

Abstract
Dipeptide cyclo[(S)-His-(S)-Phe] 1, first applied by Inoue et al. in 1981, catalyzes the hydrocyanation of aromatic aldehydes very efficiently. Enantioselective autoinduction has also been reported for the process. We have employed QM (Density Functional Theory and MP2), Molecular Mechanics (MM) and Molecular Dynamics (MD) methods to (i) derive a mechanistic picture for catalysis and (ii) reveal the origin of stereochemistry and autoinduction. A dimer is proposed to be the catalytic species, in which one imidazole group is essential for the delivery of the nucleophile and the second imidazole group acts as an acid, accompanied with π-interaction for most favorable substrate binding. H-bonding via hydroxy groups is crucial for catalysis also. MD studies indicate stability of the dimer only in non-polar media, which is consistent with the need of the experimental (heterogeneous) reaction conditions to achieve high enantioselectivities. DFT and MP2 results suggest the incorporation of the product cyanohydrin via extended edge-to-face π-interaction over three aromatic units. Transition states derived from this model are in good agreement with experimental findings and enantioselectivities.
1. Background
In 1981, Inoue and co-workers1 reported the enantioselective hydrocyanation2 of aldehydes employing the cyclic dipeptide catalyst cyclo[(R)-His-(R)-Phe] or cyclo[(S)-His-(S)-Phe] 1, illustrated in Scheme 1. This catalyst is a small molecule alternative to the enzyme hydroxynitrile lyase (oxynitrilase),3 a rare example of an industrially important enzyme. Aromatic cyanohydrins are important building blocks for the synthesis of alpha-hydroxy acids and amino alcohols.4
Scheme 1.

Several conditions were found to be crucial for high enantioselectivities: the reaction is most effective under heterogeneous conditions, using toluene5 as solvent in which catalyst 1 is insoluble. This heterogeneous state is thixotropic: greater ee’s were observed with higher stirring rate.6 Some authors state that the catalyst was precipitated or recrystallized from a protic solvent prior to usage,1,7 and that the presence of water or methanol within the solid state of the catalyst is essential for high ee’s.10 Others mention thorough drying of the catalyst.9,7 Enantioselective autoinduction was reported by Danda et al.7 As the reaction with catalyst 1 proceeded, the enantioselectivity rose gradually throughout the course of reaction as the percentage of product increased. They proposed that the product complexes with the chiral catalyst, and that the resulting species is more efficient in catalysis. When a small amount of the (R)-product was added to the (S,S)-catalyst, a constant high ee of 98% was observed throughout the course of reaction; in the absence of the (R)-product or in the presence of the (S)-product, the ee gradually increased again as the reaction proceeded.7,8 Studies by Danda et al.7 to examine the composition of the ‘autoinduction catalyst’ revealed a 1:0.8 composition of catalyst and cyanohydrin product. More recent mechanistic studies by Lipton and co-workers9 revealed that the autoinduction is general and not limited to specific aldeyde substrates. It was further concluded that the most likely active catalyst would be a dimer to which the product complexes in an unknown fashion.
Three mechanistic proposals appeared in the literature (A10, B11 and C, see Scheme 2). Shvo et al.8 proposed mechanism C based on their kinetic analyses in which a second order rate dependence on the catalyst concentration was determined.12 Structural modifications of ‘Ph’ and ‘His’-moieties of the catalyst were carried out, and further transition state proposals appeared.10,13,2a When ‘CH2-Ph’ was replaced by isopropyl [cyclo-(S)-leucyl-(S)-histidyl], introducing a sterically demanding group, catalysis was still found with high ee’s. However, the opposite stereochemistry resulted.14 NMR-studies of 1 in DMSO15 (in which 1 is fully soluble) and in the solid state16 as well as preliminary computational analyses17,8 have been performed. A clear elucidation of the mechanism is lacking, and the origin of the observed stereoselectivity is unknown. Nevertheless, 1 has opened an entire field of peptide catalysis.18 We have undertaken computational studies of hydrocyanations catalyzed by cyclic dipeptide 1, and have developed a model that explains the catalytic mechanism and stereoselectivity.
Scheme 2.

2. Computational details
All gas phase [RB3LYP/6-31G(d) and RB3LYP/6-31+G(d)] optimized stationary points were verified as minima or first-order saddle points by calculation of the full Hessian in Gaussian03.19 MP2 and SCS-MP2 treatment were performed via single-point energy calculation on the 6-31G* geometry, using thermal free energy corrections of the DFT 6-31G* optimizations. CPCM single point solvent calculations (implicit toluene, radii=UAO) were applied also. Molecular Dynamic studies (MD) [parameters: 1.0 ps (equilibrium time), 10 ps (simulation time), 10–1000 fs (timestep)] and conformational searches (MM) were performed using OPLS-AA force field20 as implemented in MacroModel 5.1.016.21
3. Results/ Discussion
(a) Isomerization of HCN
Recent theoretical studies of hydrocyanations have appeared. The bicyclic guanidine-catalyzed Strecker reaction, summarized in Figure 1, was investigated by Li et al.22 Two mechanisms were proposed. In the first (A), isohydrocyanic acid, HNC, adds to the imine; protonation of the imine and deprotonation of HNC are proposed to take place concertedly. In (B) the addition of HCN via nitrogen is followed by isomerization to the corresponding C-C bonded nitrile intermediate. Both mechanisms were suggested to be plausible, but the barrier of isomerization from HCN to HNC was not provided.
Figure 1.

Proposed reaction paths (A and B) for the bicyclic guanidine-catalyzed Strecker reaction.
Prior to Li et al.’s study, Barone and co-workers23 studied the hydocyanation of imines in the presence and absence of two explicit water molecules. In the gas phase it was found that HCN H-bonds to the imine nitrogen, isomerizes to HNC with a barrier of 27 kcal/mol, and subsequently attacks the imine carbon. In the presence of two water molecules, the picture changed completely. Several intermediates are formed, and the water served as an H-bond donor to direct the nucleophile to the imine carbon.
We have investigated the isomerization of HCN to CNH catalyzed by imidazole. Figure 2 gives the results. The B3LYP/6-31G* barrier of 36.9 kcal/mol was found to decrease to 30.3 kcal/mol in the gas phase with the inclusion of diffuse functions, solvation lowered the barrier by 11 kcal/mol. The uncatalyzed barrier was calculated to be 50.5 kcal/mol with HF 6-31G(d,p).24 Calculation of the isomerization in the presence of two methanol molecules led to essentially no barrier for the isomerization with less than 1 kcal/mol activation energy.
Figure 2.

Free energy of isomerization of HCN to HNC catalyzed by imidazole [to mimic dipeptide 1].
Kinetic analyses by Lipton et al.9 conclude that the reaction rate in the presence of cyanohydrin product does not change. In the presence of the new complexed catalytic species the rate of transformation is not increased; the reaction simply gets more selective. This suggests that no additional H-bond is formed in the catalysis involving autoinduction compared to the mechanism without autoinduction.25 Methanol (from purification/ activation of the catalyst) or water might therefore be involved in the mechanism prior to cyanohydrin incorporation. Due to the nearly 1:1 product-to-catalyst ratio in the autoinduction catalyst complex, two hydroxyl hydrogen bonds [from the cyanohydrin product] can be assumed in the course of reaction, which have been modeled with MeOH in Figure 2.
(b) Study of the potential roles of the functional groups in the catalyst
We have explored whether the imidazole or the dipeptide moiety of the catalyst might be a more effective acid catalyst. The model system for cyanide attack is shown in Figure 3. We calculated the proton affinities of both groups and found these to be nearly equal (Figure 3a). The gas phase proton affinities, are nearly identical for the imidazole-HCN and the peptidic-HCN complexes. For the transition states, imidazole-HCN protonation and cyanide attack (Figure 3b) is favored. These results are consistent with mechanism C (compare Scheme 2) that was proposed by Shvo et al.8
Figure 3.

(a): gas-phase proton affinities (at B3LYP/6-31G* and B3LYP/6-31+G* in bold). (b): relative stabilities of TS (free energy in kcal/mol, computed at B3LYP/6-31G*, gas-phase at r.t.).
A transition state for the concerted mechanism of the hydrocyanation of benzaldehyde, involving two HCN and two imidazole molecules was located (Figure 4). One imidazole group delivers the nucleophile, after isomerization of HCN, while the other imidazole-HCN complex activates benzaldehyde and functions as an acid. An activation free energy barrier of 20.8 kcal/mol in the gas phase was obtained.
Figure 4.

Reaction path involving activation and delivery of nucleophile, B3LYP/6-31G* free energies are from gas-phase calculations (kcal/mol).
(c) Effect of HCN or hydroxy groups on catalysis
No information about the kinetic order of hydrogen cyanide has been obtained experimentally. We systematically explored the role of hydroxyl groups (modeled with MeOH) on catalysis, since the observed autoinduction (as discussed above) suggests the presence of at least two hydroxyl groups in the active catalyst complex. Several combinations were explored. The corresponding activation (ΔG‡) and reaction free energies (ΔGrxn) are presented in Figure 5 with the transition state of each path [see supporting information for reactant and product complexes]. In the absence of HCN or hydroxyl, the activation barrier is relatively high (TS A).26 However, it is found to be lower with two MeOH molecules (F, G) in place of hydrogen cyanide. The reaction is now slightly exergonic and the reverse reaction less favorable compared to the path shown in Figure 4.
Figure 5.

Transition states representing the various possible reaction paths of which reaction free energies and activation energies are illustrated (free energies in kcal/mol; B3LYP/6-31G* gas-phase).
The presence of a third methanol molecule (TS H), led to even further decrease of the activation barrier. However, since the number of hydroxyl groups in the transition state is unknown, only two methanol molecules were considered further, i.e. TS F (Figure 5).
The isomerization of HCN to HNC prior to attack onto the carbonyl functionality in the pre-organized reactant complex of TS F would require an activation energy of 13.1 kcal/mol (Figure 6), essentially the same magnitude as the addition step (TS F in Figure 5). However, as illustrated in Figure 2, interaction with two hydroxyls directly, leads to essentially no barrier to isomerization.
Figure 6.
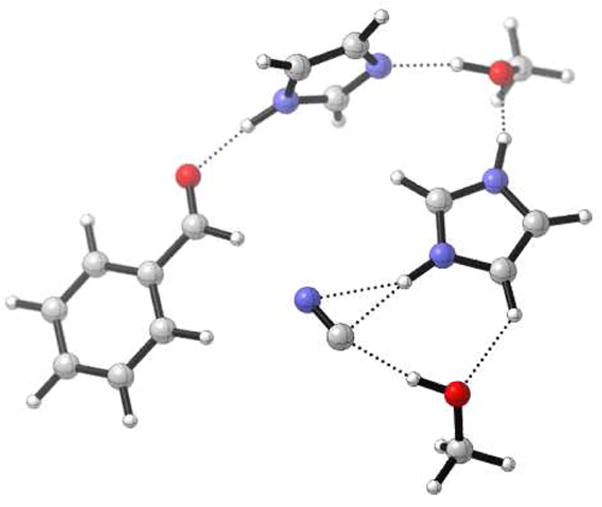
Transition state of HCN-HNC isomerization in the complex prior to HCN addition, calculated at B3LYP/6-31G* in the gas-phase.
(d) Origin of stereoselectivity and autoinduction
Kinetic data by Lipton et al.9 suggests dimer catalysis. This agrees with our proposal that two imidazole moieties participate in the step in which stereochemistry is established [TS F in Figure 5]. To explore the origin of (R)-stereochemistry in the product that is observed upon catalysis with cyclo[(S)-His-(S)-Phe] 1, the structure of a dimer was investigated. Three possible dimers exist, a head-to-head (D-1) and two head-to-tail (D-2, D-3) alignments that have the possibility of forming H-bonds between the imidazole-N-H and the C=O groups. Preliminary calculations of these dimers were performed by Shvo and co-workers8, but no information of the active catalytic dimer nor a transition state model could be obtained. With a molecular mechanics approach, using the force-field OPLS-AA27 as supported in MacroModel, conformational searches of the three hydrogen-bonded dimers (D-1, D-2, D-3) were performed. Conformers within 3 kcal/mol of the global minimum were extracted in each run and subsequently optimized with DFT B3LYP/6-31G*.
For all three initial dimer arrangements, (D-1, D-2, D-3) the identical global minimum, i.e. D-4 shown in Figure 8 was found. It consists of an extended linear H-bond from the carbonyl group via imidazole-imidazole H-bond. A high degree of symmetry is found: front and backside are identical (with the exception of the H-atom missing on imidazole). The next higher minimum, D-5 [+1.7 kcal/mol (free energy) at r.t. in the gas phase] consists of a very similar geometry. However, the central imidazole-imidazole H-bond is not linear but distorted in that case, accounting for the higher energy.
Figure 8.

Global minimum D-4 (front-view: top left; back-view: top right) and next higher conformer D-5 (bottom).
Assuming that such a dimer might constitute the active catalyst in the heterogeneous phase of the reaction mixture, molecular dynamic (MD) studies were performed to assess its flexibility. Several MD runs were carried out. Simulation in the gas phase at 5°C showed that the aromatic groups are quite flexible, and rotate freely. The central peptidic H-bond (N-H····O=C) remained stable. However, the central imidazole H-bond (N···H-N) cleaved and both imidazole groups separated from each other during the dynamic simulation.
Repeating the same run at −20°C (at which temperature greater ee’s were observed experimentally) showed a similar behavior of the dimer in terms of rotation of Ph-groups and overall stability of the dimer. However, the central imidazoles now remained in proximity and an equilibrium between (N-H····N)-hydrogen-bond formation and separation beyond H-bonding distance was observed. Performing dynamic studies in implicit chloroform (to mimic an organic, weakly H-bonding solvent) at 5°C and −20°C gave essentially the identical dynamic simulation as observed in the gas phase at the specific temperatures. In contrast, when running dynamics in water (implicit) at 5°C as well as −20°C, the dimer dissociated, and the two monomer fragments moved apart. Thus, if a dimer were indeed responsible for the observed stereochemistry in the hydrocyanation, this dynamic behavior would be consistent with the experimental observations, i.e. the loss of stereochemistry when the reaction was run in polar solvents, such as methanol.10
Based on these analyses, we have derived a model to rationalize the stereochemistry. As established with TS F in Figure 5, one monomeric unit of the dimer activates the aldehyde species via H-bonding to the imidazole moiety. The second imidazole group delivers the nucleophile, isocyanide (after the isomerization of HCN to HNC has taken place) with additional H-bonding by hydroxy groups. To find the transition states, TS F from Figure 5 was overlapped with the dimer D-4 from Figure 8. The imidazole groups from D-4 were subsequently deleted, and the imidazole groups from TS F were connected with the dimer scaffold. The monomeric part of the dimer that is presumed to deliver the nucleophile was subsequently omitted with the exception of the crucial imidazole ring (to allow calculations of transition states). The resulting structure was optimized to a minimum by constraining the critical catalytic distances,28 followed by transition state optimization of the completely relaxed structure in the absence of any constraints. The transition states TS-1, TS-2 and TS-2b (shown in Figure 9) were calculated with this methodology using the B3LYP/6-31G* and PBEPBE/6-31G* levels in the gas phase. Thermal corrections were applied for −20°C (at which temperature best ee’s are found experimentally).
Figure 9.

Favored TS-1 (top left) and disfavored TS-2 (top right) and parallel displaced TS-2b (bottom left); see Table 1 for energies.
B3LYP as well as PBEPBE give a slight free-energy preference of approximately 0.5 kcal/mol for TS-1. TS-1 has edge-to-face π-interaction and is the TS leading to the experimentally observed (R)-enantiomer (see Table 1). The parallel displaced TS-2b structure was found only at PBEPBE level and is less favorable with respect to the edge-to-face TS-1 and also TS-2. However, that edge-to-face interaction is favored at the levels employed over π-p stacking (parallel or parallel-displaced) has been discussed in the literature.29 The PBEPBE level was chosen as it has been shown to give attractive aromatic interactions, even though it is known to underestimate these.29,30 B3LYP is reported to be repulsive with respect to π–interactions in some cases, possibly explaining why TS-2b could not be located at that level. Post MP2 and SCS-MP2 treatments on the B3LYP/6-31G* geometry, diminish this 0.5 kcal/mol preference to only 0.2 and 0.0, and thus give essentially no preference of TS-1 over TS-2. We identified two possible reasons for this: (i) a close look at the transition states reveals that an additional hydrogen bond between the aromatic C-H and the carbonyl group of benzaldehyde in TS-2 exists in comparison to TS-1. This C-H···O=C interaction seems to just compensate the C-H···π interaction present in TS-1. On the other hand, (ii) DFT methods represent only the lower limit of π-interaction due to their underestimation (PBEPBE) or even possible repulsive features (B3LYP). This will be reflected in the geometries and thus also affect follow-up MP2 and SCS-MP2 energies. However, bearing in mind the possibility of enantioselective autoinduction discussed above, one would not expect a large energy preference for one stereoisomer over the other, as high ee’s were seen only if the product was present and presumably incorporated into the active catalyst.
Table 1.
Relative free energy preference of TS-1 over TS-2 in kcal/mol at various levels (at −20°C).
| Method | Relative activation free energy (kcal/mol) | ||
|---|---|---|---|
| TS-1 | TS-2 | TS-2b | |
| B3LYP/ 6-31G* | 0.0 | 0.5 | — |
| PBEPBE/ 6-31G* | 0.0 | 0.5 | 1.3 |
| MP2// B3LYP/6-31G* | 0.0 | 0.2 | — |
| SCS-MP2// B3LYP/6-31G* | 0.0 | 0.0 | — |
That π-interaction might indeed be the crucial factor that determines stereoselectivity would be consistent with experimental findings by Inoue et al. Upon replacement of the Ph-group in catalyst 1 by isopropyl, the opposite stereoselectivity of the cyanohydrin was observed.14 Considering the enantioselective autoinduction, favorable incorporation of the product cyanohydrin would be via π-interaction of the phenyl groups of the catalyst dimer with the aromatic ring of the cyanohydrin. There are two phenyl groups in the dimer and hence two product cyanohydrins could be incorporated symmetrically. This would be consistent with the almost 1:1 catalyst to product composition that was found in studies of autoinduction [see discussion above]. Accordingly, such complexes were explored by adding the appropriate groups (i.e. CN and Ph) on the methanol in TS-1 and TS-2 from Figure 9 (only one product molecule was considered for simplification in terms of computational cost), followed by geometry optimization of the structures to minima with constrained catalytic distances.28 The geometries were then relaxed (removal of constraints), and the transition states were calculated with B3LYP/6-31G* followed by MP2 and SCS-MP2 single point treatments. The results are shown in Figure 10.
Figure 10.

Model for enantioselective autoinduction: product molecule-catalyst complex - favored TS-3 (left), disfavored TS-4 (right).
Transition state TS-3, that leads to the experimentally observed enantiomer, is now favored by 1.8 kcal/mol over TS-4. This drastic improvement is based on the triple edge-to-face π-interaction, as can be seen in Figure 10 and Table 2. The C-H-π distances are ca. 3.2 Å in the stacking arrangement. At the same time, the aromatic C-H···O=C interaction that was encountered in TS-2 (see Figure 9) is now absent in TS-4, showing that aromatic C-H···π interaction is favored over C-H···O=C. According to the experimental results (98% ee), there should be a free energy difference of 2.3 kcal/mol between TS-3 and TS-4. B3LYP shows a rather good performance. MP2 overestimated the binding slightly. Using SCS-MP2 we obtained an energy difference of 2.5 kcal/mol which is very close to the expected one (2.3 kcal/mol). However, under experimental conditions, additional intermolecular interactions, whether repulsive or attractive, can be expected in the reaction mixture; these are likely to influence the TS preferences also.
Table 2.
Relative free energy preference of TS-3 over TS-4 in kcal/mol at various levels (at −20°C).
| Method | Relative activation free energy difference (kcal/mol) | |
|---|---|---|
| TS-3 | TS-4 | |
| B3LYP/ 6-31G* | 0.0 | 1.8 |
| MP2// B3LYP/6-31G* | 0.0 | 2.9 |
| SCS-MP2// B3LYP/6-31G* | 0.0 | 2.5 |
It remains to be explained why constant high ee throughout the reaction is seen only if the (R)-enantiomer is added to the (S,S)-catalyst. If the opposite product enantiomer (S) is added, the ee incresed gradually throughout the course of the reaction.7 It could be that the (S)-enantiomer does not incorporate into the (S,S)-catalyst complex or, if it does, that the resulting complex is not as efficient in catalysis as the one resulting from the (R)-enantiomer. We calculated the transition states using the (S)-enantiomer of the cyanohydrin with the methodology described above (i.e. adding CN and Ph to TS-1 and TS-2, then applying constrained optimization, followed by relaxed transition state optimization). This resulted in TS-5 and TS-6, shown in Figure 11.
Figure 11.

Model for enantioselective autoinduction: product molecule-catalyst complex with the opposite product enantiomer (S) being incorporated: favored TS-5 (left), disfavored TS-6 (right).
With the (S)-product cyanohydrin incorporated, the preference of TS-5 [that leads to the observed (R)-stereoisomer] is now smaller than in the case of the (R)-product being incorporated. The energies are shown in Table 3. In particular SCS-MP2 treatment on the B3LYP geometry predicts low enantioselectivity. This can be rationalized when comparing TS-6 with TS-4. The incorporated (S)-product now forces the catalyst phenyl ring in a different orientation than the (R)-enantiomer does. Thus, TS-6 now benefits from C-H···O=C interaction, decreasing therefore the stereoselectivity in the case of the catalyst complex that contains the (S)-product.
Table 3.
Relative free energy preference of TS-5 over TS-6 in kcal/mol at various levels (at −20°C).
| Method | Relative activation free energy difference (kcal/mol) | |
|---|---|---|
| TS-5 | TS-6 | |
| B3LYP/ 6-31G* | 0.0 | 1.1 |
| MP2// B3LYP/6-31G* | 0.0 | 0.4 |
| SCS-MP2// B3LYP/6-31G* | 0.0 | 0.2 |
Furthermore, TS-5 is overall 1 kcal/mol higher in energy (at SCS-MP2) than TS-3 and thus, the (R)-product incorporated catalyst complex will, once formed (slowly over the course of reaction), outcompete the (S)-enantiomer complex. This explains why in the presence of added (S)-product the same reaction behavior is seen as in the absence of any added product, i.e. enantioselective autoinduction.
That TS-5 is slightly higher in energy than TS-3 possibly originates from the slightly different aromatic C-H···π interaction of the two complexes. Figure 11 illustrates the electrostatic potential surfaces of both transition states. In TS-3, in which the (R)-product is complexed, the aromatic C-H points into the region of greater electron density, resulting therefore possibly in slightly greater interaction.
(e) Addressing the reason for lower enantioselectivites when electron-poor and aliphatic aldehydes are employed
Having derived a model for stereochemistry and autoinduction, we addressed the scope of reaction. It is reported that electron-rich aldehydes are converted efficiently by dipeptide catalyst 1, where electron-poor or aliphatic analogues are converted with low ee values (see Figure 12).
Figure 12.

Scope of reaction with catalyst 1 and potential deprotonation.
One explanation for this could be that enantioselectivity throughout the catalysis is high; however, the product might racemize, if it is exposed to basic residues. This possibility was discussed previously by Inoue et al.31 To gain information about the C-H and O-H acidities in the products, we calculated the gas phase proton affinities at r.t. ( H) as well as reaction free energies for the protonation of the anions ( G at −20°C in toluene) using different levels of theory (see Table 4).
Table 4.
Proton affinities and relative free energies for transformations A and B, using
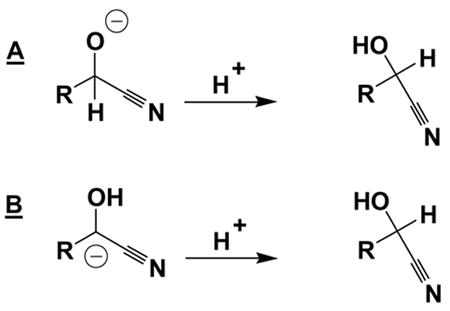
|
R | ΔH(1) | ΔH(2) | ΔG(3) | |
| (in kcal/mol) | |||||
| A1) | Ph | 353.9 | 341.2 | 327.7 | |
| A2) | 4-CN-Ph | 342.6 | 330.4 | 320.8 | |
| A3) | C6H13 | 358.8 | 345.3 | 331.7 | |
| B1) | Ph | 354.5 | 344.0 | 330.0 | |
| B2) | 4-CN-Ph | 334.9 | 325.4 | 314.7 | |
| B3) | C6H13 | 381.4 | 367.3 | 352.0 | |
: B3LYP/6-31G*, gas-phase, r.t.;
: B3LYP/6-31+G*, gas-phase, r.t.;
: B3LYP/6-31G*, CPCM toluene, −20°C.
The product derived from the electron-poor aromatic aldehyde (R=CN) shows indeed a slightly greater acidity of the C-H (entry B2) over the O-H (entry A2) position, which would agree with the hypothesis of racemization of the product after stereoselective generation. In addition, the edge-to-face π-interaction that was found to be crucial for stereoselectivity changes with different electron density of the aromatic ring; this could be the reason for lower stereoselectivity. The aliphatic cyanohydrin that also shows poor enantioselectivity (entry 8, Figure 12) is most acidic in the O-H rather than C-H (B3) position. Racemization would not be consistent with these findings.
To gain further insight, we also explored the reaction of propanal (see supporting information). Propanal shows a barrier of ΔG‡= 7.7 kcal/mol for the reaction with HCN, which is considerably lower than the barrier with benzaldehyde. The non-catalyzed (racemic) reaction might therefore be in competition with the dipeptide-catalyzed reaction, possibly accounting for the loss of stereoselectivity. Similar arguments might also be applicable to the electron-poor (and more electrophilic) aromatic aldehydes. Additionally, the lack of π-interaction could account for the low enantioselectivity in reactions with aliphatic aldehydes.
In conclusion, a mechanistic picture was derived, consistent with a dimer as the catalytic species. One imidazole group is essential for delivery of the nucleophile and the second imidazole group acts as an acid, accompanied with π-interaction for most favorable substrate binding and hydrogen bonding from hydroxy groups. The heterogeneous conditions are crucial for the dimer stability (intermolecular H-bonding between the monomers) and stereoselectivity. To be able to carry out enantioselective hydrocyanations under homogeneous conditions, favorable intramolecular interactions within the catalyst molecule must be present to favor a specific conformation. This has been accomplished in peptide catalysis of similarly or more complex structures, such as Miller’s peptide32,15a or thiourea catalysis33,15b in hydrocyanations and related nucleophile-electrophile catalyses.
Supplementary Material
The full reference 15, absolute energies, thermochemistry and the Cartesian coordinates of all stationary points are provided in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 7.

Possible hydrogen-bonded dimer arrangements: head-to-head D-1, head-to-tail alignments D-2 and D-3 prior to conformational search.
Figure 12.

MP2 electrostatic potential mapped on the electron-density surface [Iso-value: 0.004] of the transition states TS-3 (R-cyanohydrin/S,S-catalyst) and TS-5 (S-cyanohydrin/S,S-catalyst), Color code: [−0.0275 au; +0.0410 au] with red corresponding to electron-rich and blue to electron-poor region.
Acknowledgments
We thank the Alexander von Humboldt foundation for a Feodor Lynen fellowship to F.S. and the National Institute of General Medical Sciences, National Institutes of Health for financial support of research. We are grateful to Guillaume Bollot, Saron Catak and Paul Cheong for technical information. Computations were performed on the National Science Foundation Terascale Computing System at the National Center for Super computing Applications (NCSA), on the California NanoSystems Institute clusters and UCLA Hoffman2 cluster.
References
- 1.Inoue S, Oku JI. J Chem Soc, Chem Commun. 1981:229. [Google Scholar]
- 2.(a) See also: North M. Tetrahedron: Asymmetry. 2003;14:147–176.(b) for an early example of hydrocyanation, see: Lapworth A. J Chem Soc. 1903;83:995–1005.
- 3.Becker W, Pfeil E. J Am Chem Soc. 1966;88:4299–4300. [Google Scholar]
- 4.Effenberger F, Stelzer U. Angew Chem Int Ed. 1991;30:873. and references therein. [Google Scholar]
- 5.(a) Tanaka K, Mori A, Inoue S. J Org Chem. 1990;55:181. [Google Scholar]; (b) Matthews BR, Jackson WR, Jayatilake GS, Wilshire C, Jacobs H. Aust J Chem. 1988;41:1997. [Google Scholar]
- 6.Danda H. Synlett. 1991:263–264. [Google Scholar]
- 7.Danda H, Nishikawa H, Otaka K. J Org Chem. 1991;56:6740–6741. [Google Scholar]
- 8.Shvo Y, Gal M, Becker Y, Elgavi A. Tetrahedron: Asymmetry. 1996;7:911–924. [Google Scholar]
- 9.Kogut EF, Thoen JC, Lipton MA. J Org Chem. 1998;63:4604. [Google Scholar]
- 10.Jackson WR, Jayatilake GS, Matthews BR, Wilshire C. Aust J Chem. 1988;41:203–213. [Google Scholar]
- 11.Hogg DJP, North M, Stokoe RB. Tetrahedron: Asymmetry. 1996;7:911–924. [Google Scholar]
- 12.For further studies that are consistent with the proposed mechanism, see: Prelog V, Wilhelm M. Helvetica Chimica Acta. 1954;37:1634–60.
- 13.For a study of further derivatives and a transition state proposal, see: Xie L, Hua W, Chan ASC, Leung YC. Tetrahedron: Asymmetry. 1999;10:4715–4728.
- 14.Mori A, Ikeda Y, Kinoshita K, Inoue S. Chem Letters. 1989:2119–2122. [Google Scholar]
- 15.North M. Tetrahedron. 1992;48:5509–5522. [Google Scholar]
- 16.Apperley D, North M, Stokoe RB. Tetrahedron: Asymmetry. 1995;6:1869–1872. [Google Scholar]
- 17.Callant D, Coussens B, vdMaten T, de Vries NK. Tetrahedron: Asymmetry. 1992;2:401–414. [Google Scholar]
- 18.Recent reviews: Colby Davie EA, Mennen SM, Xu Y, Miller SJ. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w.Taylor MS, Jacobsen EN. Angew Chem Int Ed. 2006;45:1520 – 1543. doi: 10.1002/anie.200503132.
- 19.Gaussian03, Revision C.02, M. J.Frisch et al. [see supporting information for full reference].
- 20.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 21.© 2001 Schrödinger, Inc. All rights reserved. RV7200801 September 2001
- 22.Li J, Jiang WY, Han KL, He GZ, Li C. J Org Chem. 2003;68:8786–8789. doi: 10.1021/jo034891f. [DOI] [PubMed] [Google Scholar]
- 23.Arnaud R, Adamo C, Cossi M, Milet A, Vallée Y, Barone V. J Am Chem Soc. 2000;122:324–330. [Google Scholar]
- 24.Kumeda Y, Minami Y, Takano K, Taketsugu T, Hirano T. J Mol Structure (Theochem) 1999;458:285. [Google Scholar]
- 25.For the corresponding enzyme (oxynitrilase) mechanism, it was found that the addition step of the nucleophile on the aldehyde carbonyl is the rate determining step [see Ching WM, Kallen RG. J Am Chem Soc. 1978;100:6119–6124. ]. This seems to be the case in the catalysis by dipeptide 1 also, since electron-poor aldehydes are reported to react in shorter reaction times than their electron-rich counterparts (compare Figure 12).
- 26.The N···N-H distance of 2.99 Å between the imidazole groups was frozen.
- 27.(a) Kaminski G, Jorgensen WL. J Phys Chem. 1996;100:18010–18013. [Google Scholar]; (b) Price MLP, Ostrovsky D, Jorgensen WL. J Comp Chem. 2001;22:1340–1352. [Google Scholar]; (c) Kaminski G, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 28.With ‘catalytic distances’ we mean the distances highlighted with dotted lines in the transition states in Figure 9 and Figure 10 (excluding π-interaction).
- 29.Grimme S. J Comput Chem. 2004;25:1463–1473. doi: 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- 30.Grimme S. Chem Eur J. 2004;10:3423–3429. doi: 10.1002/chem.200400091. [DOI] [PubMed] [Google Scholar]
- 31.(a) Oku J, Ito N, Inoue S. Makromol Chem. 1982;183:579. [Google Scholar]; (b) Asada S, Kobayashi Y, Inoue S. Makromol Chem. 1985;186:1755. [Google Scholar]
- 32.For example: Sculimbrene BR, Miller SJ. J Am Chem Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+.Fierman MB, O’Leary DJ, Steinmetz WE, Miller SJ. J Am Chem Soc. 2004;126:6967–6971. doi: 10.1021/ja049661c.Guerin DJ, Miller SJ. J Am Chem Soc. 2002;124:2134–2136. doi: 10.1021/ja0177814.Vasbinder MM, Jarvo ER, Miller SJ. Angew Chem Int Ed. 2001;40:2824–2827. doi: 10.1002/1521-3773(20010803)40:15<2824::aid-anie2824>3.3.co;2-a.
- 33.For example: Zuend SJ, Jacobsen EN. J Am Chem Soc. 2007;129:15872–15883. doi: 10.1021/ja0735352.Huang H, Jacobsen EN. J Am Chem Soc. 2006;128:7170–7171. doi: 10.1021/ja0620890.Lalonde MP, Chen Y, Jacobsen EN. Angew Chem Int Ed. 2006;45:6366–6370. doi: 10.1002/anie.200602221.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The full reference 15, absolute energies, thermochemistry and the Cartesian coordinates of all stationary points are provided in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.