

Abstract
Familial pulmonary arterial hypertension (FPAH) is a progressive, fatal disease caused by mutations in the bone morphogenetic protein receptor type 2 gene (BMPR2). FPAH is inherited as an autosomal dominant trait and shows incomplete penetrance in that many with BMPR2 mutations do not develop FPAH suggesting a role for, as yet unidentified, modifier genes in disease penetrance. We hypothesized that variable level of expression of the wild type (WT) BMPR2 allele could act as a modifier and influence penetrance of FPAH. WT BMPR2 levels were determined by real-time PCR analysis in lymphoblastoid (LB) cell lines derived from normal controls and individuals with FPAH. The FPAH kindreds analyzed carried mutations that result in the activation of nonsense mediated decay (NMD) pathway, which leads to the degradation of the mutant RNA thus ensuring that only the WT BMPR2 transcripts will be detected in the real-time assay. Our data show that WT and mutant BMPR2 levels can be reproducibly measured in patient derived LB cell lines and that unaffected mutation carrier derived LB cell lines have higher levels of WT BMPR2 transcripts than FPAH patient derived LB cell lines (p≤0.005). Our findings suggest that the levels of expression of WT BMPR2 allele transcripts is important in the pathogenesis of FPAH caused by NMD+ mutations. Furthermore, our study illustrates a novel application of lymphoblastoid cell lines in the study of PAH, especially important because the affected site, i.e. lung is not available for unaffected mutation carriers.
Keywords: BMPR2, Penetrance, Pulmonary arterial hypertension, Modifier, FPAH, PPH, NMD
Introduction
Pulmonary arterial hypertension (PAH) is a progressive, fatal disease characterized by vascular remodeling of the small pulmonary arteries, which results in increased vascular resistance and subsequent right heart failure (Chan and Loscalzo, 2008; Pietra, et al., 2004). Familial PAH (FPAH) has an autosomal dominant mode of inheritance with reduced penetrance, variable age of onset and a 2.4:1 (female to male) sex ratio (Newman, et al., 2001). Mutations in the BMPR2 gene (MIM# 600799), a member of the transforming growth factor (TGF-β) super family, are found in the majority of cases of FPAH and constitute the largest known risk for the development of FPAH (Cogan, et al., 2006; Deng, et al., 2000; Lane, et al., 2000). While both haploinsufficient (HI) and dominant negative (DN) mechanisms of disease have been proposed (Machado, et al., 2001; Rudarakanchana, et al., 2002), reduced penetrance and variable age at diagnosis suggest there are additional modifiers of disease expression (Newman, et al., 2008).
RNA studies have shown that some BMPR2 mutations produce stable transcripts while others contain premature termination codons (PTC) and are rapidly degraded through the nonsense mediated decay (NMD) pathway (Cogan, et al., 2006). NMD is an mRNA surveillance system that degrades transcripts containing PTCs to prevent translation of unnecessary or harmful transcripts (Gonzalez, et al., 2001; Maquat, 2004). We have recently demonstrated that FPAH patients with BMPR2 mutations subject to NMD (NMD+) tend to have milder disease (later age of diagnosis) than individuals with mutations that produce stable transcripts (NMD−)(Austin, et al., 2008). Failure to eliminate PTC containing mRNAs can result in synthesis of abnormal proteins that can be toxic to cells through dominant negative (DN) or gain of function effects. Thus, individuals with PAH and NMD+ BMPR2 mutations have disease due to HI whereas patients whose mutations are NMD− may have disease due to a DN mechanism. Affected individuals with BMPR2 NMD+ mutations have a 12 year later age of onset than those with NMD− mutations, making BMPR2 NMD status an important modifier in the disease process (Austin, et al., 2008).
One of the major impediments to identify genes that cause or modify FPAH pathogenesis is the lack of easy availability of the tissue of interest (the smallest pulmonary arteries). It has been hypothesized that vascular smooth muscles of the small arteries are the disease initiating tissue in FPAH but uncertainty remains and It is now believed to be likely that multiple cell types in the pulmonary arterial wall and pulmonary arterial circulation contribute to the development of vessel remodeling (for a review see (Chan and Loscalzo, 2008; Pietra, et al., 2004). Also, the isolation and eventual study of the pulmonary vascular smooth muscle cells remains difficult if not essentially impossible due to the sporadic availability of tissue from affected patients at the time of lung transplant or post mortem and the poor condition secondary to disease related pathogenesis rendering them less than ideal for laboratory use. Furthermore the tissue of interest cannot be obtained from carriers and non-affected individuals secondary to the significant risks associated with lung biopsy.
In order to overcome these issues we have used patient-derived lymphoblastoid cell (LB) lines to study disease pathogenesis and gene expression. LB lines offer several advantages such as unlimited amount of tissue carrying the mutation, elimination of possible effects on gene expression form drugs and disease pathogenesis. While it is true that the culture of cells may introduce changes in cell physiology these alterations are uniform across the samples. Moreover LB cell lines have been successfully used in gene expression studies for a wide variety of disease processes including schizophrenia (Kakiuchi, et al., 2007), drug resistance (Huang, et al., 2007), autism (Baron, et al., 2006), and asthma (Dixon, et al., 2007). Lastly, we believe that comparing expression between affected and unaffected BMPR2 mutation carriers is the most effective way to identify and study genetic modifiers associated with FPAH and LB cell lines are the most practical way to perform these studies.
Based on our studies demonstrating that NMD+ BMPR2 mutations tend to be later onset and follow HI model of inheritance, we reasoned that the level of expression of the wild type (WT) BMPR2 allele could also influence disease development. To determine the possible role of the WT BMPR2 allele as a modifier of disease, we measured expression levels of WT BMPR2 in patient-derived LB lines from selected NMD+ FPAH kindreds, by real time RT-PCR, and compared the expression of BMPR2 transcripts from the normal alleles of mutation carriers with and without disease. Our study demonstrated the utility of LB cell lines in the study of PAH and suggested that higher expression levels of WT alleles correlate with non-penetrant carrier status while lower levels predicted penetrance.
MATERIALS and METHODS
FPAH Kindreds
Study subjects included individuals with FPAH and BMPR2 mutation carriers without disease from 4 different kindreds (Cogan, et al., 2006; Machado, et al., 2001). All subjects are heterozygous for BMPR2 mutations (Table 1) that resulted in frame shifts leading to PTCs that were shown to be subject to NMD as described previously (Cogan, et al., 2006; Cogan, et al., 2005). The BMPR2 mutation in family-20 had deletion of exons 4–5 (c.419-?_621+?del) causing an out of frame deletion beginning at codon 140 with a PTC at codon 151. Families 59 & 80 were heterozygous for an IVS8 C to G transversion at the –3 position of the acceptor splice site (c.1129-3C>G) which resulted in deletion of exon 9 with a frameshift at codon 377 and generation of a PTC at codon 424. Family 127 had an insertion of a single adenosine in exon 9 (c.1141_1142insA) altering the predicted amino acid sequence starting at codon 381 and causing a PCT at codon 398. Nucleotide numbering reflects cDNA numbering, where +1 corresponds to the A of the ATG translation initiation codon in the reference sequence (GenBank NM_001204.6), according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Table 1.
BMPR2 DNA Mutations Studied
| Family | Location | Nucleotide Change |
Amino Acid change |
|---|---|---|---|
| 20 | Exons 4–5 | c.419-?_621+?del | p.S140fsX12 |
| 59 & 80 | Intron 8 | c.1129-3C>G | p.V377fsX48 |
| 127 | Exon 9 | c.1141_1142insA | p.R381fsX18 |
Nucleotide numbering reflects cDNA numbering, where +1 corresponds to the A of the ATG translation initiation codon in the reference sequence (GenBank NM_001204.6), according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Establishment of LB cell lines
Lymphocytes were isolated from anticoagulated whole blood within 48 hrs of collection and exposed to Epstein-Barr Virus (EBV) to induce cell immortalization as previously described (Oh, et al., 2003) (Bird, et al., 1981).
Analysis of BMPR2 transcripts by real-time
cDNA was synthesized from total cellular RNA (1 µg) using (Superscript III cDNA Synthesis Kit (Invitrogen). Taqman real-time assay were designed for exon-9-deletion and wild type (WT) BMPR2 transcripts (NM_001204.6) using the Primer Express software package. Primer and probe sequences are as follows. WT-forward primer 5-TTAGTGACTTTGGACTGTCCATGAG-3′, WT-reverse primer 5′-TCTAGCACTTCTGGTGCCATATATCT-3′, WT-Taqman probe 5′-FAM-TAAGCGAGGTTGGCACT-3′, exon-9-deletion forward primer 5′-TTATTAGTGACTTTGGACTGTCCATGA-3′, exon-9-deletion reverse primer 5′-GAGAACCTGCATATCCTCAAAAGTC-3′, exon-9-deletion Taqman probe 5′-FAM-ATAAGCAAGGGGAATCC-3′. Assays were optimized using the standard curve method as previously described (Bustin, 2002; Livak and Schmittgen, 2001) and all had amplifications efficiencies of > than 99.5%. Real-time PCR analysis was carried out using Taqman Universal Master Mix and a 7500 Real-Time PCR system according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA). We used the TaqMan human endogenous control plate (catalog number 4309199) to analyze 13 genes as potential house keeping genes for data normalization as previously described (Hamid, et al., 2008). Amplification parameters consisted of initial denaturation at 95°C for 10 minutes followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing and extension at 60°C for 1 minute. Relative expression levels were calculated using the comparative Ct method (Bustin, 2002; Livak and Schmittgen, 2001).
Western analysis
Membranes were probed with primary antibody ab-10862 (Abcam, Cambridge, MA) for one hour and with secondary antibody 111-035-003 (Jackson ImmunoResearch, West Grove, PA) for an additional one hour. Detection was done using the Immobilon Chemiluminescent HRP substrate (Millipore, Billerica, Massachusetts). β-Actin was used as a loading control.
Cell culture
Control LB cell lines were obtained from the Coriell Cell Repositories (Camden, N.J). All LB cell lines were grown in 15% fetal bovine serum in RPMI 1640 with 2mM L-Glutamine.
Statistics
Data are expressed as means ± SD. Comparisons between groups were performed with one-way analysis of variance (ANOVA) as well as Mann-Whitney U (Wilcoxon rank-sum) test. All experiments were repeated 3 times and done in replicates of 3 or more. A P value less than 0.05 was considered significant. JMP software package version 6.0 (SAS Institute Inc. Cary NC) was used for all analysis.
RESULTS
BMPR2 transcripts can be detected in LB cell lines
To determine if LB cell lines express BMPR2 we used Taqman-based relative real-time PCR analysis on RNA isolated from a number of LB cell lines from normal individuals. Our results showed that BMPR2 transcripts can be detected in LB cell lines and that our real-time PCR assay detected relative differences in expression between samples (Fig. 1A). We then determined if similar differences in BMPR2 transcript expression can also be detected in PAH patient LB cell lines. Our data showed that BMPR2 RNA could be detected in LB cell lines derived from FPAH samples (Fig. 1B). Importantly, as was the case with the controls, we detected differences in total BMPR2 levels between different patients.
Fig. 1.
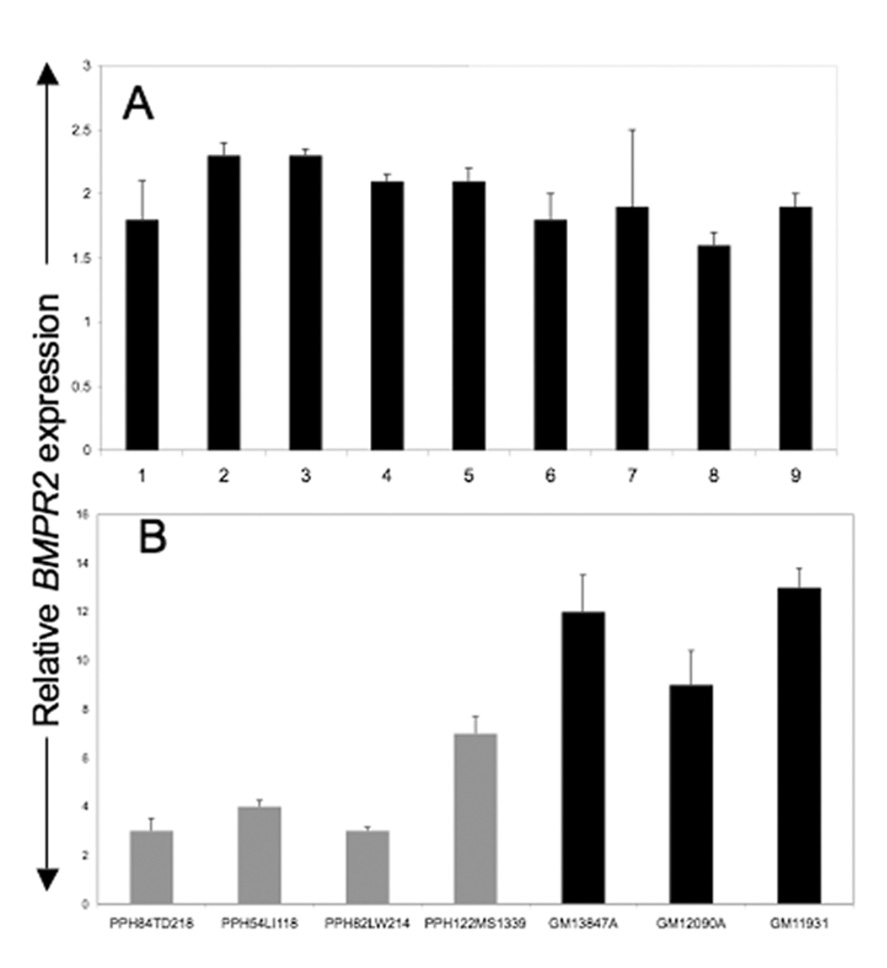
BMPR2 transcript levels, as measured by relative real-time PCR analysis, in CEPH control LB samples (A) and FPAH derived LB samples (B). Samples from control individuals (non-mutation carriers) are shown in black and from affected individuals in grey. Each experiment was done at least three times in replicates of 3. Error bars represent SD of the replicates.
Variation in BMPR2 expression is not secondary to LB immortalization
Our finding that FPAH patient derived LB cell lines had different levels of BMPR2 transcripts could either be secondary to the LB immortalization process or, more likely, represented true variation in BMPR2 transcripts expression across patient samples. To test the first we established multiple LB cell lines using lymphocytes isolated from a single patient but with different stocks of EBV viruses in three separate experiments. After immortalization total cellular RNA was isolated and used in a cDNA reaction and then subjected to real-time PCR analyses for total cellular BMPR2 transcript levels. The results showed that BMPR2 transcript expression did not vary between the three LB cell lines established from a single patient (Fig. 2). This suggests that the differences seen in BMPR2 transcript levels between patients with either SPAH or FPAH were not an artifact of LB immortalization process but likely represent true differences in transcript levels.
Fig. 2.
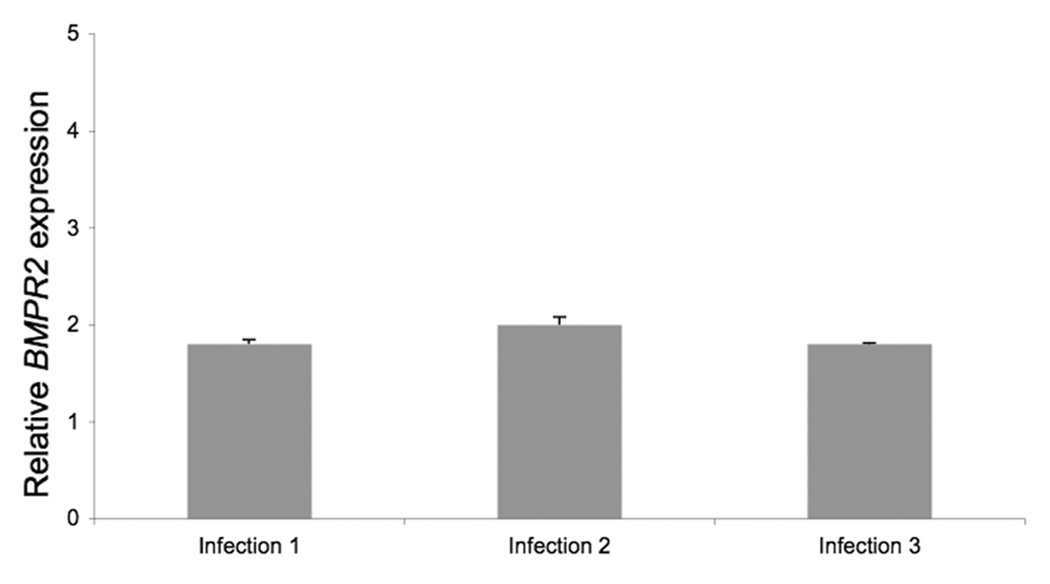
BMPR2 transcript amounts as measured by relative real-time PCR analysis from three LB cell lines established from three separate infections of lymphocytes from one affected FPAH patient from kindred 80. Each experiment was done at least three times in replicates of 3. Error bars represent SD of the replicates.
NMD can be detected in LB cell lines by real-time PCR
We have shown previously that certain mutations in BMPR2 can generate PTCs that result in NMD of the mutation carrying mRNA (Cogan, et al., 2006; Cogan, et al., 2005). We then determined if real-time PCR could detect NMD+ transcripts in patient derived LB cell lines. We examined LB cells from two cohorts (family 80 and family 59) of patients in whom BMPR2 exon 9 is deleted resulting in a frame shift and generation of a PTC. RNA was isolated from LB cells grown with or without puromycin. Real-time PCR assays were then used to quantitate relative levels of wild-type (WT) or exon-9 deleted BMPR2 transcripts (c.1129-3C>G). Our results showed we can detect transcripts that are subjected NMD in LB cell lines (Fig. 3A). Growth of LB cell lines in puromycin containing media [which inhibits NMD and thus would stabilize the mutant transcript (Carter, et al., 1995; Noensie and Dietz, 2001)] resulted in very significant increases (112 fold in LB cell line derived from patient 80-1409 and 23 fold in patient 80-1312; p<.0001 in each case) in the mutant mRNAs (exon-9 deleted BMPR2 mRNA; c.1129-3C>G) which otherwise are present in very low quantities in LB cells grown without puromycin (Fig. 3A). As expected, puromycin did not have a statistically significant effect on the expression of WT mRNA (data not shown). We then compared the levels of mutant (c.1129-3C>G) and WT mRNA in non-puromycin treated LBs derived from the same two kindreds (PPH-80 and PPH-59) to determine the relative contribution of either to the total cellular BMPR2 message. The results show that, when LB cells are not grown with puromycin, the levels of WT allele are by far the major determinant of the total cellular BMPR2 transcripts (Fig. 3B). The mutant transcript (c.1129-3C>G) contributes 2.3%, 0.06%, 2.1% and 2.5% to the total BMPR2 levels in case in LB cell lines derived from patients 80-1409A, 80-1872, 80-1881, 80-1312 and 59-139 respectively (all with p<.0001). The real-time expression data correlated with western blot analysis which showed that carriers have higher BMPR2 protein expression than affected individuals in these two kindreds (Fig. 3C).
Fig. 3.
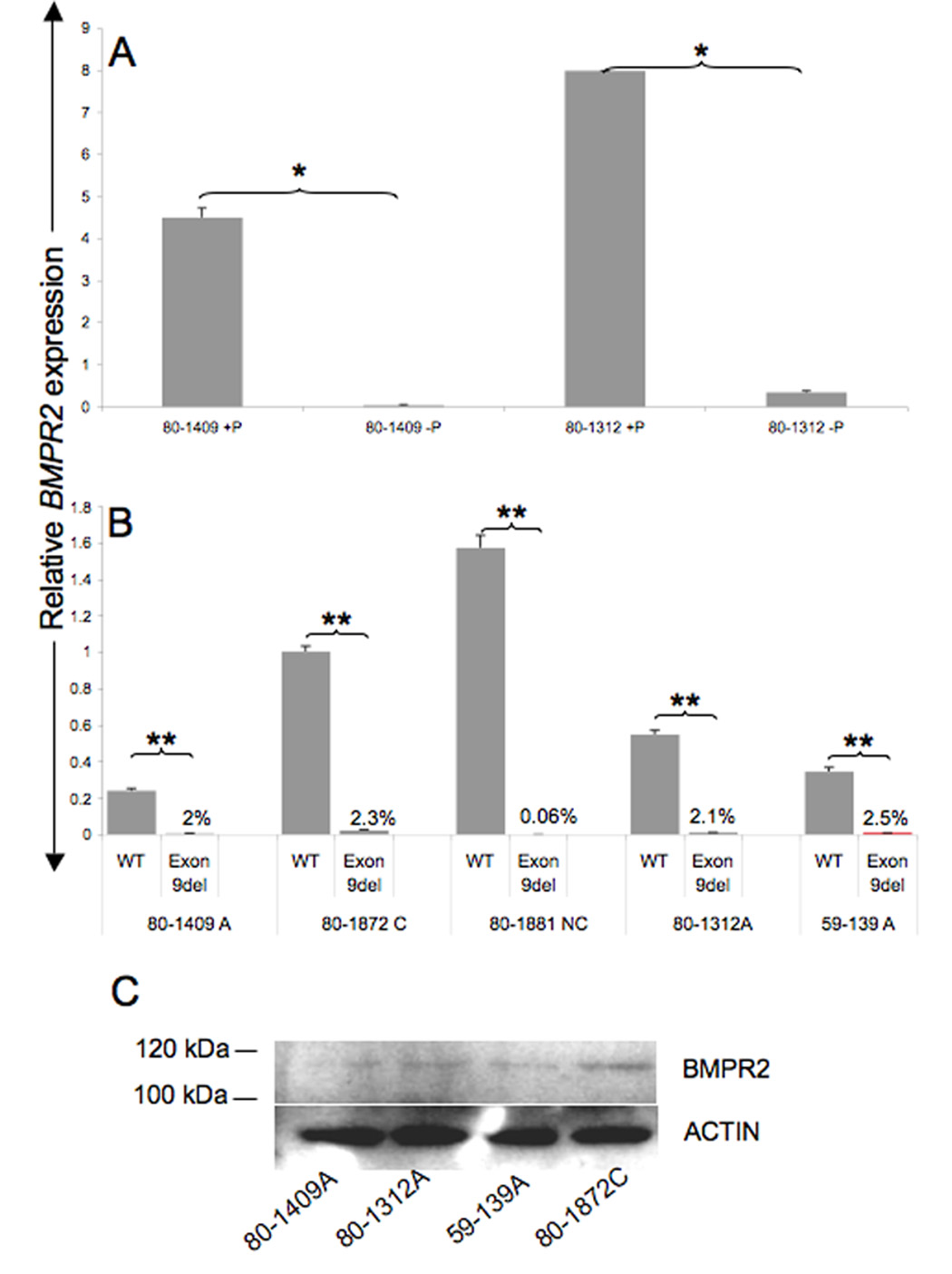
Real-time PCR analysis of the relative amounts of NMD+-mutant BMPR2 transcripts (exon 9del resulting in frame shift and PTC) in LB cell lines derived from kindred 80, with (+P) and without puromycin (−P). Puromycin stabilizes the mutant product, resulting in 112 fold increase in case of patient 80-1409 (*p<.0001) and 23 fold in case of patient 80-1312 (*p<.0001) and thus allowing for detection by real-time PCR (A). Without puromycin the bulk of the BMPR2 transcript is WT transcript (B) (**p<.0001). The percentage numbers represent the contribution of the mutant transcript to the total BMPR2 level in the absence of puromycin treatment. Western blot analysis shows that the BMPR2 transcript differences are also present at the protein level (C). A (Affected)= mutation positive and clinically affected, C (carriers)=mutation positive but clinically unaffected, NC (non carriers) non-mutation carriers and not clinically affected. Each experiment was done at least three times in replicates of 3. Error bars represent SD of the replicates.
BMPR2 expression levels are different between patients and carriers
After characterizing and validating levels of BMPR2 transcripts, we tested our hypothesis that the amounts of the wild type (WT) BMPR2 transcripts may influence FPAH pathogenesis by affecting penetrance. To do this we analyzed all the available LB cell lines (grown without puromycin and thus expected to show transcripts only from the WT allele) derived from blood samples from four kindreds (PPH, 20, 59, 80 and 127). that have NMD+ mutations (Table 1). There were two affected and one carrier from kindred 80 (c.1129-3C>G), three affected and two carriers from kindred 20 (c.419-?_621+?del), two affected and 5 carriers from kindred 127 (c.1141_1142insA) and one affected from kindred 59 (c.1129-3C>G). Total BMPR2 transcript amounts were determined using our real-time PCR assay. Expression data from all four kindreds (PPH 20, 59, 80 and 127) were analyzed together and showed that the expression of WT BMPR2 transcripts was significantly lower in patients with PAH than individuals who are unaffected mutation carriers (Fig. 4) (p<0.005). We also determine the level of statistical significance in a more conservative manner by reducing the data to remove redundant variables from the data, replacing the data file with a smaller number of uncorrelated variables. To do this, we calculated the average value of WT BMPR2 transcript levels for each kindred in the affected group, and the average value of WT BMPR2 transcript levels for each kindred in the unaffected group. As a result, each kindred was represented by one value that contributes to the overall data file for each group: affected versus unaffected. Data reduction to the average contribution of each kindred results in a more conservative statistical approach due to a loss of power. The data was reanalyzed for statistical significance using the conservative nonparametric approach for continuous variables: Mann-Whitney U (Wilcoxon rank-sum) test. Expression of WT BMPR2 transcripts was still significantly lower in patients affected with PAH compared to unaffected mutation carriers (p value 0.034). These results thus suggest that the level of expression of the normal WT BMPR2 allele may contribute to the clinical development of FPAH in individuals who carry a mutant BMPR2 allele.
Fig. 4.
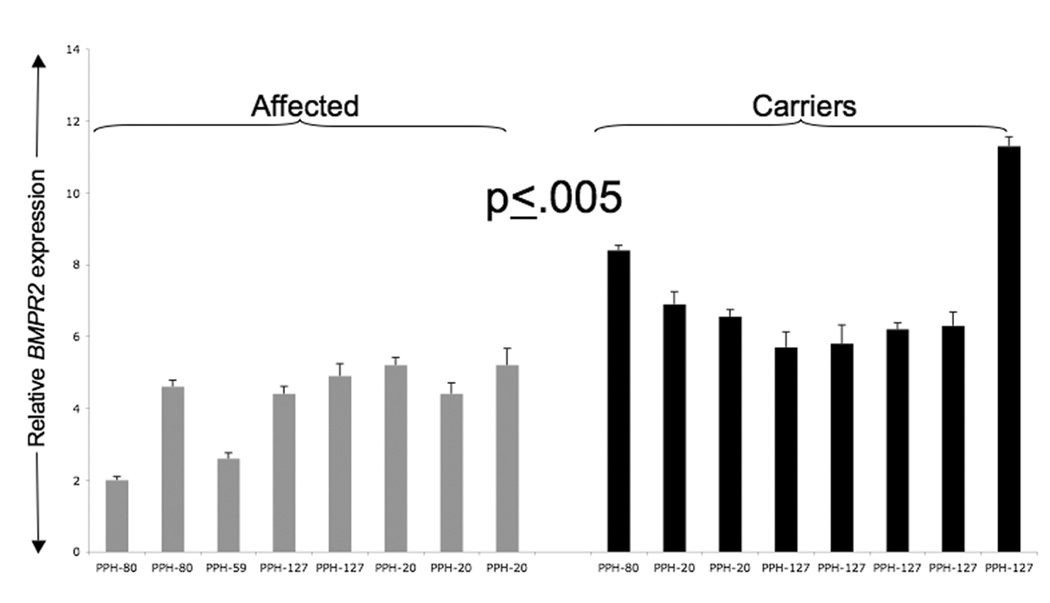
BMPR2 WT transcript levels in affected (grey) and carrier (black) individuals as detected by relative real-time PCR analysis. Affected individuals (grey) have lower levels of expression of WT allele as compared to carriers (black) of the same mutation. Analysis was done on all the available LB cells from 4 kindreds (numbers under the bars represent the kindred number in our registry database; PPH-20, PPH-59, PPH-80, PPH-127). Please see Table 1 for list of mutation present in each kindred. Each experiment was done at least three times in replicates of 3. Error bars represent SD of the replicates. One-way analysis of variance (ANOVA) and Mann-Whitney U (Wilcoxon rank-sum) test was used to determine significance with the help of JMP software (SAS Institute Inc. Cary NC) package.
DISCUSSION
While all the members of FPAH kindreds with NMD+ BMPR2 mutations could potentially develop disease due to HI, only 20% actually develop FPAH (Austin and Loyd, 2007). Thus, the mutation-induced HI alone is not sufficient to cause FPAH in most individuals. We hypothesized that the levels of expression of normal (WT) transcripts could modify the degree of HI and contribute to penetrance in individuals, carrying the NMD+ mutations.
To test our hypothesis we first characterized WT and mutant BMPR2 transcript levels in LB cell lines to show that LB cell lines can be used to study BMPR2 expression. This was important since as explained in the introduction tissues from the affected site, i.e. lung are not available for study, particularly, from unaffected mutation carriers. Utilizing LBs derived from normal controls and PAH patients, we showed that WT BMPR2 transcripts can be detected and reproducibly quantitated by real-time PCR and that expression levels vary between individuals. We further show that these differences in transcript amounts were not secondary to the immortalization process and that we could quantify and differentially detect WT BMPR2 and mutant (NMD+) transcripts in LB cells derived from affected and non-affected BMPR2 mutation carriers. Further more our data show that in the absence of puromycin, the WT transcripts constitute the great bulk of the detected transcripts.
After validation of the LB cell line as a reproducible model for BMPR2 expression we then compared WT BMPR2 expression in four kindreds with NMD+ mutations and found that LB cells from the affected family members have lower levels of WT BMPR2 transcripts compared to those found in LB cells from unaffected relatives who carry the same BMPR2 mutation. This association of transcript levels with penetrance is not limited to a single NMD+ mutation since all four of the kindreds analyzed have different NMD+ mutations. Our data thus support the notion that variation in WT BMPR2 transcript amounts may act as a modifier that influences the penetrance of FPAH at least in individuals who carry HI causing mutations.
There is growing body of work that suggests that modifier genes play an important role in complex disease pathogenesis (Nadeau, 2001). In lung disease the best studied example for the role of modifiers is in cystic fibrosis (for a review see (Boyle, 2007)). However, in many other chronic lung diseases, including PAH, these modifiers are poorly understood. The findings from our study suggest that the levels of WT transcripts may be one such modifier of PAH. Clues as to how levels of WT transcripts might affect PAH penetrance come from the finding that HI (secondary to NMD+ mutations) of BMPR2 is responsible for nearly 50% FPAH. Since BMPR2 receptor forms a heteromeric complex with BMPR1a and BMRP1b the degree of deficiency of normal BMPR2 receptor could affect the stoichiometric imbalance in the BMPR2 heteromeric complex on the cell surface leading to decreased signaling through the receptor and thus disease. Thus, decreased expression of the normal allele in a HI background (caused by the heterozygosity for NMD+ mutation) may further lower BMPR2 expression, below a critical threshold, needed for proper receptor stoichiometry and function thus impacting clinical expression of the disease. Such modulation of disease penetrance by WT transcripts is thought to be a rare phenomenon that has been previously reported in only three genetic disorders such as dominantly inherited Erythropoietic Protoporphyria, Hereditary Elliptocytosis and autosomal dominant Retinitis Pigmentosa (Gouya, et al., 2002; Gouya, et al., 2004; Vithana, et al., 2003; Wilmotte, et al., 1993). Such a hypothetical threshold effect for BMPR2 in FPAH pathogenesis suggests novel therapeutic approaches such as the use of agents that promote read-through of the PTCs of the mutant BMPR2 transcripts resulting in an increase in expression, enough, to exceed the threshold of HI and prevent FPAH may have some clinical utility. Preliminary data from ongoing research suggest that aminoglycosides allow read through of the PTCs in LB cell lines (Hamid et al. unpublished data).
The mechanisms underlying variable expression of the WT BMPR2 allele are not known at this time. Altered expression could be secondary to a cis effect i.e. variation in regulatory regions, or a trans effect i.e. variation of expression of genes or factors that control BMPR2 expression. Thus, our findings suggest the importance of detailed characterization of the proximal regulatory regions BMPR2 and of WT BMPR2 alleles that have different expression levels.
In summary, our data show the validity of using LB cell lines to study BMPR2 expression and importantly, show for the first time that variations in the amounts of WT BMPR2 transcripts may modulate the HI that causes FPAH pathogenesis in all four kindreds studied. A future expanded study (already underway in our laboratory) is to determine if this association is seen consistently in all FPAH kindreds with NMD+/HI BMPR2 mutations.
Acknowledgments
Supported by: National Institutes of Health grant P01 HL072058 to J. E. Loyd and National Institutes of Health GCRC grant to Vanderbilt University- RR000095
References
- Austin E, Phillips J, Cogan J, Stanton K, Phillips C, Yu C, Wheeler L, Newman J, Loyd J. Functinal genetic variations in multiple signaling pathways modify clinical expression of familial pulmonary arterial hypertension. 2008 In review. [Google Scholar]
- Austin ED, Loyd JE. Genetics and mediators in pulmonary arterial hypertension. Clin Chest Med. 2007;28(1):43–57. doi: 10.1016/j.ccm.2006.11.007. vii–viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron CA, Liu SY, Hicks C, Gregg JP. Utilization of lymphoblastoid cell lines as a system for the molecular modeling of autism. J Autism Dev Disord. 2006;36(8):973–982. doi: 10.1007/s10803-006-0134-x. [DOI] [PubMed] [Google Scholar]
- Bird AG, McLachlan SM, Britton S. Cyclosporin A promotes spontaneous outgrowth in vitro of Epstein-Barr virus-induced B-cell lines. Nature. 1981;289(5795):300–301. doi: 10.1038/289300a0. [DOI] [PubMed] [Google Scholar]
- Boyle MP. Strategies for identifying modifier genes in cystic fibrosis. Proc Am Thorac Soc. 2007;4(1):52–57. doi: 10.1513/pats.200605-129JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29(1):23–39. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem. 1995;270(48):28995–29003. doi: 10.1074/jbc.270.48.28995. [DOI] [PubMed] [Google Scholar]
- Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44(1):14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, Stanton KC, Wheeler LA, Phillips JA, 3rd, Loyd JE, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(5):590–598. doi: 10.1164/rccm.200602-165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan JD, Vnencak-Jones CL, Phillips JA, 3rd, Lane KB, Wheeler LA, Robbins IM, Garrison G, Hedges LK, Loyd JE. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med. 2005;7(3):169–174. doi: 10.1097/01.gim.0000156525.09595.e9. [DOI] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, Taylor J, Burnett E, Gut I, Farrall M, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39(10):1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- Gonzalez CI, Wang W, Peltz SW. Nonsense-mediated mRNA decay in Saccharomyces cerevisiae: a quality control mechanism that degrades transcripts harboring premature termination codons. Cold Spring Harb Symp Quant Biol. 2001;66:321–328. doi: 10.1101/sqb.2001.66.321. [DOI] [PubMed] [Google Scholar]
- Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, Da Silva V, Grandchamp B, Deybach JC. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet. 2002;30(1):27–28. doi: 10.1038/ng809. [DOI] [PubMed] [Google Scholar]
- Gouya L, Puy H, Robreau AM, Lyoumi S, Lamoril J, Da Silva V, Grandchamp B, Deybach JC. Modulation of penetrance by the wild-type allele in dominantly inherited erythropoietic protoporphyria and acute hepatic porphyrias. Hum Genet. 2004;114(3):256–262. doi: 10.1007/s00439-003-1059-5. [DOI] [PubMed] [Google Scholar]
- Hamid R, Patterson J, Brandt SJ. Genomic structure, alternative splicing and expression of TG-interacting factor, in human myeloid leukemia blasts and cell lines. Biochim Biophys Acta. 2008;1779(5):347–355. doi: 10.1016/j.bbagrm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci U S A. 2007;104(23):9758–9763. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi C, Ishiwata M, Nanko S, Ozaki N, Iwata N, Umekage T, Tochigi M, Kohda K, Sasaki T, Imamura A, et al. Up-regulation of ADM and SEPX1 in the lymphoblastoid cells of patients in monozygotic twins discordant for schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2007 doi: 10.1002/ajmg.b.30643. [DOI] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26(1):81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68(1):92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5(2):89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2(3):165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- Newman JH, Phillips JA, 3rd, Loyd JE. Narrative review: the enigma of pulmonary arterial hypertension: new insights from genetic studies. Ann Intern Med. 2008;148(4):278–283. doi: 10.7326/0003-4819-148-4-200802190-00006. [DOI] [PubMed] [Google Scholar]
- Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, 3rd, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345(5):319–324. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- Noensie EN, Dietz HC. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat Biotechnol. 2001;19(5):434–439. doi: 10.1038/88099. [DOI] [PubMed] [Google Scholar]
- Oh HM, Oh JM, Choi SC, Kim SW, Han WC, Kim TH, Park DS, Jun CD. An efficient method for the rapid establishment of Epstein-Barr virus immortalization of human B lymphocytes. Cell Prolif. 2003;36(4):191–197. doi: 10.1046/j.1365-2184.2003.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11(13):1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- Vithana EN, Abu-Safieh L, Pelosini L, Winchester E, Hornan D, Bird AC, Hunt DM, Bustin SA, Bhattacharya SS. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci. 2003;44(10):4204–4209. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]
- Wilmotte R, Marechal J, Morle L, Baklouti F, Philippe N, Kastally R, Kotula L, Delaunay J, Alloisio N. Low expression allele alpha LELY of red cell spectrin is associated with mutations in exon 40 (alpha V/41 polymorphism) and intron 45 and with partial skipping of exon 46. J Clin Invest. 1993;91(5):2091–2096. doi: 10.1172/JCI116432. [DOI] [PMC free article] [PubMed] [Google Scholar]