

Abstract
Like many behavioral phenotypes, generalized vulnerability to substance dependence in adolescence has a complex etiology; it is influenced by both genetic and environmental risks, with a heritability of approximately 0.40 (Button et al., 2006). However, the extent to which the magnitudes of genetic and environmental risk for substance dependence are contextually moderated is unclear. The aim of the current study was to determine whether the etiology of substance dependence vulnerability (DV; total lifetime symptom count of dependence criteria endorsed across numerous substances divided by the number of substances used) varies depending on the extent of affiliation with delinquent peers as perceived by the adolescent. Results show that affiliation with delinquent peers moderates both the unstandardized (absolute) and the relative contribution of genetic, shared, and non-shared environmental risks to the variance of DV. The genetic variance was estimated to be higher among subjects who perceived their peers to be least delinquent and among those who considered their peers to be the most delinquent. The magnitude of both shared and non-shared environmental influences were negligible among those who perceived their peers to be least delinquent and were greater among those with higher levels of perceived peers’ delinquency.
Keywords: substance dependence, peer delinquency, gene-environment moderation, gene-environment correlation
1. Introduction
The prevalences of substance dependence in the general population are estimated to be around 6-8% for alcohol dependence and 7-14% for marijuana dependence (Kandel et al., 1997a; Young et al., 2002). Substance use and dependence in adolescents confers substantial health risks to the adolescent, including physical problems and psychopathological problems (Kandel et al., 1997b; McGue and Iacono, 2005). Furthermore, drug use and dependence have a significant negative effect on society, given associated crime, reduced workplace productivity, and increased health care costs. These were estimated to cost the United States $143.4 billion in 1998, with a predicted 5.9% increase annually (Office of National Drug Control Policy, 2001). Therefore, adolescent substance dependence is a critical personal and public health concern.
1.1 Moderation of genetic risk
To develop successful prevention and intervention techniques, it is important to understand the etiology of substance dependence and identify those at most risk. Numerous studies have demonstrated that, regardless of the substance of interest, both genetic predisposition and environmental influences contribute to the development of substance dependence in adolescents (Button et al., 2007b; Dick and Bierut, 2006; Heath et al., 1997; Rhee et al., 2003; Tsuang et al., 1996). Using a subsample of the participants described for the current study, we previously demonstrated that the heritability for a generalized vulnerability to develop dependence on drugs (dependence vulnerability; DV) is 0.40 (Button et al., 2006). However, recent research has demonstrated that the extent to which genes affect both substance use and associated problems may be moderated by exposure to different environments. For example, urbanicity (Dick et al., 2001; Rose et al., 2001) and marital status (Dick et al., 2006) have both been found to affect the heritability of alcoholism, with higher heritability for alcohol use in urban (versus rural) communities and unmarried women. Therefore, previously reported heritabilities for substance use, abuse, and dependence probably represent an average heritability across a range of environmental backgrounds rather than genetic variance that is consistent across heterogeneous environments.
Such contextually dependent variation in genetic risk is sometimes referred to as genotype-environment interaction (GxE). It is important to note that the term genotype-environment interaction is also used to refer to situations in which the genotype confers susceptibility to an environmental risk (or vice versa). This type of GxE affects the mean scores of a trait, as demonstrated using adoption studies (Cadoret et al., 1995) and molecular techniques (Caspi et al., 2003). In contrast, the interactions discussed in the current paper refer to moderation of the variance and variance components of the traits. As a result, any conclusions drawn from one type of study are not directly applicable to the other. Furthermore, the term “E” as used here may not strictly refer to an environmental factor. Instead, it refers to anything that moderates the magnitude of variance components. This could be a shared environment effect, a non-shared environment effect, or even another phenotype. To avoid confusion with the GxE described above, and to incorporate the concept of a moderator variable that may or may not be truly environmental, we will use the term GxM to refer to contextual moderation of genetic risk, where M refers to the moderating variable. Similarly, CxM refers to contextual moderation of shared environmental risk, and ExM refers to contextual moderation of non-shared environmental risks.
1.2 Social mechanisms of moderation
This paper draws upon the paradigm developed by Shanahan and Hofer (2005), whose social control model anticipates that the genetic variance for particular behaviors will be reduced within social settings characterized by high levels of social control. In their words, “in circumstances marked by high levels of social control, a large percentage of the sample –irrespective of their genetic diversity- exhibits the same phenotype; in settings marked by low social control, people’s choices and behaviors are more apt to reflect their genotype.” Social control can be characterized by both institutional control and social network control. Institutional control involves the introduction of policies to reduced substance use by illegalizing substances, and introducing fines for use. For legal substances, controls include limits on the locations in which smoking or drinking is permitted, limits on the sale of these products, or taxes placed on tobacco or alcohol purchases. These policies influence mean levels of use (Kandel et al., 2004) but they may also be particularly effective in reducing both the variance as well as genetic tendencies to use and develop dependence on substances. As an example, Boardman (2008) shows that the heritability of daily smoking among adolescents is significantly attenuated within states that have the highest taxes per pack of cigarettes. Social network (meso-level) control emphasizes the norms, values, and sanctions developed and maintained by actors within a particular environment, i.e., the population itself develops norms and standards, rather than government or other institutions. Two previous studies have found support for the social network control model for the heritability of substance use. Kendler, Thornton, and Pedersen (2000) compared reported tobacco use among same-sex twin pairs across three birth cohorts (1910-1924; 1925-1939; and 1940-1958). According to their results, genetic factors account for fifty to sixty percent of the variance in regular tobacco use for men, regardless of birth cohort. Among women, however, they show that none of the variance is due to genetic factors in the early cohort but there is a consistent convergence in these estimates such that by the later cohort, there is no significant gender difference in the heritability of regular tobacco use. They argue that this is due to changes in the social restrictions on women’s tobacco use across these periods. As society became more permissive, it appears women were able to act in accordance with their own genetic predisposition, thereby increasing heritability. Timberlake et al. (2006) show that self-rated religiosity mutes the additive genetic component for smoking onset; they show an average heritability estimate of roughly sixty percent but this estimate drops to nearly zero among adolescents who report that religion is “more important than anything else” to them. In both cases, the heritability estimates are the smallest among those who are socialized in more controlled social environments with clearly established norms and corresponding sanctions against using substances. Low levels of PPD may also characterize environments with greater levels of social control because they are more stable, integrated, and supportive environments with greater resources to monitor adolescents’ behaviors effectively. Similar to other social groups, peer groups may also impose restrictions on members of the group, thereby regulating their behavior, and in a group made up of non-deviant individuals, such restrictions would likely act to ensure group members adhere to the same non-deviant values, thereby restricting behaviors such as drug use.
Putative environmental characteristics may also increase the relative influence of genes on the risk of substance use. According to the social expression model, latent (genetic) tendencies to use substances are most likely to differentiate between individuals within environments in which there are social pressures to use various substances; the social environment triggers genetic expression. Therefore, the genetic variance of substance use should increase with increasing prevalence of substance use in the population, decreased social sanctions against using substances, and increased expectations to use substances. Evidence for this perspective was demonstrated by Boardman and colleagues (2008), who show that the heritability of daily smoking is significantly higher in schools where the most popular students were also the heaviest smokers, compared to schools with less clear pro-smoking norms. Importantly, this same association was less apparent in schools with established anti-smoking norms. In other words, in this context, the social expression model was more relevant than the social control model for this particular outcome.
In some instances, genetic factors become more relevant within contexts in which the trait is rare. That is, according to the social distinction model, genetic vulnerability to substance use may manifest more clearly within environments in which smoking is uncommon. Support for this perspective is found in work by Button et al. (2005) who show that the genetic risks for antisocial behavior were the highest among adolescents from families with the lowest levels of family dysfunction. In other words, regardless of genetic risk, children from families with high levels of dysfunction exhibited a greater risk of antisocial behavior, but the defining feature of those exhibiting high levels of antisocial behavior from stable, integrating, and functional families may have been common genetic risk. Raine (2002) describes a variant of this model that is called the “social push perspective” in which it is claimed that contexts lacking social factors that either encourage or discourage drug use (i.e. benign environments) are the most relevant contexts in which to examine genetic associations. Therefore, we would expect greater genetic variance at the most normative levels of PPD in which drug use behavior is not likely to be either encouraged or discouraged against, and is therefore considered benign. Smaller genetic variances would be expected in both lower levels of PPD, in which drug use behavior is more likely to be discouraged against, and higher PPD, in which drug use behavior is more likely to be encouraged. However, unlike the social distinction model, which focuses on extremes of the “environmental” context, the social push model focuses on changes within the normal range of behavior. Diagrammatic representation of these different models can be seen in Figure 1.
Figure 1.
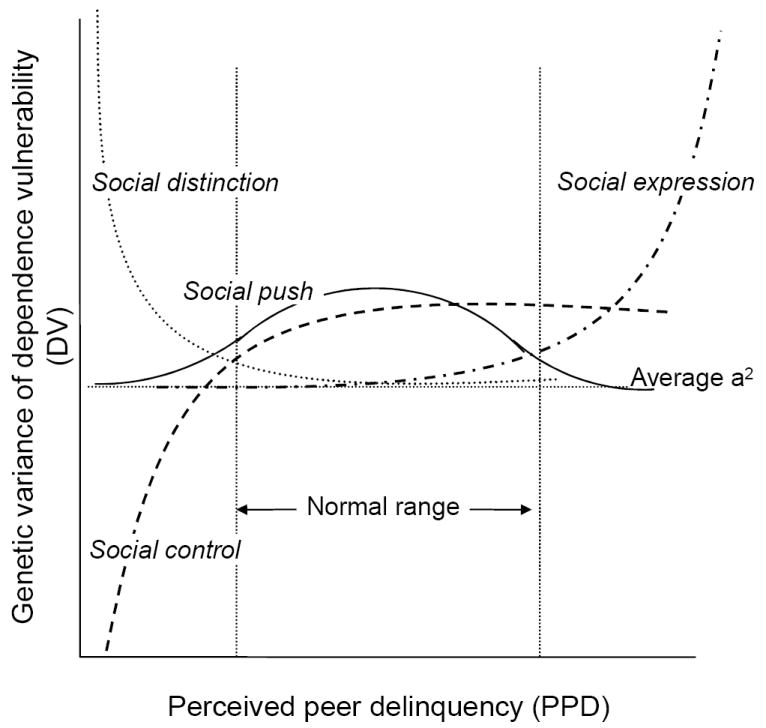
Diagrammatic representation of the theoretical models explaining changes in genetic variance of dependence vulnerability at different levels perceived peer delinquency.
These models need not be mutually exclusive; because moderation in these models is a function of the extreme range of perceived peer delinquency (PPD), it is possible that both social control and social expression forces are acting and the moderation function would be cubic in nature. This may also be the case if social distinction and social push were operating jointly. Similarly, both social distinction and social expression forces could be at play in which the association would be quadratic and positive (as opposed to quadratic and negative with the social control/social push model). Although social control here has been applied to substance use, its relevance to dependence is also intrinsic; given use is a pre-requisite for dependence. Very little research of this nature has been conducted in relation to substance dependence.
1.3 Gene-environment correlation
One problem associated with testing for environmental moderation of genetic risk is that many so-called environmental risks do not occur at random to people. People often seek out or create their environments, possibly based upon genetically predisposed characteristics. This is often referred to as genotype-environment correlation (rGE) (Plomin and Bergeman, 1991; Scarr and McCartney, 1983), although again, it may be inaccurate to assume that the environmental component is purely environmental. In biometrical analyses of moderation, it is often difficult to disentangle GxM from rGE. For example, the higher genetic variance for alcoholism in adults from urban communities than those from rural communities (Rose et al., 2001) may occur because urbanicity influences the expression of genetic risk (GxM), or because people who are genetically predisposed to alcoholism are more inclined to seek out environments in which alcohol is more readily available (rGE). Therefore, without controlling for the existence of rGE, one cannot attribute evidence for moderating effects to GxM.
1.4 Delinquent peers as a risk for substance dependence
One of the strongest predictors of substance use in adolescents is affiliation with delinquent peers (Ary et al., 1999; Dishion and Loeber, 1985; Duncan et al., 1998; Fergusson et al., 2002; Fergusson and Horwood, 1997), which correlates approximately 0.24 with alcohol use and 0.40 with marijuana use (Duncan et al., 1998). Furthermore, adolescents’ perceptions of their peers’ behavior may be an even more salient risk for their own behavior than the peers’ actual behavior (Iannotti and Bush, 1992). The nature of the association between substance use and delinquent peer affiliation in adolescents is unclear. The relationship may be causal, mediated by social learning, facilitation (e.g. providing the alcohol or the marijuana), and reinforcement (Fergusson and Horwood, 1996), or it may be due to common risk factors predisposing to both affiliation with delinquent peers and substance use problems. For example, there has been some evidence that genetic factors influence a person’s tendency to affiliate with deviant peers (Manke et al., 1995; Rowe and Osgood, 1984); possibly through selection or attraction of delinquent peers, and that the same genetic factors may also contribute to one’s own behavior (Rowe and Osgood, 1984). This is an example of active and evocative rGE. In reality, it is likely that causation, selection, and attraction all contribute to the correlation between affiliations with delinquent peers and substance use problems. We have previously demonstrated that affiliation with peers perceived to be delinquent moderates the genetic variance for conduct problems in adolescents, and both absolute genetic variance and heritability were greater at higher levels of exposure to delinquent peers (Button et al., 2007a). However, this is the first study to examine the mediating role of perceived peer delinquency on generalized substance dependence vulnerability.
The current study aimed to identify the extent to which the genetic variance of generalized dependence vulnerability in adolescents was dependent on the adolescents’ perceived levels of their peers’ delinquency while simultaneously controlling for factors common to both variables. Furthermore, we investigated how the etiology of the association between perceived peer delinquency and dependence vulnerability varied with changes in levels of peer delinquency.
2. Method
2.1 Participants
Participants were twin pairs from two community based twin samples participating in the NIDA-funded Center on Antisocial Drug Dependence (CADD; DA-11015): the Colorado Longitudinal Twin Sample (LTS, Emde and Hewitt, 2001; Rhea et al., 2006) and the Community Twin Sample (CTS) of the Colorado Twin Registry (CTR) (Young et al., 2002). The LTS twins were recruited through the Colorado Department of Health’s Division of Vital Statistics (Young et al., 2000) and were included in the CADD sample as they reached their 12th birthday. Twins from the CTR were recruited through the Department of Health and 170 (of 176 eligible) school districts in Colorado (Young et al., 2000). Written informed consent from parents or guardians of participants and written informed assent from the minor participants was obtained. The full twin sample comprised 1377 twin pairs (data for only 1 twin was available for 5 of these “pairs”), between 12 and 18 years of age (mean = 14.52, SD = 2.10). Of these, 657 were identical or monozygotic (MZ) pairs (362 female and 295 male) and 720 were fraternal or dizygotic (DZ) pairs (222 female, 223 male, and 275 opposite sex).
Zygosity for same sex twin pairs was determined using a 9-item assessment of physical characteristics completed by interviewers (Nichols and Bilbro, Jr., 1966), and by genotyping a minimum of 11 informative short tandem repeat polymorphisms (STRPs) using DNA obtained from buccal cells. Twin pairs with similar physical characteristics and concordant markers were categorized as MZ, and twin pairs with dissimilar physical characteristics and discordant markers were categorized as DZ. Discrepancies between the zygosity determination by interviewer ratings and genotyping were re-evaluated and resolved.
2.2 Measures
2.2.1 Dependence Vulnerability
Drug dependence was assessed using the Composite International Diagnostic Interview-Substance Abuse Module (CIDI-SAM: Cottler et al., 1989), a structured diagnostic interview that assesses DSM-IV abuse and dependence criteria for tobacco, alcohol, marijuana, cocaine, amphetamines, sedatives, inhalants, hallucinogens, opiates, and phencyclidine (PCP). For the purpose of these analyses, we examined nonspecific substance dependence (Stallings et al., 2003), given the evidence that use of multiple substances, rather than specializing in any single substance, is typical in adolescents, especially among those with substance dependence (Glantz and Leshner, 2000; Johnston et al., 2001; Young et al., 2002). Moreover, there is increasing empirical evidence that common genetic influences are responsible for the comorbidity across use of and dependence on different substances, and even across substance use and other externalizing problems (Button et al., 2006; Kendler et al., 2003; Krueger et al., 2002; Slutske et al., 1998; Stallings et al., 2003; Stallings et al., 2005; Young et al., 2000; Young et al., 2006). Stallings et al. (2003) considered 10 alternative phenotypes that might quantify an adolescent’s vulnerability to develop substance dependence. Of these 10 alternatives, an index of generalized, or non-specific, substance dependence vulnerability (DV) best met their criteria for a phenotype that was clinically valid, familial, and heritable. DV was calculated by taking a total lifetime symptom count of DSM dependence criteria endorsed across all classes of substances, and then dividing by the number of substances used. Use was defined by the CIDI-SAM as using almost daily for at least 30 days for tobacco, having six or more drinks during one’s lifetime for alcohol, and using more than five times during one’s lifetime for illegal drugs (Corley et al., 2001; Stallings et al., 2003). Participants who did not meet the definitions of use were assigned a DV score of zero.
2.2.2 Perceived Peer Delinquency
Perceived Peer Delinquency (PPD) was measured using the Exposure to Delinquent Peers Measure from Wave 6 of the National Youth Survey (Elliott, Huizinga, & Menard, 1989), with a single item (sold or given alcohol to kids under 18) excluded from our measure (as it was considered to have a different meaning for this age group). Predictive validity for this measure has been established (Elliott et al., 1989). The individual items from the PPD measure have a Cronbach’s α of 0.84, indicating good internal consistency. Respondents reported how many of their friends had participated in 13 delinquent behaviors, including alcohol and drug use, stealing, and violence, in the 6 months prior to the interview. Each item was scored according to whether none (1), very few (2), some (3), most (4), or all (5) of their friends engaged in each activity. Consequently, the PPD score provides information regarding the individuals’ perceptions of the relative proportion of their peers who have engaged in a variety of delinquent behaviors. A total PPD score was derived by multiplying the mean score of the individual items by the number of items, resulting in a maximum possible score of 65. This method ensures that the total peer scores for participants with missing item scores are not artificially reduced. Participants who responded to less than 60% of the individual peer items (8 items) were dropped from our analyses (132 individuals), as the mean from such few responses is unreliable (Elliott et al., 1989). The PPD measure was only included in our questionnaire after January 10, 1998; participants tested prior to this date did not have responses for the PPD measure. Consequently, a further 173 participants were excluded. Five participants were excluded because they claimed not to have any friends, and so were not questioned further on this subject.
2.3 Analyses
There were significant correlations between age and both DV (r = 0.366) and PPD (0.485). Therefore, prior to model-fitting analyses, both DV and PPD scores were regressed on age and age2 within each sex separately, and the residual scores were standardized. Because the scores were positively skewed, they were then log-transformed to approximate a normal distribution using the equation (y) = ln(2+x), where x is the original age corrected, standardized score, and (y) is the transformed score. The resulting scores were approximately normally distributed (DV: skew = 0.776, kurtosis = 2.812; PPD: = skew = -0.598, kurtosis = 2.180).
Twin data were analyzed with Mx (Neale, 2004) using structural equation modeling. This method compares the similarities between twins of different genetic relatedness to partition phenotypic variances (VP) into genetic (VA), shared environment (environment that makes members of a family more similar to one another, VC), and non-shared environment (environment that is uncorrelated among family members, VE) variance components. Identical, or monozygotic (MZ), twin pairs share 100% of their alleles identical by descent and the shared environment whereas fraternal, or dizygotic (DZ), twin pairs share on average 50% of their alleles identical by descent and the shared environment. Assuming equal shared environmental influences for MZ and DZ twins, if MZ twin pairs are more similar to one another for a phenotype than DZ twin pairs, there is evidence of a genetic contribution to the variance of that trait. If the MZ twin pairs are less than twice as similar as the DZ twin pairs, shared environment influence is implied; however, if the MZ twin pairs were at least twice as similar as DZ twin pairs it would indicate that the additive effects of genes are sufficient to explain their similarity. Finally, if the correlation between MZ twin pairs is less that 1, there must be an influence of non-shared environment on the phenotypic variance, since this is the only component that accounts for MZ twin pairs being different from one another. If MZ twin pairs correlation more than twice the DZ correlation, there is evidence of non-additive genetic effects, for instance dominance (interaction of genes at a single locus). However, in twins reared together the effects of shared environment and non-additive genetic effects are confounded and cannot be estimated simultaneously.
Many of the studies of contextual moderation of the variance components of substance use problems have examined dichotomous environmental variables (e.g. married vs. unmarried status, urban vs. rural environment). However, many identified risks for substance use are continuously distributed (e.g. family dysfunction, problem behavior). Therefore, we utilized a model that allows for a continuous moderator (Purcell, 2002) to examine the data for evidence of genotypic moderation in the presence of genotype-environment correlation. This model (presented in Figure 2) accounts for the covariance between PPD and DV by estimating both the magnitudes of genetic, shared environment, and non-shared environmental influences on the moderator (aM, cM, and eM, respectively in Figure 2) and the genetic, shared environment, and non-shared environmental influences shared by the two variables (aC, cC, and eC, respectively). A genetic component to PPD is evidence for a genetic contribution to a putative environmental risk. Furthermore, if this genetic risk is shared in common with DV, there is evidence that the same genes that predispose a person to affiliate with individuals they perceive as being delinquent also predispose one to substance dependence, thereby accounting for at least some of the correlation observed between them.
Figure 2.
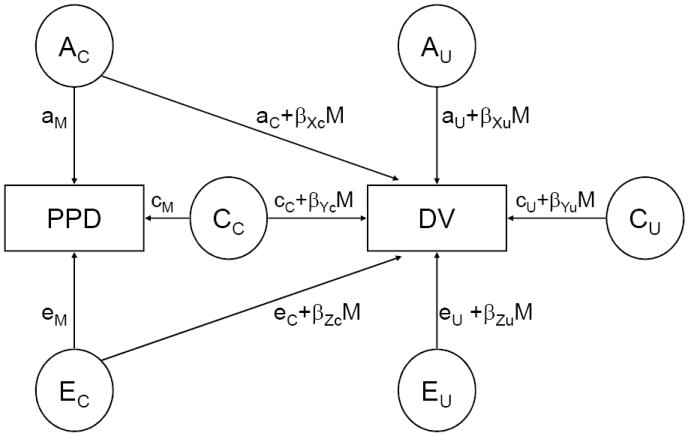
The moderation / correlation model.
PPD: Perceived Peer Delinquency; DV: Dependence Vulnerability; AC: genetic effects common to PPD and DV; CC: shared environment effects common to PPD and DV; EC: non-shared environment effects common to PPD and DV; AU: genetic effects unique to DV; CU: shared environment effects unique to DV; EU: non-shared environment effects unique to DV ; aM: influence of AC on PPD; cM: influence of CC on PPD; eM: influence of EC on PPD; aC: main effect of AC on DV; cC: main effect of CC on DV; eC: main effect of EC on DV; aU: main effect of AU on DV; cU: main effect of CU on DV; eU: main effect of EU on DV; βXc: interaction between PPD and aC; βYc: interaction between PPD and cC; βZc: interaction between PPD and eC; βXu: interaction between PPD and aU; βYu: interaction between PPD and cU; βZu: interaction between PPD and eU.
To test for a moderating effect of PPD on the variance components of DV (GxM), a moderation term is included on all pathways that contribute to the variance of DV (both those shared with PPD and those specific to DV). By including these moderation terms, each path can be expressed both in terms of a main effect of that component as well as also being dependent on levels of exposure to the moderator (M). Therefore, the genetic (ADV), shared environmental (CDV), and non-shared environmental (EDV) variances for DV are:
where the C and U subscript refers to common and unique effects, respectively.
The total variance of DV is potentially attributable to genetic, shared environment and non-shared environment influences in common with peer delinquency (aC, cC, eC), those influences unique to DV (aU, cU, eU), and the interaction of each of these pathways with the PPD moderator variable, represented by the path coefficients βxC, βxU, βyC, βyU, βzC, and βzU, respectively.
The significance of moderations was tested by first dropping all moderation terms, then testing all possible nested models. The fit of all models was assessed using Akaike’s Information Criterion (AIC; Akaike, 1987). AIC is derived as minus twice the log likelihood (-2ll) minus twice the degrees of freedom (df; AIC = -2ll-2df). The fit of nested models was assessed using both AIC and the chi-square (χ2) comparison test. The χ2 comparison test involves calculating the χ2 of the nested model as the difference between the -2ll’s of the full and the nested model. We also calculate the difference in degrees of freedom (calculated as the change in number of estimated parameters) between the full and nested models. A p < 0.05 indicates a significant deterioration in the fit of the nested model compared with the full model, indicating that the dropped parameters should remain in the model. If p > 0.05, we have evidence that the parameters can be dropped to create a more parsimonious model.
Moderation terms that could be dropped without a significant change in χ2 were dropped from the full model to produce the most parsimonious solution that adequately explained the data.
3. Results
3.1 Descriptive and univariate results
The PPD scores ranged from 13 to 58, with a mean PPD score of 18.24 and a standard deviation of 6.62. Results of a t-test demonstrated that males perceive their peers to be significantly more delinquent than females (male: mean = 18.58, S.D. = 6.61; female mean = 17.94, S.D. = 6.62; t = 2.378, p = 0.02). However, there were no significant sex differences in the magnitude of genetic and environmental influences on PPD. For both males and females the heritability of PPD was estimated at 0.21 (0.04-0.39), shared environment 0.40 (0.23-0.55) and non-shared environment 0.39 (0.34-0.44). Button et al. (2006) reported a heritability of 0.40, a shared environment estimate of 0.19, and a non-shared environment estimate of 0.41 for DV (i.e. assuming no rGE or GxM). However, the current study has been expanded to include 12-year-olds (the previous analysis only included those aged 13 years and older), resulting in slightly different standardized parameter estimates of 0.32 (0.15-0.50) for a2, 0.28 (0.11-0.53) for c2, and 0.41 (0.38-0.46) for e2. As there was insufficient power with this sample to detect differences in parameter estimates across sex for the DV and PPD variables, males and females were combined for the moderation analyses.
3.2 Genetic and environmental moderation
Results of model fitting are presented in Table 1. Dropping the interaction terms on the pathways shared in common by PPD and DV did not result in a significant worsening in the fit of the model, indicating that these components were not moderated by levels of PPD. The best fitting model was one in which there were no interaction terms on any of the common pathways. However, dropping the interaction terms on each of the unique pathways resulted in a significant deterioration in fit when compared with the full model in which all interactions were estimated freely, indicating that PPD does moderate the extent to which these components influence DV. Therefore, the magnitude of genetic, shared environment, and non-shared environment components specific to DV are moderated by levels of PPD.
Table 1.
Model fitting results for the gene-environment correlation and moderation model and nested models.
| -2LL | d.f. | A.I.C. | Δχ2 | Δd.f. | p | |
|---|---|---|---|---|---|---|
| Full model | 8157.337 | 4611 | -1064.663 | - | - | - |
| Drop βaC | 8158.189 | 4612 | -1065.811 | 0.852 | 1 | 0.356 |
| Drop βaU | 8163.715 | 4612 | -1060.285 | 6.378 | 1 | 0.012 |
| Drop βcC | 8158.848 | 4612 | -1065.152 | 1.511 | 1 | 0.219 |
| Drop βcU | 8161.927 | 4612 | -1062.073 | 4.590 | 1 | 0.032 |
| Drop βeC | 8158.512 | 4612 | -1065.488 | 1.176 | 1 | 0.287 |
| Drop βeU | 8322.088 | 4612 | -901.912 | 164.752 | 1 | 0.000 |
| Best-fitting* | 8160.716 | 4614 | -1067.284 | 3.380 | 3 | 0.337 |
-2LL: minus twice the log likelihood fit of the model; d.f.: degrees of freedom; A.I.C.: Akaike information criterion (calculated as -2LL minus 2d.f.); Δχ2: the chi-square difference between the full and nested models; p: probability
Best fitting model is one which βaC, βcC, and βeC have all been dropped
Although some moderating effects could be dropped, indicating a non-significant effect, this may be the result of lack of power at the extremes of the moderator, and results from the full model should be presented (Purcell, 2002). Therefore, we present results derived from the full model only (see Table 2). Changes in absolute variance estimates (based on the full model) for the genetic, shared, and non-shared environmental influences on DV with increasing PPD score, along with confidence intervals are presented in Figure 3. The number of participants (N) at each level of PPD is provided on the right-side vertical axes to provide additional information regarding power (i.e. there is lower power at the extremes of PPD, where the N is lower). As seen in Figure 3a, the ADV was highest among adolescents with low PPD, decreased at more common levels of PPD, and increased slightly among adolescents with higher PPD. Figures 3b and 3c show the change in shared environmental (CDV) and non-shared environmental (EDV) variance respectively, as levels of PPD increase. Both shared environmental and non-shared environmental variance increase with increasing PPD
Table 2.
Path and interaction coefficients for the full moderation/interaction model
| Genetic | Shared Environment | Non-Shared Environment | |||
|---|---|---|---|---|---|
| Parameter | Estimate (95% CI) | Parameter | Estimate (95% CI) | Parameter | Estimate (95% CI) |
| aM | 0.28 (0.21-0.34) | cM | 0.24 (0.16-0.30) | eM | 0.30 (0.29-0.32) |
| aC | 0.47 (0.28-0.69) | cC | 0.28 (0.09-0.48) | eC | 0.02 (-0.03-0.08) |
| aU | -0.54 (-0.66--0.26) | cU | 0.34 (0.15-0.39) | eU | 0.32 (0.30-0.35) |
| βaC | -0.12 (-0.41- 0.14) | βcC | 0.1 5 (-0.09-0.39) | βeC | 0.05 (-0.04-0.13) |
| βaU | 0.87 (0.66-1.05) | βcU | 0.24 (0.02-0.45) | βeU | 0.33 (0.31-0.48) |
PPD: Perceived Peer Delinquency; DV: Dependence Vulnerability; aM: influence of AC on PPD; cM: influence of CC on PPD; eM: influence of EC on PPD; aC: main effect of AC on DV; cC: main effect of CC on DV; eC: main effect of EC on DV; aU: main effect of AU on DV; cU: main effect of CU on DV; eU: main effect of EU on DV; βXc: interaction between PPD and aC; βYc: interaction between PPD and cC; βZc: interaction between PPD and eC; βXu: interaction between PPD and aU; βYu: interaction between PPD and cU; βZu: interaction between PPD and eU.
Figure 3.

A series of graphs showing the change in the absolute values of genetic variance (a), shared environmental variance (b), and non-shared environmental variance (c) for dependence vulnerability conditional on levels of perceived peer delinquency, derived from the full model. Estimates are represented by the black line. The grey lines either side of the black line are the confidence intervals. The bars represent the number of people at each level of PPD. PPD scores are log-transformed z-scores.
3.3 Results of a follow-up analysis
To assess the reliability of the results obtained from the complex model used here, we also stratified the sample into 3 equal categories according to levels of PPD (Low, Moderate, and High), and estimated the total variance, as well as ADV, CDV, and EDV for DV in each PPD category independent of the genetic moderation model. For this analysis, twin pairs were included if they scored in different categories, e.g. one scored low and the co-twin scored high, by using a model that accounts for this by utilizing the ‘definitional variable’ options in Mx. Consistent with the findings reported for the full model described above, we found that the total variance was high in the low PPD group (0.74), decreased in the moderate PPD group (0.50), and increased in the high PPD group (1.49). Furthermore, estimates of ADV, CDV, and EDV showed similar patterns to those reported for the full model described above. The ADV was highest among adolescents with low PPD, low among those with moderate PPD and increased among adolescents with high PPD (low PPD: 0.63; moderate PPD: 0.10; high PPD: 0.50). The CDV initially increased from the low to moderate PPD group, but decreased in those with the highest levels of PPD (low PPD: 0.00; moderate PPD: 0.36; high PPD: 0.06). Finally, the EDV showed a general increase with increasing PPD (low PPD: 0.11; moderate PPD: 0.04; high PPD: 0.93). The consistency between this traditional method of testing for moderations and the results described above provide further evidence for a moderating effect of PPD on the variance components for DV as well as demonstrating the robustness of complex model for this type of analysis.
3.4 Moderation of the covariance between PPD and DV
In addition, we estimated the extent to which the genetic, shared environmental, and non-shared environmental influences shared in common by DV and PPD differed as a function of PPD. The results from model fitting provide evidence that the common influences were not significantly moderated by PPD, as indicated by the small and non-significant moderation on the common genetic pathway.
4. Discussion
The primary goal of the current study was to investigate whether the magnitude of the variance components for dependence vulnerability varied as a function of perceived peer delinquency. In terms of the models proposed in the introduction it appears that social distinction is important in adolescents with low PPD scores, as the genetic factors are more salient when perceptions of peers’ delinquency is very low. Conversely, social expression appears to play a role in those with extremely high PPD scores, as the genetic variance is smaller at normative levels of PPD, and increases as adolescents perceive their peers to be more delinquent. In both instances, little change in the genetic variance over the normal range would be expected, and this is evident from the current results. Therefore, a combination of the social distinction and social expression models appears to explain the pattern of results seen here. The mechanisms behind this are unknown but can be speculated about. For instance, in those adolescents who have non-delinquent peers, we would anticipate that the adolescents must have some other predisposition, i.e. a genetic risk for substance use. In other words, individuals who do not perceive their peers to be delinquent are less likely to use and develop dependence to drugs because of their peers’ behavior, for example, by peer encouragement, or social learning etc., and thus are more likely to display such vulnerability because of an underlying genetic predisposition. At the other extreme, the substance use among delinquent peers is likely to be rewarded and reinforced by environmental influences (hence the expression), but only after a certain point. This conclusion implies that the adolescents already have a genetic vulnerability; however, they only act upon this propensity in environments that do encourage negative behavior. This could happen for a number of reasons. For example, delinquent peers might be more likely to provide illicit substances, leading the vulnerable person to be more inclined to become dependent, whereas a person with fewer delinquent peers may be less likely to be offered illicit substances, and therefore, their genetic risk remains un-expressed. Thus, adolescents exposed to those with behavioral problems are more likely to have any underlying genetic risk for associated behaviors triggered. Our second aim was to examine how the association between PPD and DV changed with levels of PPD. Consistent with previous studies, there was a strong positive correlation between PPD and DV, demonstrating that exposure to peers one perceives to be delinquent does increase one’s risk for substance dependence in both male and female adolescents. The results also provide evidence that there is a significant genetic covariation between PPD and DV, and this covariation is not dependent on the levels of PPD. Therefore, the same genetic liability, and possibly genes, that drive the adolescents own vulnerability to substance dependence also contributed to either their choice of peers (selecting and attracting peers with similar characteristics to their own), their perception of their peers (those who are predisposed to substance dependence may also perceive their peers to behave badly, irrespective of their peers actual behavior), or even directly affecting the behavior of their peers (those who are genetically predisposed to negative behaviors may also encourage such behavior in their peers).
The findings from this study contribute further to the growing literature demonstrating both that exposure to certain “environmental” variables is not random, but is in part influenced by one’s own genotype, and that the “environment” may act to moderate the genetic and environmental etiology of a phenotype. Furthermore, these findings demonstrate that failing to consider either correlations or moderations of the kind described here may result in failure to fully understand the mechanisms by which the genes and the environment influence the phenotype.
A number of implications can be drawn from this study. For example, the genetic correlation between PPD and DV indicates that an adolescent’s desire to affiliate with deviant peers is indicative of a potential risk for use and dependence. Furthermore, results of studies such as these can aid in the identification of environments to select participants for molecular studies. For example, the current study showed that genetic variance is greater among individuals with fewer delinquent peers, and those with extremely high levels of PPD. Thus, the phenotypic variance explained by each contributing QTL might increase in populations at either extreme and thus be easier to detect.
4.1 Limitations
These results need to be considered within the context of a number of methodological limitations. First, the cross sectional nature of this sample makes it difficult to evaluate the possible causal relationship between PPD and DV. Although it is possible to test causal relationships between 2 variables, it is often difficult to distinguish the correct model among alternative causal models using a cross-sectional dataset. Moreover, for the purposes of this analysis, where both causation and correlation are likely to both play a role, such causal analyses would not prove valuable. Furthermore, in this analysis, we only considered the role of PPD as a moderator and DV as the phenotypic outcome. This was the logical order, as affiliating with delinquent peers can provide an environmental background within which genetic and environmental risk might vary, whereas one’s own behavior (i.e. DV) seems to be the logical outcome. However, this may not be the only scenario; it is also be possible that levels of DV moderate the magnitude of genetic, shared environmental, and non-shared environmental variance of PPD.
A further limitation to this analysis is the use of substance dependence without consideration of substance use. Since the development of dependence requires both initiation of drug use, and regular use, both of which are heritable, it is possible that some GxM effects are taking place at the initiation and regular use level. To differentiate between these is beyond the scope of the current paper; however, it is a potential area for future research.
Finally, Eaves (2006) demonstrated that evidence of interactions between genes and “environments” can be an artifact of the scale used, rather than providing evidence of a true interaction between persons and environments, even when genotypes and environments are directly assessed. The same caveat might apply to studies of moderation, and thus we must accept that the GxM found here, while controlling for the confounding effects of rGE, may be also be subject to the same limitations and therefore specific to the measures used, thus any conclusions drawn from these results need to be treated with caution.
4.2 Summary
Despite the caveats described here, this study provides further evidence that the genetic risk for a phenotype, in this case for vulnerability to display dependence towards multiple substances, is contextually dependant. However, the level of genetic risk between DV and PPD was not dependent on levels of PPD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akaike H. Factor analysis and AIC. Psychometrika. 1987;52:317–332. [Google Scholar]
- Ary DV, Duncan TE, Duncan SC, Hops H. Adolescent problem behavior: the influence of parents and peers. Behav Res Ther. 1999;37:217–230. doi: 10.1016/s0005-7967(98)00133-8. [DOI] [PubMed] [Google Scholar]
- Boardman Jason D. State-level moderation of genetic tendencies to smoke. American Journal of Public Health. 2009:99. doi: 10.2105/AJPH.2008.134932. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Saint Onge JM, Haberstick BC, Timberlake DS, Hewitt JK. Do schools moderate the genetic determinants of smoking? Behav Genet. 2008;38:234–46. doi: 10.1007/s10519-008-9197-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Button TM, Corley RP, Rhee SH, Hewitt JK, Young SE, Stallings MC. Delinquent peer affiliation and conduct problems: A twin study. J Abnorm Psychol. 2007a;116:554–564. doi: 10.1037/0021-843X.116.3.554. [DOI] [PubMed] [Google Scholar]
- Button TM, Hewitt JK, Rhee SH, Young SE, Corley RP, Stallings MC. Examination of the causes of covariation between conduct disorder symptoms and vulnerability to drug dependence. Twin Res Hum Genet. 2006;9:38–45. doi: 10.1375/183242706776402993. [DOI] [PubMed] [Google Scholar]
- Button TM, Rhee SH, Hewitt JK, Young SE, Corley RP, Stallings MC. The role of conduct disorder in explaining the comorbidity between alcohol and illicit drug dependence in adolescence. Drug Alcohol Depend. 2007b;87:46–53. doi: 10.1016/j.drugalcdep.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Button TMM, Scourfield J, Martin N, Purcell S, McGuffin P. Family Dysfunction Interacts with Genes in the Causation of Antisocial Symptoms. Behav Genet. 2005;35:115–120. doi: 10.1007/s10519-004-0826-y. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Yates WR, Troughton E, Woodworth G, Stewart MA. Genetic-environmental interaction in the genesis of aggressivity and conduct disorders. Arch Gen Psychiatry. 1995;52:916–924. doi: 10.1001/archpsyc.1995.03950230030006. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Corley RP, Hewitt JK, Stallings MC, Young SE. Using unselected community samples to derive optimal multiple substance dependence phenotypes for genetic analysis. Drug Alcohol Depend. 2001;63:S32–S33. [Google Scholar]
- Cottler LB, Robins LN, Helzer JE. The reliability of the CIDI-SAM: a comprehensive substance abuse interview. Br J Addict. 1989;84:801–814. doi: 10.1111/j.1360-0443.1989.tb03060.x. [DOI] [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Schuckit MA, Bierut L, Hinrichs A, Fox L, Mullaney J, Cloninger CR, Hesselbrock V, Nurnberger JI, Jr, Almasy L, Foroud T, Porjesz B, Edenberg H, Begleiter H. Marital status, alcohol dependence, and GABRA2: evidence for gene-environment correlation and interaction. J Stud Alcohol. 2006;67:185–194. doi: 10.15288/jsa.2006.67.185. [DOI] [PubMed] [Google Scholar]
- Dick DM, Bierut LJ. The genetics of alcohol dependence. Curr Psychiatry Rep. 2006;8:151–157. doi: 10.1007/s11920-006-0015-1. [DOI] [PubMed] [Google Scholar]
- Dick DM, Rose RJ, Viken RJ, Kaprio J, Koskenvuo M. Exploring gene-environment interactions: socioregional moderation of alcohol use. J Abnorm Psychol. 2001;110:625–632. doi: 10.1037//0021-843x.110.4.625. [DOI] [PubMed] [Google Scholar]
- Dishion TJ, Loeber R. Adolescent marijuana and alcohol use: the role of parents and peers revisited. Am J Drug Alcohol Abuse. 1985;11:11–25. doi: 10.3109/00952998509016846. [DOI] [PubMed] [Google Scholar]
- Duncan SC, Duncan TE, Biglan A, Ary D. Contributions of the social context to the development of adolescent substance use: a multivariate latent growth modeling approach. Drug Alcohol Depend. 1998;50:57–71. doi: 10.1016/s0376-8716(98)00006-4. [DOI] [PubMed] [Google Scholar]
- Eaves LJ. Genotype x environment interaction in psychopathology: Fact or artifact? Twin Res Hum Genet. 2006;9:1–8. doi: 10.1375/183242706776403073. [DOI] [PubMed] [Google Scholar]
- Elliott DS, Huizinga D, Menard S. Multiple Problem Youth: Delinquency. Drugs and Mental Health Problems Springer; New York, NY: 1989. [Google Scholar]
- Emde RN, Hewitt JK. Infancy to early childhood: Genetic and environmental influences on developmental change. Oxford University Press; Oxford: 2001. [Google Scholar]
- Fergusson DM, Horwood LJ. The role of adolescent peer affiliations in the continuity between childhood behavioral adjustment and juvenile offending. J Abnorm Child Psychol. 1996;24:205–221. doi: 10.1007/BF01441485. [DOI] [PubMed] [Google Scholar]
- Fergusson DM, Horwood LJ. Early onset cannabis use and psychosocial adjustment in young adults. Addiction. 1997;92:279–296. [PubMed] [Google Scholar]
- Fergusson DM, Swain-Campbell NR, Horwood LJ. Deviant peer affiliations, crime and substance use: a fixed effects regression analysis. J Abnorm Child Psychol. 2002;30:419–430. doi: 10.1023/a:1015774125952. [DOI] [PubMed] [Google Scholar]
- Glantz MD, Leshner AI. Drug abuse and developmental psychopathology. Dev Psychopath. 2000;12:795–814. doi: 10.1017/s0954579400004120. [DOI] [PubMed] [Google Scholar]
- Heath AC, Bucholz KK, Madden PA, Dinwiddie SH, Slutske WS, Bierut LJ, Statham DJ, Dunne MP, Whitfield JB, Martin NG. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol Med. 1997;27:1381–1396. doi: 10.1017/s0033291797005643. [DOI] [PubMed] [Google Scholar]
- Iannotti RJ, Bush PJ. Perceived vs. actual friends’ use of alcohol, cigarettes, marijuana, and cocaine: Which has the most influence? Journal of Youth and Adolescence. 1992;21:375–389. doi: 10.1007/BF01537024. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG. The Monitoring the Future national survey results on adolescent drug use: Overview of key findings, 2000. NIH Publication No. 01-4923. National Institute on Drug Abuse; Bethesda, MD: 2001. [Google Scholar]
- Kandel D, Chen K, Warner LA, Kessler RC, Grant B. Prevalence and demographic correlates of symptoms of last year dependence on alcohol, nicotine, marijuana and cocaine in the U.S. population. Drug Alcohol Depend. 1997a;44:11–29. doi: 10.1016/s0376-8716(96)01315-4. [DOI] [PubMed] [Google Scholar]
- Kandel DB, Johnson JG, Bird HR, Canino G, Goodman SH, Lahey BB, Regier DA, Schwab-Stone M. Psychiatric disorders associated with substance use among children and adolescents: findings from the Methods for the Epidemiology of Child and Adolescent Mental Disorders (MECA) Study. J Abnorm Child Psychol. 1997b;25:121–132. doi: 10.1023/a:1025779412167. [DOI] [PubMed] [Google Scholar]
- Kandel DB, Kiros GE, Schaffran C, Hu MC. Racial/ethnic differences in cigarette smoking initiation and progression to daily smoking: a multilevel analysis. Am J Public Health. 2004;94:128–135. doi: 10.2105/ajph.94.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am J Psychiatry. 2003;160:687–695. doi: 10.1176/appi.ajp.160.4.687. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Thornton LM, Pedersen NL. Tobacco consumption in Swedish twins reared apart and reared together. Arch Gen Psychiatry. 2000;57:886–892. doi: 10.1001/archpsyc.57.9.886. [DOI] [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WG, McGue M. Etiologic connections among substance dependence, antisocial behavior, and personality: modeling the externalizing spectrum. J Abnorm Psychol. 2002;111:411–424. [PubMed] [Google Scholar]
- Manke B, McGuire S, Reiss D, Hetherington EM, Plomin R. Genetic contributions to adolescents’ extrafamilial social interactions: Teachers, best friends, and peers. Social Development. 1995;4:238–256. [Google Scholar]
- McGue M, Iacono WG. The association of early adolescent problem behavior with adult psychopathology. Am J Psychiatry. 2005;162:1118–1124. doi: 10.1176/appi.ajp.162.6.1118. [DOI] [PubMed] [Google Scholar]
- Neale MC. Mx: Statistical modeling. 6. Richmond VA, Box 980126: MCV; 2004. [Google Scholar]
- Nichols RC, Bilbro WC., Jr The diagnosis of twin zygosity. Acta Genet Stat Med. 1966;16:265–275. doi: 10.1159/000151973. [DOI] [PubMed] [Google Scholar]
- Office of National Drug Control Policy. The economic cost of drug abuse in the United States, 1992-1998. NCJ-190636. Washington, DC: Executive Office of the President; 2001. [Google Scholar]
- Plomin R, Bergeman CS. The nature of nurture: Genetic influences on “environmental” measures. Behav Brain Sci. 1991;14:373–427. [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Res. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Raine A. Biosocial studies of antisocial and violent behavior in children and adults: A review. J Abnorm Child Psychol. 2002;30:311–326. doi: 10.1023/a:1015754122318. [DOI] [PubMed] [Google Scholar]
- Rhea SA, Gross AA, Haberstick BC, Corley RP. Colorado twin registry. Twin Res Hum Genet. 2006;9:941–949. doi: 10.1375/183242706779462895. [DOI] [PubMed] [Google Scholar]
- Rhee SH, Hewitt JK, Young SE, Corley RP, Crowley TJ, Stallings MC. Genetic and environmental influences on substance initiation, use, and problem use in adolescents. Arch Gen Psychiatry. 2003;60:1256–1264. doi: 10.1001/archpsyc.60.12.1256. [DOI] [PubMed] [Google Scholar]
- Rose RJ, Dick DM, Viken And RJ, Kaprio J. Gene-environment interaction in patterns of adolescent drinking: regional residency moderates longitudinal influences on alcohol use. Alcohol Clin Exp Res. 2001;25:637–643. [PubMed] [Google Scholar]
- Rowe DC, Osgood DW. Heredity and sociological theories of development: A reconsideration. Am Soc Rev. 1984;49:526–540. [Google Scholar]
- Scarr S, McCartney K. How people make their own environments: a theory of genotype greater than environment effects. Child Dev. 1983;54:424–435. doi: 10.1111/j.1467-8624.1983.tb03884.x. [DOI] [PubMed] [Google Scholar]
- Shanahan MJ, Hofer SM. Social context in gene-environment interactions: retrospect and prospect. Journals of Gerontology Series B: Psychological Sciences and Social Sciences. 2005;60(Spec No 1):65–76. doi: 10.1093/geronb/60.special_issue_1.65. [DOI] [PubMed] [Google Scholar]
- Slutske WS, Heath AC, Dinwiddie SH, Madden PA, Bucholz KK, Dunne MP, Statham DJ, Martin NG. Common genetic risk factors for conduct disorder and alcohol dependence. J Abnorm Psychol. 1998;107:363–374. doi: 10.1037//0021-843x.107.3.363. [DOI] [PubMed] [Google Scholar]
- Stallings MC, Corley RP, Dennehey B, Hewitt JK, Krauter KS, Lessem JM, Mikulich-Gilbertson SK, Rhee SH, Smolen A, Young SE, Crowley TJ. A genome-wide search for quantitative trait Loci that influence antisocial drug dependence in adolescence. Arch Gen Psychiatry. 2005;62:1042–1051. doi: 10.1001/archpsyc.62.9.1042. [DOI] [PubMed] [Google Scholar]
- Stallings MC, Corley RP, Hewitt JK, Krauter KS, Lessem JM, Mikulich SK, Rhee SH, Smolen A, Young SE, Crowley TJ. A genome-wide search for quantitative trait loci influencing substance dependence vulnerability in adolescence. Drug Alcohol Depend. 2003;70:295–307. doi: 10.1016/s0376-8716(03)00031-0. [DOI] [PubMed] [Google Scholar]
- Timberlake DS, Rhee SH, Haberstick BC, Hopfer C, Ehringer M, Lessem JM, Smolen A, Hewitt JK. The moderating effects of religiosity on the genetic and environmental determinants of smoking initiation. Nicotine Tob Res. 2006;8:123–133. doi: 10.1080/14622200500432054. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Lyons MJ, Eisen SA, Goldberg J, True W, Lin N, Meyer JM, Toomey R, Faraone SV, Eaves LJ. Genetic influences on DSM-III-R drug abuse and dependence: a study of 3 372 twin pairs. Am J Med Genet. 1996;67:473–477. doi: 10.1002/(SICI)1096-8628(19960920)67:5<473::AID-AJMG6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Young SE, Corley RP, Stallings MC, Rhee SH, Crowley TJ, Hewitt JK. Substance use, abuse and dependence in adolescence: prevalence, symptom profiles and correlates. Drug Alcohol Depend. 2002;68:309–322. doi: 10.1016/s0376-8716(02)00225-9. [DOI] [PubMed] [Google Scholar]
- Young SE, Rhee SH, Stallings MC, Corley RP, Hewitt JK. Genetic and environmental vulnerabilities underlying adolescent substance use and problem use: general or specific? Behav Genet. 2006;36:603–615. doi: 10.1007/s10519-006-9066-7. [DOI] [PubMed] [Google Scholar]
- Young SE, Stallings MC, Corley RP, Krauter KS, Hewitt JK. Genetic and environmental influences on behavioral disinhibition. Am J Med Genet. 2000;96:684–695. [PubMed] [Google Scholar]