

Abstract
Mutations in the DMD gene result in two common phenotypes associated with progressive muscle weakness: the more severe Duchenne Muscular Dystrophy (DMD) and the milder Becker Muscular Dystrophy (BMD). We have previously identified a nonsense mutation (c.9G>A; p.Trp3X) within the first exon of the DMD gene, encoding the unique N-terminus of the 427 kDa muscle isoform of the dystrophin protein. Although this mutation would be expected to result in severe disease, the clinical phenotype is very mild BMD, with ambulation preserved into the 7th decade. We identify the molecular mechanism responsible for the amelioration of disease severity to be initiation of translation at two proximate AUG codons within exon 6. Analysis of large mutational data sets suggests that this may be a general mechanism of phenotypic rescue for point mutations within at least the first two exons of the DMD gene. Our results directly demonstrate, for the first time, the use of alternate translational initiation codons within the DMD gene, and suggest that dystrophin protein lacking amino acids encoded by the first five exons retains significant function.
Keywords: Duchenne Muscular Dystrophy, DMD, Becker Muscular Dystrophy, BMD, dystrophinopathy, translation, truncating mutations
Introduction
Mutations in DMD (MIM# 300377) result in the predominantly skeletal muscle degenerative diseases Duchenne muscular dystrophy (DMD; MIM# 310200) and Becker muscular dystrophy (BMD; MIM# 300376), and in the relatively rare X-linked dilated cardiomyopathy (MIM# 301045); collectively, these are the dystrophinopathies. DMD is characterized by complete or near complete absence of functional dystrophin, the DMD gene product, whereas BMD is associated with the presence of dystrophin, albeit of reduced size or reduced amount. Consistent with this, DMD is more severe than BMD, with onset of symptoms by the age of five and loss of ambulation generally occurring by age 12 years. In contrast, BMD is characterized by broad spectrum of phenotypes, ranging from a “mild DMD”, with loss of ambulation in the early teens, to a nearly asymptomatic state into late adulthood.
In addition to the muscle promoter, which results in the transcription of the most abundant isoform (Dp427m) found in skeletal and cardiac muscle, there are two more promoters that drive expression of their own first exon and give rise to a predicted full-length 427 kiloDalton (kDa) protein: Dp427c, found primarily in cortical neurons; and Dp427p, abundant in cerebellar Purkinje cells. The unique first exons of both the Dp427c and Dp427p transcripts are spliced directly to the second exon that is found in common with the muscle isoform. As a result, the three full-length dystrophin proteins are all encoded by 79 exons, differ only by few N-terminal amino acids, and are proposed to be functionally equivalent.
In skeletal and cardiac muscle, dystrophin binds via an N-terminal domain (proposed to be in exons 2-8) to filamentous actin. This actin binding domain consists of two calponin homology domains (CH1 and CH2), which localize approximately to amino acids p.Arg13 to p.Val 120 (CH1) and p.Asn135 to p.Val238 (CH2) (Norwood, et al., 2000). A central rod domain consists of 24 spectrin-like repeats, and includes a second actin-binding domain. Deletion of this second actin binding domain appears to have little effect on dystrophin function, whereas deletion of the N-terminal domain leads to a mild BMD phenotype (Banks, et al., 2007). A cysteine-rich domain near the C-terminus of dystrophin binds to β-dystroglycan, part of the dystrophin-associated glycoprotein complex that extends through the sarcolemma to the extracellular matrix. Dystrophin thus flexibly connects the basal lamina of the extracellular matrix to the inner cytoskeleton.
The majority of patients (approximately 90%) with either DMD or BMD have DMD mutations which follow the “reading frame rule” in relating genotype to phenotype(Monaco, et al., 1988). Mutations that disrupt the translational open reading frame result in the production of little or no dystrophin, and as a consequence are associated with the DMD phenotype. In contrast, mutations that result in the preservation of an open reading frame that encodes the C-terminal and dystroglycan-binding region of the protein are associated with BMD. This holds true even for extended in-frame exonic deletion mutations that give rise to relatively large internal truncations but a partially functional dystrophin protein and a BMD phenotype.
The molecular mechanisms leading to exceptions to the reading frame rule are being defined. One mechanism is alternative splicing of the DMD gene such that the mutation-containing exon is spliced out but the open reading frame is maintained. This has been demonstrated in the setting of some cases of BMD associated with nonsense mutations, which might be expected to result in DMD by the reading frame rule (Tuffery-Giraud, et al., 2005). In these cases, nonsense mutations alter exonic splice enhancer or suppressor sequences, resulting in the altered splicing pattern (Disset, et al., 2006). A second mechanism is operative in splice site mutations, in which point mutations within (or nearby to) consensus splice sites have only a partial effect on splicing. This is also evident in the case of intronic point mutations that create novel splice signals within the intron, resulting in the inclusion of intronic sequence as a “pseudoexon” in the mature mRNA in which the reading frame is disrupted; in this case, the efficiency of the new splice signal correlates with disease severity (Gurvich, et al., 2008). A third mechanism has been inferred from patients with out-of-frame deletions of exons 3 through 7, which has been reported in patients with both the DMD and BMD phenotypes. Epitope mapping using antibodies that recognize the 5’ and 3’ end of the region encoded by exon 8 led to the inference that translation initiation can occur at a methionine codon within exon 8, although direct evidence of translational initiation from that AUG was not presented (Winnard, et al., 1995).
Here we present for the first time direct evidence for alternative translational initiation as a mechanism of amelioration of the severity of DMD nonsense mutations. The mutation c.9G>A results in a change of the third codon of the muscle exon 1 from a tryptophan-encoding UGG to a UGA stop codon (p.Trp3X). This mutation is associated with a very mild phenotype; the proband in the original report (Flanigan, et al., 2003), now 66 years old, stopped walking at age 62 years, and his brother (who also carries the mutation) presently has only minimal difficulty climbing stairs at age 62. Since we first reported this mutation and phenotype (Flanigan, et al., 2003), we have identified it independently in five more BMD families, in which all probands were males within their first two decades who only had elevated serum creatine kinase levels, sometimes with exertional myalgias. Based upon detailed analysis of the pattern of linked single-nucleotide polymorphisms, we have now shown this to be the first founder allele described in the DMD gene (Weiss et al, manuscript in submission). The maintenance of this allele in the population is most likely due to its exceptionally mild phenotype as explained by the mechanism of alternate translation initiation in exon 6 described here.
In addition to the c.9G>A allele, two other exon 1 point mutations that disrupt the translational reading frame have been identified. One, reported from the Leiden database as BMD (but without detailed phenotypic information) is c.11G>A (p.Trp4X). The second (c.14_15delAAinsT/p.Glu5ValfsX3; but previously reported in the Leiden database as c.14delA) was found in a BMD patient who was ambulant until 42 years of age and is now aged 54 years.
The existence of other patients with amino-terminal truncating mutations that result in a similar mild BMD phenotype suggests a common auxiliary pathway of dystrophin expression in the presence of such mutations. We investigated the initiation of translation from downstream methionine codons in the DMD gene, and demonstrate this to be a novel mechanism of dystrophin expression that ameliorates the predicted DMD phenotype in the presence of truncating mutations in exon 1.
Material and Methods
Immunofluorescent analysis
Cryosections (11 micron) from frozen muscle biopsy samples were rehydrated in phosphate-buffered saline (PBS), pH 7.4, and blocked with 5% non-immune goat serum/1% BSA in PBS for 30 minutes. Sections were then incubated with primary antibodies diluted with wash buffer (1% goat serum/1% BSA in PBS) for 1-2 hours. Working dilutions of antibodies were 1:100 for Mandra I (Sigma) and 1:10 for Manex 1A, Manex 7B (both obtained from Dr. Glenn Morris) and NCL-dys3 (Vector Labs). Following three 5 minute washes, sections were incubated for 30 minutes with the secondary antibody AlexaFluor 488 (Invitrogen) diluted 1:500 with wash buffer, then washed three times (5 minutes each) and mounted in ProLong Gold antifade (Invitrogen). Immunofluorescent images were obtained on FluoView 300 Olympus IX81 confocal microscope with 20x objective. Z-series images were captured through the section at 1 micron intervals. Image acquisition parameters were the same for all samples stained with the same antibody. Photomultiplier tube (PMT) gain settings were adjusted for each antibody: 650V for Mandra I; 620V for NCL-dys3; 647V for Manex7B; and 620V for Manex 1A.
Gel Electrophoresis and Western Blotting
Eleven micron muscle biopsy sections were incubated in lysis buffer (4% SDS, 40% glycerol, 100mM DTT, 125mM Tris-HCl pH 8.7, and complete protease inhibitors [Roche]) at 90°C for 5 minutes. Samples were loaded on 3-8% Tris-Acetate gels (Invitrogen); electrophoresis and blotting onto PVDF or nitrocellulose membrane was carried out according to manufacturer’s instructions. Membranes were blocked either with 5% milk or 1% casein (Novagen) and probed with Mandra I (Sigma-Aldrich), anti-GAPDH (monoclonal, Abcam) or anti-alpha-Actinin (monoclonal, Sigma-Aldrich), all diluted 1:20,000. Bound antibodies were visualized using SuperSignal West Femto Maximum Sensitivity kit (Pierce) according to manufacturer’s instructions.
Reporter Constructs
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the DMD cDNA reference sequence (GenBank NM_004006.1). For protein numbering, the initiation codon is codon 1. Dystrophin cDNA was amplified from muscle RNA using primer A (ctcgctagcTCCTGGCATCAGTTACTGTGTTGACTCACTCAGTGTTG), in which the upper case letters are complementary to nucleotides c.-244_-207, and primer B (gtgctcgggTGACTTGTCTTCAGGAGCTTCC), in which the upper case letters are complementary to nucleotides c.966_987. The product was digested with NheI and AvaI and cloned into the corresponding sites of phRL-CMV (Promega). Further codon changes were introduced by site-directed mutagenesis using the Phusion Hot Start High-Fidelity DNA Polymerase Kit (New England Biolabs). Changes were confirmed by DNA sequencing.
In vitro expression
For in vitro expression of the reporter constructs, T7 transcription and translation in the presence of 35S-methionine (Amersham) was performed using PROTEINscript II T7 kit (Ambion) according to the manufacturer’s protocol. Morpholino anti-sense oligonucleotides were added directly to the transcription reaction. The synthesized proteins were separated on 10% NuPage Bis-Tris gels (Invitrogen) in MOPS buffer (Invitrogen). After fixation in 30% methanol/5% glycerol gels were dried and exposed overnight on a PhosphorImager screen.
Renilla luciferase assays
A C2C12 mouse muscle cell (ATCC) line was cultured in DMEM supplemented with 20% fetal bovine serum. 104 cells were seeded per well on a 96-well plate and 100 ng of plasmid DNA was transfected in suspension using Lipofectamine 2000 (Invitrogen). The next day renilla luciferase activity was assayed using the Dual Luciferase Reporter Assay System (Promega). Each construct was assayed in quadruplicate, and the luciferase activity was averaged and normalized to that of the wild type (WT) construct.
Results
Analysis of dystrophin protein in patients muscle samples
Archived clinical muscle biopsy samples were available from two patients with the c.9G>A/p.Trp3X mutation (patients 1 and 2) and one patient with c.14_15delAAinsT/p.Glu5ValfsX3 (patient 3). Control samples consisted of muscle biopsy samples from a patient with a wild-type dystrophin gene, and a DMD patient with a stop codon mutation in exon 5 (c.355C>T; p.Gln119>X). Although the DMD negative control patient carries a premature stop codon upstream of the exon 6 translation initiation sites, it is a null allele (see Discussion). Immunofluorescent analysis was performed using four antibodies that recognize various domains of dystrophin protein: MANEX1A (specific for the peptide encoded by exon 1 of the muscle isoforms); MANEX7B (specific to exons 7/8); NCL-Dys3 (exon 10-12); and Mandra I (C-terminus, encoded by exons 63-79).
The results of staining with a panel of anti-dystrophin antibodies are seen in Figure 1. Sections of both the p.Trp3X and frameshift patients (pts. 1, 2 and 3) show staining of nearly all fibers with the Mandra I and dys3 antibodies, although the staining is diminished compared to normal muscle, and varies in comparison between the patient specimens. Nevertheless, staining suggests the presence of peptide sequence corresponding to exons 12-79 in all three patients. MANEX7B (exons 7/8) weakly stains both p.Trp3X patient sections, whereas staining of patient 3 is extremely weak. Staining with the muscle exon 1-specific MANEX1A antibody, however, is completely negative for all samples except wild-type. The immunofluorescence studies thus suggest that the dystrophin protein expressed in the muscle fibers of the exon 1 point mutation patients has an altered or absent N-terminal sequence but possesses the amino acid sequence
Figure 1.

Immunofluorescent analysis of patients’ tissue sections with dystrophin antibodies. Antibody epitopes are encoded by the following exons: exon 1 for Manex 1A; exons 7/8 for Manex 7B; exons 10-12 for NCL-dys3 and exons 67-79 for Mandra I. Images represent projections of z-series stacks of confocal sections.
encoded in exons 7-79 of the DMD gene
Expression of dystrophin in the patients’ muscle was also analyzed by immunoblotting with Mandra I antibody. The immunoblot shows decreased amounts of dystrophin protein in muscle from patients 1 and 3 to about 5-15% of the level in wild-type muscle (Figure 2A). When a longer electrophoresis is performed, a decreased size of the dystrophin protein expressed in patients’ samples can be seen; the size of dystrophin from both the exon 1 nonsense and frameshift patient specimens appears to be the same (Figure 2B).
Figure 2.
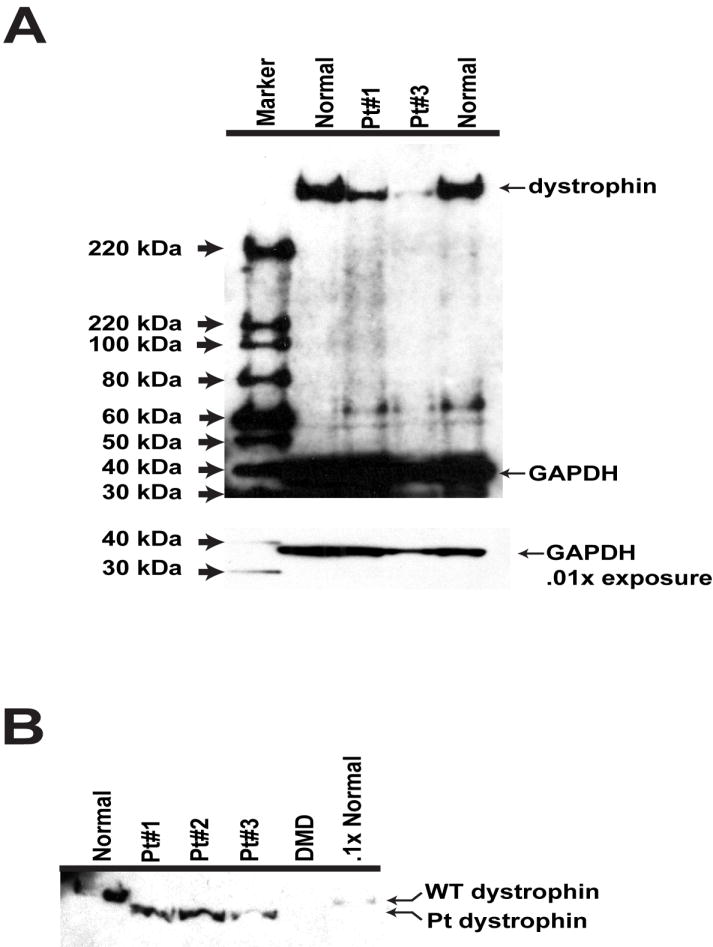
Immunoblot analysis of patients’ tissues for dystrophin protein with Mandra I antibody. (A) Electrophoresis showing decreased amount of dystrophin in the muscles of Trp3X patient (#1) and frameshift patient (#3). Antibody to glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Electrophoresis for a longer time indicating decreased size of dystrophin protein of the Trp3X patients (#1 and #2) and frameshift patient (#3) compared to wild-type dystrophin in a normal control sample. The sample labeled “DMD” is from an exon 5 nonsense mutation patient (c.355C>T; p.Gln199X).
Translation initiation studies with reporter constructs
The decreased size of the dystrophin protein as revealed by immunoblot suggested translational initiation may occur at a downstream codon. To investigate this hypothesis, reporter constructs were made by cloning sequence encompassing exons 1 to 9 of the muscle isoform into the phRL-CMV vector, upstream of and in-frame with the renilla luciferase (RL) coding sequence (schematically represented in Figure 3A). Transcription of this reporter can be driven either from the T7 promoter for in vitro studies, or from the CMV promoter for in vivo expression. Translation of the reporter cassette can occur only from the initiation codons in the dystrophin sequence (Figure 3A) and can be assayed either by labeling with 35S-methionine in vitro or by measuring activity of the expressed luciferase in transfected cells.
Figure 3.

Translation initiation studies using reporter constructs. (A) Scheme of the reporter. Transcription of the cassette is driven either from T7, in vitro, or CMV, in vivo, promoters. Translation of dystrophin-Renilla Luciferase fusion protein can occur only from initiation codons in dystrophin sequence. Positions of the most likely initiation codons, AUG, are indicated. The number after the AUG indicates its numerical order, and the number in brackets indicates the position of this codon in the dystrophin coding sequence. (B) and (C) in vitro translations of the reporter constructs labeled with 35S-Methionine. (D) Renilla Luciferase assays of constructs expressed in C2C12 cells.
Initial reporter constructs contained either wild-type (WT), p.Trp3X (W3X), or p.Glu5ValfsX3 (FS) exon 1-9 dystrophin sequences (Table 1). When expressed in vitro, WT construct produced a major product of about 72 kDa, consistent with what would be predicted if translation occurs from the conventional first methionine (AUG1) in muscle isoform cDNA, as well as a minor product of approximately 60 kDa (Figures 3B and 3C). In contrast, in vitro translation of the W3X and FS constructs did not show expression of the 72kDa product, but resulted in synthesis of the shorter 60kDa product (lane 2 in Figure 3B, and lanes 3 and 7 in Figure 3C).
Table 1.
Reporter constructs used in this study.
| Name | Description |
|---|---|
| WT | exon 1-9 of human WT dystrophin sequence placed upstream and in-frame with Renilla luciferase coding sequence of the phRL-CMV vector |
| W3X | WT; c.9G>A |
| FS | WT; c.14_15delAAinsT |
| E8X | WT; c.22G>T |
| K18X | WT; c.52A>T |
| Q28X | WT; c.82C>T |
| L74X | WT; c.220_222CTG>TAA |
| V92X | WT; c.274_276GTG>TAA |
| L110X | WT; c329T>A |
| L131X | WT; c.392T>A |
| WT_AUG1 | WT; c.1_3ATG>TAC |
| W3X_AUG2 | WT; c.9G>A, c.370_372ATG>TAC |
| W3X_AUG3 | WT; c9G>A, c.382_384ATG>TAC |
| W3X_AUG2&3 | WT; c.9G>A, c.370_372ATG>TAC, c.382_384ATG>TAC |
The size of this 60 kDa protein suggested that the translational start site for it is likely to be located in exons 5-7. To narrow down the position of this start codon, a series of constructs was made in which stop codons were introduced into the dystrophin coding sequence. This allows mapping of translation initiation of the 60 kDa protein; if it were to occur upstream of an introduced stop codon, translation would be terminated resulting in loss of the 60 kDa protein. Sense codons throughout the first 6 exons of dystrophin were changed to stop codons at positions 8, 18, 28, 72, 94, 110, and 131, resulting in the constructs E8X, K18X, Q28X, L74X, V92X, L110X and L131X (Table 2). As seen in lanes 3-9 of Figure 3B, in vitro translation from all these constructs still resulted in translation of the 60 kDa protein, except in the case of L131X. These results indicated that the initiation codon for the 60 kDa product is located between codons 110 and 131.
Table 2.
Sequences of morpholino anti-sense oligonucleotides (mAONs) used in this study.
| mAONs designation | Sequence |
|---|---|
| M1 | 5’-ACCAAAGCATTTTGAAAAGTGTATA-3’ |
| M1A | 5’-AAAACCTGGTAAAAGTTCTTCAAAC-3’ |
| 2/3 | 5’-AGCCATGATATTTTTCATTACATTT-3’ |
| C | 5’-TTCATCTTCCATGCCAGCTGTTTTT-3’ |
There are two AUG codons found in this region that potentially could serve as initiation codons and give rise to a 60kDa protein. Both are found in exon 6: AUG2 (position 124) and AUG3 (position 128). Therefore, these AUG codons were changed to non-initiation codons, both separately and simultaneously, within the context of the W3X reporter construct. Changing AUG2 (construct W3XAUG2; lane 4 in Figure 3C) diminished translation of the 60 kDa protein partially, while changing AUG3 (construct W3XAUG3; lane 5 in Figure 3C) decreased translation more significantly. Changing both codons simultaneously (construct W3XAUG2&3; lane 6 in Figure 3C) precluded translation of the 60 kDa protein almost entirely.
In order to confirm these results, translation from these reporter constructs was tested in vivo in a C2C12 mouse muscle cell line, and the results are shown in Figure 3D. Constructs were transfected into C2C12 cells and luciferase activity was assayed 24 hours post-transfection. Expression from Trp3X and FS were each around 10% of expression from WT. Expression from the W3XAUG2 construct was about half of that from W3X, and expression from the W3XAUG3 construct was about one third of that from the W3X. This data correlates with the results from the in vitro translation experiment, which also suggests that AUG3 is in a better initiation context. Expression from the W3XAUG2&3 construct was about the same as from the W3XAUG3 construct. However, the same amount of translation was observed with the L131X construct, which suggests that a difference in translation between W3XAUG3 and W3XAUG2&3 might be masked by some degree of background translation of renilla luciferase. This background translation probably occurs from minor initiation codons downstream of codon 131. As can be seen in Figures 3B, 3C, 4B and 4C, minor protein products in the 40 kDa range are produced from the reporter constructs in the in vitro translation assays. These products are larger than the translation products seen with the luciferase-containing parental vector alone (data not shown), and suggest that initation may occur from methionine codons in exon 8 (AUG4, AUG5, and AUG6, in Figure 3A), consistent with a previous model (Winnard, et al., 1995). The background translation measured in the cell-based assay (Figure 3D) may be due to initiation at methionine codons within DMD exon 8, or within the luciferase sequence itself.
Figure 4.

Morpholino anti-sense oligonucleotide inhibition of translation in vitro. (A) Relative position of mAONs (~) to the sequence of the dystrophin cDNA. (B) and (C) Inhibition of translation by morpholino addition to in vitro translation reactions of WT and W3X RNA, respectively. (-) denotes no treatment; C – control mAON, complementary to the translation initiation site of the C-isoform of dystrophin.
Notably, when we changed the conventional start site AUG1 to a non-initiation codon in the wild-type reporter construct, and expressed the resultant construct WT_AUG1 both in vitro and in vivo (Figures 3C and 3D), this change did not abolish synthesis of the 60 kDa product either in the in vitro translation system or in transfected cells.
Translation initiation inhibition studies using morpholino oligonucleotides
Morpholino antisense oligonucleotides (mAONs) bind to target RNA sequences, and as a result sterically hinder binding sites for protein factors and obstruct cellular processes. They are commonly used to alter pre-mRNA splicing, to block miRNA expression or their targets, and to preclude initiation of translation. Morpholino oligonucleotides interfere with translation initiation by precluding scanning of the mRNA by 40S subunit, but do not impede translation elongation.
To verify results obtained using targeted mutagenesis in the reporter constructs, mAONs (Table 2) were designed to inhibit translation from either AUG1, or AUG2 and AUG3. To inhibit translation from the AUG1 two mAONs were designed. One (M1A) is complementary to the stretch of nucleotides in the 5’UTR of dystrophin mRNA; the second (M1) binds the last 15 nucleotides of the 5’UTR and the first 10 nucleotides of the coding sequence of dystrophin (Figure 4A). To inhibit translation from AUG2 and AUG3 one mAON was designed (2/3, in Figure 4), because these two codons are only 9 nucleotides apart. As a control, a mAON complementary to the translation initiation site of the C-isoform of dystrophin was designed. To test translation inhibition, mAONs were added to the in vitro transcription-translation reactions with WT and W3X constructs. Translation from the WT construct was diminished overall in the presence of M1 and M1A morpholino oligos (Figure 4B). Both of these oligos diminished translation from W3X only slightly if at all (Figure 4C). However, adding the 2/3 mAON to translation reaction resulted in a decreased expression of the 60 kDa product from both the WT and W3X constructs. The control mAON did not affect translation with either of these two constructs.
Discussion
The results presented indicate that the mild BMD phenotype associated with truncating mutations in the first exon of DMD can be attributed to translation initiation in exon 6, and that the resultant protein retains enough function to significantly ameliorate the predicted DMD phenotype. Similar rescue by downstream initiation was previously reported for disease causing mutations in the genes DAX1 (Ozisik, et al., 2003), SMPD1 (Pittis, et al., 2004), ATRX (Howard, et al., 2004), ATP7A (Paulsen, et al., 2006), and RB1 (Sanchez-Sanchez, et al., 2007). Our observations in DMD raise several key questions; these include how the PTC containing mRNA avoids degradation through nonsense mediated decay (NMD), what is the mechanism by which initiation downstream of the premature termination codon (PTC) occurs, and what is the biological relevance of the amino-terminal truncated form of the protein in unaffected (wildtype) individuals.
The NMD pathway in mammalian cells is linked to splicing-dependent deposition of a protein complex the exon-junction complex, or EJC, located 20-24 nucleotides 5’ of each exon-exon junction. These EJCs are removed from the mRNA during a “pioneer” round of translation (Ishigaki, et al., 2001). If the mRNA contains a PTC located more than 50-54 nt 5’ of at least one EJC, ribosomes will terminate translation without removing the EJC. Retention of the EJC triggers the NMD response. Some studies suggested that the mechanism of downstream initiation would allow the ribosome to displace EJCs located 3’ of the nonsense mutation and prevent NMD (Buisson, et al., 2006; Perrin-Vidoz, et al., 2002; Zhang and Maquat, 1997). Of direct relevance to this study is the finding that when nonsense mutations are located in close proximity to an initiation codon, NMD is not triggered irrespective of the presence of downstream initiation codons (Inacio, et al., 2004). Presumably, when nonsense codons are near the translation start site, ribosomes arrive at the stop codon before dissociation from initiation factors and the polyA binding protein (Silva, et al., 2008). By a mechanism which is still poorly defined, such mRNAs are not-competent to trigger the NMD pathway (Silva, et al., 2006). Thus, DMD mutations resulting in premature stop codons very near the initiation codon may be resistant to NMD by this mechanism and resistance to NMD may affect the associated DMD phenotype, as previously hypothesized (Kerr, et al., 2001). This mechanism may explain why the exon 5 stop codon mutation (c.355C>T; p.Gln119>X) is associated with a DMD phenotype, with no dystrophin expression (Figure 2B).
Analysis of the sequence surrounding AUG2 and AUG3 reveals they are found in a context consistent with translational initiation. The optimal context for recognition of the AUG codon as a start codon in mammals by a scanning 40S ribosomal subunit is GCCRCCaugG, of which purine (R) in position -3 is functionally the most important, with A more preferable than G (Kozak, 1986). For both AUG2 and AUG3, a purine in the -3 position is highly conserved among vertebrate sequences (Figure 5), but the -3 nucleotide is G for AUG2 versus A for AUG3. Another highly important determinant is the G nucleotide 3’ of the AUG start (+4 position) (Kozak, 1997); this is conserved in the case of AUG3 (except in D. rerio) but is not present 3’ of AUG2. Thus, AUG3 appears to be overall in a better context for translation initiation by scanning ribosomes. This is in agreement with the results of our reporter experiments, where a greater decrease in translation efficiency was seen when AUG3 was changed to a UAC codon.
Figure 5.

Conservation of the exon 6 alternate initiation codons is demonstrated by alignment of dystrophin exon 6 sequences from different vertebrates. Conserved nucleotides are capitalized. Highlighted are the positions corresponding to the AUG codons in the human dystrophin exon 6. Although the frog sequence has a UUG codon in place of AUG2, the UUG codon can be utilized for translation initiation as well.
Translation initiation at a methionine codon downstream of the conventional initiator codon can occur due to any of three events. One is termination-dependent initiation or reinitiation, which can occur when the ribosome terminates translation shortly after initiation at a conventional start site, and then resumes scanning until the next initiation codon. The second is leaky scanning, which occurs when a fraction of ribosomes fail to initiate at the conventional start codon, and instead continue scanning down the mRNA until the next potential initiation codon. The third is the direct entry of the ribosome at the internal site.
Our data argue against the possibility of translation-dependent reinitiation due to the p.Trp3X mutation itself. When AUG1 of DMD was changed to a UAC codon to abolish initiation, the 60 kDa protein was still produced from AUG2 and AUG3, and synthesis of the 60 kDa protein occurred during translation of the WT construct, indicating that termination of translation at an earlier stop codon is not required for translation initiation. In addition, morpholino treatment that diminished translation from AUG1 had no significant effect on translation from the downstream AUG2 and AUG3 (although in Figure 4, the effect of morpholinos directed to AUG1 is not as clear as the effect of morpholino 2/3, and an alternate interpretation is that the morpholinos directed to AUG1 simply did not work well). Leaky scanning also seems unlikely: ten more AUG codons are found in alternate reading frames between AUG1 and AUG2, two of which are found in an optimal translation initiation context. One would expect that scanning ribosomes would not pass 10 potential translational initiation codons and arrive competent for translation at the AUG codons in exon 6. It remains a formal possibility that ribosomes initiate translation at one of these out-of-frame AUGs, and translation at AUG2 and AUG3 is dependent upon a translation termination event at a stop codon in this new frame. (Translation of these intervening short open reading frames may explain the lower renilla activity of the construct WT_AUG1 as compared to the W3X construct (Figure 3D): in the absence of the AUG1, ribosomes initiating translation at downstream out-of-frame AUGs may impede ribosome initiation at AUG2 and AUG3, whereas early termination at W3X causes release of ribosomes leaving the intervening mRNA more accessible for initiation at AUG2 and AUG3.) The simplest interpretation of all of our results is that the method of translational initiation is due to internal ribosome entry, a hypothesis supported indirectly by the degree of species conservation of AUG2 and AUG3, and their surrounding context. Experiments to directly address this hypothesis are underway.
To determine if initiation at AUG codon(s) in exon 6 could rescue all patients with truncating mutations close to the AUG1 start codon, phenotypes of amino-terminal truncating mutations reported in the Leiden database (www.dmd.nl) were analyzed. Aside from the exon 1 truncating mutations discussed above that gives rise to a BMD phenotype (p.Trp3X, p.Trp4X, and p.Glu5ValfsX3), available reports suggest that the length of the AUG1-initiated ORF can extend at least into exon 2 sequence. Patients with truncating mutations in the proximal part of exon 2 (c.40_41delGA/p.Glu14ArgfsX17 and c.53delA/p.Lys18ArgfsX8) are reported in the Leiden database as associated with BMD. One patient with a mutation in the more distal part of exon 2 (c.58delA/p.Thr20HisfsX5) is reported to have intermediate phenotype. Several patients with similar mutations in exon 3 are reported as DMD (or a few as “B/DMD”, indicating incomplete phenotypic information) in the Leiden data set. Interestingly, a patient with c.32-2A>T mutation (which disrupts the acceptor splice site of intron 1 and resulting in skipping of exon 2 and a disrupted coding sequence) has BMD and a dystrophin protein of a somewhat reduced molecular weight (Hamed, et al., 2005). Although the authors speculated that the milder phenotype might result from translational initiation in exon 3, there are no in-frame AUG codons in exon 3. We propose that the more likely explanation for the BMD phenotype and smaller dystrophin protein in their reported patient is translation initiation in exon 6.
The p.3Trp>X mutation represents the first true founder allele in BMD, and we infer that it has no effect on reproductive fitness (Weiss et al, in submission). The mild clinical syndrome with which it is associated can be attributed to the mechanism we describe here, and confirms that the domains encoded within the first five exons of dystrophin are not necessary in order to maintain significant function of the protein. Upregulation of translation from the exon 6 AUG codons may thus provide a potential route of therapy for those patients with mutations within the first exons of the DMD gene.
Acknowledgments
The authors wish to thank Glenn Morris for Manex 1A and Manex 7B antibodies, and acknowledge assistance of Dr. Christopher Rodesch of the University of Utah Fluorescence Microscopy Core Facility. This work was supported by the National Institute of Neurologic Diseases and Stroke (R01 NS043264 [KMF, MTH, RBW] and T32 NS07493 [O.G.]), and by the Association Francaise Contre les Myopathies (KMF).
References
- Banks GB, Gregorevic P, Allen JM, Finn EE, Chamberlain JS. Functional capacity of dystrophins carrying deletions in the N-terminal actin-binding domain. Hum Mol Genet. 2007;16(17):2105–13. doi: 10.1093/hmg/ddm158. [DOI] [PubMed] [Google Scholar]
- Buisson M, Anczukow O, Zetoune AB, Ware MD, Mazoyer S. The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mutat. 2006;27(10):1024–9. doi: 10.1002/humu.20384. [DOI] [PubMed] [Google Scholar]
- Disset A, Bourgeois CF, Benmalek N, Claustres M, Stevenin J, Tuffery-Giraud S. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum Mol Genet. 2006;15(6):999–1013. doi: 10.1093/hmg/ddl015. [DOI] [PubMed] [Google Scholar]
- Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72(4):931–9. doi: 10.1086/374176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich OL, Tuohy TM, Howard MT, Finkel RS, Medne L, Anderson CB, Weiss RB, Wilton SD, Flanigan KM. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Ann Neurol. 2008;63(1):81–9. doi: 10.1002/ana.21290. [DOI] [PubMed] [Google Scholar]
- Hamed S, Sutherland-Smith A, Gorospe J, Kendrick-Jones J, Hoffman E. DNA sequence analysis for structure/function and mutation studies in Becker muscular dystrophy. Clin Genet. 2005;68(1):69–79. doi: 10.1111/j.1399-0004.2005.00455.x. [DOI] [PubMed] [Google Scholar]
- Howard MT, Malik N, Anderson CB, Voskuil JL, Atkins JF, Gibbons RJ. Attenuation of an amino-terminal premature stop codon mutation in the ATRX gene by an alternative mode of translational initiation. J Med Genet. 2004;41(12):951–6. doi: 10.1136/jmg.2004.020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio A, Silva AL, Pinto J, Ji X, Morgado A, Almeida F, Faustino P, Lavinha J, Liebhaber SA, Romao L. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem. 2004;279(31):32170–80. doi: 10.1074/jbc.M405024200. [DOI] [PubMed] [Google Scholar]
- Ishigaki Y, Li X, Serin G, Maquat LE. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106(5):607–17. doi: 10.1016/s0092-8674(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Kerr TP, Sewry CA, Robb SA, Roberts RG. Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum Genet. 2001;109(4):402–7. doi: 10.1007/s004390100598. [DOI] [PubMed] [Google Scholar]
- Kozak M. Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc Natl Acad Sci U S A. 1986;83(9):2850–4. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16(9):2482–92. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco AP, Bertelson CJ, Liechti Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–5. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- Norwood FL, Sutherland-Smith AJ, Keep NH, Kendrick-Jones J. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000;8(5):481–91. doi: 10.1016/s0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]
- Ozisik G, Mantovani G, Achermann JC, Persani L, Spada A, Weiss J, Beck-Peccoz P, Jameson JL. An alternate translation initiation site circumvents an amino-terminal DAX1 nonsense mutation leading to a mild form of X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab. 2003;88(1):417–23. doi: 10.1210/jc.2002-021034. [DOI] [PubMed] [Google Scholar]
- Paulsen M, Lund C, Akram Z, Winther JR, Horn N, Moller LB. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am J Hum Genet. 2006;79(2):214–29. doi: 10.1086/505407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet. 2002;11(23):2805–14. doi: 10.1093/hmg/11.23.2805. [DOI] [PubMed] [Google Scholar]
- Pittis MG, Ricci V, Guerci VI, Marcais C, Ciana G, Dardis A, Gerin F, Stroppiano M, Vanier MT, Filocamo M et al. Acid sphingomyelinase: identification of nine novel mutations among Italian Niemann Pick type B patients and characterization of in vivo functional in-frame start codon. Hum Mutat. 2004;24(2):186–7. doi: 10.1002/humu.9263. [DOI] [PubMed] [Google Scholar]
- Sanchez-Sanchez F, Ramirez-Castillejo C, Weekes DB, Beneyto M, Prieto F, Najera C, Mittnacht S. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Hum Mutat. 2007;28(2):159–67. doi: 10.1002/humu.20394. [DOI] [PubMed] [Google Scholar]
- Silva AL, Pereira FJ, Morgado A, Kong J, Martins R, Faustino P, Liebhaber SA, Romao L. The canonical UPF1-dependent nonsense-mediated mRNA decay is inhibited in transcripts carrying a short open reading frame independent of sequence context. RNA. 2006;12(12):2160–70. doi: 10.1261/rna.201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AL, Ribeiro P, Inacio A, Liebhaber SA, Romao L. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. RNA. 2008;14(3):563–76. doi: 10.1261/rna.815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuffery-Giraud S, Saquet C, Thorel D, Disset A, Rivier F, Malcolm S, Claustres M. Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. Eur J Hum Genet. 2005;13(12):1254–60. doi: 10.1038/sj.ejhg.5201478. [DOI] [PubMed] [Google Scholar]
- Winnard AV, Mendell JR, Prior TW, Florence J, Burghes AH. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: mechanisms of dystrophin production. Am J Hum Genet. 1995;56(1):158–66. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Maquat LE. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997;16(4):826–33. doi: 10.1093/emboj/16.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]