:e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jobe AH. Antenatal factors and the development of bronchopulmonary dysplasia. Semin Neonatol 2003;8:9–17. [DOI] [PubMed] [Google Scholar]
- 12.Speer CP. Inflammation and bronchopulmonary dysplasia. Semin Neonatol 2003;8:29–38. [DOI] [PubMed] [Google Scholar]
- 13.Merritt TA, Cochrane CG, Holcomb K, Bohl B, Hallman M, Strayer D, Edwards DD, Gluck L. Elastase and alpha 1-proteinase inhibitor activity in tracheal aspirates during respiratory distress syndrome: role of inflammation in the pathogenesis of bronchopulmonary dysplasia. J Clin Invest 1983;72:656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogden BE, Murphy SA, Saunders GC, Pathak D, Johnson JD. Neonatal lung neutrophils and elastase/proteinase inhibitor imbalance. Am Rev Respir Dis 1984;130:817–821. [DOI] [PubMed] [Google Scholar]
- 15.Watterberg KL, Carmichael DF, Gerdes JS, Werner S, Backstrom C, Murphy S. Secretory leukocyte protease inhibitor and lung inflammation in developing bronchopulmonary dysplasia. J Pediatr 1994;125:264–269. [DOI] [PubMed] [Google Scholar]
- 16.Bruce MC, Schuyler M, Martin RJ, Starcher BC, Tomashefski JF Jr, Wedig KE. Risk factors for the degradation of lung elastic fibers in the ventilated neonate: implications for impaired lung development in bronchopulmonary dysplasia. Am Rev Respir Dis 1992;146:204–212. [DOI] [PubMed] [Google Scholar]
- 17.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 2000;23:320–326. [DOI] [PubMed] [Google Scholar]
- 18.McCurnin DC, Pierce RA, Chang LY, Gibson LL, Osborne-Lawrence S, Yoder BA, Kerecman JD, Albertine KH, Winter VT, Coalson JJ, et al. Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 2005;288:L450–L459. [DOI] [PubMed] [Google Scholar]
- 19.Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: disordered pulmonary elastin deposition. Am J Physiol 1997;272:L452–L460. [DOI] [PubMed] [Google Scholar]
- 20.Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: disordered respiratory tract development. Am J Respir Crit Care Med 1999;159:945–958. [DOI] [PubMed] [Google Scholar]
- 21.Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics 2000;106:1452–1459. [DOI] [PubMed] [Google Scholar]
- 22.Cederqvist K, Haglund C, Heikkila P, Sorsa T, Tervahartiala T, Stenman UH, Andersson S. Pulmonary trypsin-2 in the development of bronchopulmonary dysplasia in preterm infants. Pediatrics 2003;112:570–577. [DOI] [PubMed] [Google Scholar]
- 23.Danan C, Jarreau PH, Franco ML, Dassieu G, Grillon C, Abd Alsamad I, Lafuma C, Harf A, Delacourt C. Gelatinase activities in the airways of premature infants and development of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2002;283:L1086–L1093. [DOI] [PubMed] [Google Scholar]
- 24.Ekekezie II, Thibeault DW, Simon SD, Norberg M, Merrill JD, Ballard RA, Ballard PL, Truog WE. Low levels of tissue inhibitors of metalloproteinases with a high matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio are present in tracheal aspirate fluids of infants who develop chronic lung disease. Pediatrics 2004;113:1709–1714. [DOI] [PubMed] [Google Scholar]
- 25.Wolters PJ, Chapman HA. Importance of lysosomal cysteine proteases in lung disease. Respir Res 2000;1:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buhling F, Rocken C, Brasch F, Hartig R, Yasuda Y, Saftig P, Bromme D, Welte T. Pivotal role of cathepsin K in lung fibrosis. Am J Pathol 2004;164:2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michallet MC, Saltel F, Flacher M, Revillard JP, Genestier L. Cathepsin-dependent apoptosis triggered by supraoptimal activation of T lymphocytes: a possible mechanism of high dose tolerance. J Immunol 2004;172:5405–5414. [DOI] [PubMed] [Google Scholar]
- 28.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000;106:1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med 2000;192:1587–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med 1999;160:1333–1346. [DOI] [PubMed] [Google Scholar]
- 31.Altiok O, Yasumatsu R, Riese R, Stahlman M, Weber E, Dwyer W, Cataltepe S. Cysteine proteases are up-regulated in a baboon model of bronchopulmonary dysplasia [abstract]. Proc Am Thorac Soc 2005;2:A24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cataltepe S, Schick C, Luke CJ, Pak SC, Goldfarb D, Chen P, Tanasiyevic MJ, Posner MR, Silverman GA. Development of specific monoclonal antibodies and a sensitive discriminatory immunoassay for the circulating tumor markers SCCA1 and SCCA2. Clin Chim Acta 2000;295:107–127. [DOI] [PubMed] [Google Scholar]
- 33.Storm van's Gravesande K, Layne MD, Ye Q, Le L, Baron RM, Perrella MA, Santambrogio L, Silverman ES, Riese RJ. IFN regulatory factor-1 regulates IFN-gamma-dependent cathepsin S expression. J Immunol 2002;168:4488–4494. [DOI] [PubMed] [Google Scholar]
- 34.Cataltepe S, Gornstein ER, Schick C, Kamachi Y, Chatson K, Fries J, Silverman GA, Upton MP. Co-expression of the squamous cell carcinoma antigens 1 and 2 in normal adult human tissues and squamous cell carcinomas. J Histochem Cytochem 2000;48:113–122. [DOI] [PubMed] [Google Scholar]
- 35.Mariani TJ, Arikan MC, Pierce RA. Fibroblast tropoelastin and alpha-smooth-muscle actin expression are repressed by particulate-activated macrophage-derived tumor necrosis factor-alpha in experimental silicosis. Am J Respir Cell Mol Biol 1999;21:185–192. [DOI] [PubMed] [Google Scholar]
- 36.Buhling F, Reisenauer A, Gerber A, Kruger S, Weber E, Bromme D, Roessner A, Ansorge S, Welte T, Rocken C. Cathepsin K: a marker of macrophage differentiation? J Pathol 2001;195:375–382. [DOI] [PubMed] [Google Scholar]
- 37.Lyle RE, Tryka AF, Griffin WS, Taylor BJ. Tryptase immunoreactive mast cell hyperplasia in bronchopulmonary dysplasia. Pediatr Pulmonol 1995;19:336–343. [DOI] [PubMed] [Google Scholar]
- 38.Subramaniam M, Sugiyama K, Coy DH, Kong Y, Miller YE, Weller PF, Wada K, Wada E, Sunday ME. Bombesin-like peptides and mast cell responses: relevance to bronchopulmonary dysplasia? Am J Respir Crit Care Med 2003;168:601–611. [DOI] [PubMed] [Google Scholar]
- 39.Buhling F, Gerber A, Hackel C, Kruger S, Kohnlein T, Bromme D, Reinhold D, Ansorge S, Welte T. Expression of cathepsin K in lung epithelial cells. Am J Respir Cell Mol Biol 1999;20:612–619. [DOI] [PubMed] [Google Scholar]
- 40.Lecaille F, Kaleta J, Bromme D. Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002;102:4459–4488. [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa T, Roth W, Wong P, Nelson A, Farr A, Deussing J, Villadangos JA, Ploegh H, Peters C, Rudensky AY. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 1998;280:450–453. [DOI] [PubMed] [Google Scholar]
- 42.Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, De Sanctis GT, Ploegh HL, Chapman HA. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest 1998;101:2351–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci USA 1998;95:13453–13458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou WS, Bromme D, Zhao Y, Mehler E, Dushey C, Weinstein H, Miranda CS, Fraga C, Greig F, Carey J, et al. Characterization of novel cathepsin K mutations in the pro and mature polypeptide regions causing pycnodysostosis. J Clin Invest 1999;103:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guttentag S, Robinson L, Zhang P, Brasch F, Buhling F, Beers M. Cysteine protease activity is required for surfactant protein B processing and lamellar body genesis. Am J Respir Cell Mol Biol 2003;28:69–79. [DOI] [PubMed] [Google Scholar]
- 46.Brasch F, Ten Brinke A, Johnen G, Ochs M, Kapp N, Muller KM, Beers MF, Fehrenbach H, Richter J, Batenburg JJ, et al. Involvement of cathepsin H in the processing of the hydrophobic surfactant-associated protein C in type II pneumocytes. Am J Respir Cell Mol Biol 2002;26:659–670. [DOI] [PubMed] [Google Scholar]
- 47.Johnson DA, Barrett AJ, Mason RW. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J Biol Chem 1986;261:14748–14751. [PubMed] [Google Scholar]
- 48.Taggart CC, Lowe GJ, Greene CM, Mulgrew AT, O'Neill SJ, Levine RL, McElvaney NG. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem 2001;276:33345–33352. [DOI] [PubMed] [Google Scholar]
- 49.Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB Jr, O'Neill S, McElvaney NG. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol 2003;171:931–937. [DOI] [PubMed] [Google Scholar]
- 50.Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome C. J Clin Invest 2000;106:1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications. Am J Pathol 2001;159:2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman HJ, Munger JS, Shi GP. The role of thiol proteases in tissue injury and remodeling. Am J Respir Crit Care Med 1994;150:S155–S159. [DOI] [PubMed] [Google Scholar]
- 53.Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc Natl Acad Sci USA 1995;92:3849–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Punturieri A, Filippov S, Allen E, Caras I, Murray R, Reddy V, Weiss SJ. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med 2000;192:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi GP, Sukhova GK, Grubb A, Ducharme A, Rhode LH, Lee RT, Ridker PM, Libby P, Chapman HA. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest 1999;104:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koblinski JE, Dosescu J, Sameni M, Moin K, Clark K, Sloane BF. Interaction of human breast fibroblasts with collagen I increases secretion of procathepsin B. J Biol Chem 2002;277:32220–32227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.