

Abstract
Mice lacking hepatocyte IKKβ (IkkβΔhep) are defective in TNFα-activation of hepatocellular transcription factor NF-κB, and highly susceptible to hepatotoxicity. Following diethylnitrosamine (DEN) exposure, IkkβΔhep mice develop more hepatocellular carcinoma (HCC) than control mice due partly to enhanced DEN-induced hepatocyte death. Here we show that IkkβΔhep hepatocytes display growth advantages over normal hepatocytes consisting of precocious PCNA and cyclin D1 expression during liver regeneration (shortened hepatocyte G0 → G1 transitions), and enhanced recovery efficiency, cyclin D1 expression and cell proliferation after plating. Ex vivo deletion of Ikkβ also accelerates hepatocyte growth. IkkβΔhep hepatocyte proliferative responses show heightened sensitivity to TGFα and TNFα, and heightened expression of fibronectin, collagens I/III, nidogen, β-actin and integrin β1 mRNAs. These findings suggest that altered mitogen signaling and expression of extracellular matrix and its associated components underlie growth advantages. Increased HCC development in IkkβΔhep mice may also be caused by growth advantages of surviving Ikkβ-deleted hepatocytes.
Keywords: Growth-suppressor, Precocious S-phase onset, Mitogen sensitivity, Extracellular matrix
IκB kinase β (IKKβ) is required for activation of NF-κB [1,2], a transcription factor that regulates liver inflammation and protection from injury [2,3]. Although its liver growth-controlling role is controversial [4], current evidence suggests that NF-κB activation is unnecessary for partial hepatectomy(PH)-induced liver regeneration [5–7]. Our results also suggested slightly accelerated regeneration in IkkβΔhep mice which carry Cre-mediated deletions of hepatocyte Ikkβ exon 3 [7]. In correlation with defective NF-κB activation and failed survival gene induction, IkkβΔhep mice also show enhanced susceptibility to concanavalin-A(ConA)-induced liver failure [6] and to diethylnitrosamine(DEN)-induced hepatocellular carcinoma (HCC [7]).
In trying to understand the basis for the enhanced HCC susceptibility, we found that IkkβΔhep mice showed increased liver cell death and cytokine-driven compensatory hepatocyte proliferation [7]. Indeed, further work with IkkβΔhep mice showed that HCC-development was partly due to IL-1α, released from damaged hepatocytes, which induced Kupffer cell IL-6-expression and activated IL-1 receptors [8–10]. Here, we extend these investigations by comparing growth properties of hepatocytes cultured from IkkβΔhep and IkkβF/F mice. We find that Ikkβ deletion confers direct growth advantages to hepatocytes, which consist of higher recovery efficiency (R.E.) and enhanced cell proliferation, possibly related to increased mitogen sensitivities and to augmented extracellular matrix (ECM), basement membrane, cytoskeletal, and integrin mRNA expression. Both advantages implicate a growth-suppressor role of IKKβ, and should be considered along with the cell death hypothesis under conditions of induced hepatotoxicity and hepatocarcinogenesis.
Materials and methods
Animals
IkkβF/F and IkkβF/F: Alb-Cre (referred to as IkkβΔhep) mice were maintained per NIH guidelines [6]. Homogeneous backgrounds were obtained by backcrossing IkkβF/F with C57BL/6J mice for ⩾ 8 generations. IkkβF/F: Alb-Cre ♂ mice were bred with IkkβF/F ♀ mice to generate IkkβF/F and IkkβΔhep ♂ littermates; these and C57BL/6J mice were used at 6–8 weeks.
Liver regeneration and tissue processing
Seventy percent PH and sham hepatectomy, and liver analyses (histology; DNA, RNA, and proteins) were described elsewhere [7]. Apoptotic cells were quantified by TUNEL assays [7].
Genotyping, deletion analysis, PCR and real time Q-PCR
Cre and F/F alleles, and Ikkβ deletions were assessed by PCR assays of tail, liver and cultured hepatocyte DNA [6,7]. Q-PCR was performed on liver, and freshly isolated and cultured hepatocyte RNA (Suppl. Table 1 lists primers); results were normalized to glyceral-dehyde phosphate dehydrogenase mRNA levels [11].
Primary culture
Hepatocytes were isolated by intraportal perfusion using a 23G-needle with 5 ml digestion buffer containing 0.5 mg Sigma Type I collagenase/ml [12; Appendix A]. Cells were plated (N0 = initial cell concentrations) into uncoated plastic 35 mm-diameter tissue culture dishes or 10 mm-diameter 24-multiwell plates (NUNC) containing 2 ml or 1 ml plating medium [12], respectively. Human TGFα and mouse TNFα (R&D) were aliquoted into serum-free medium and stored at −20 °C until use. Attached cells were trypsinized from cultures washed 3× with PBS and Coulter-counted.
Adenoviruses
Ad-CMV-Cre adenovirus (Vector Biolabs) was amplified in HEK293 cells. Ad-CMV-GFP was constructed by inserting GFP cDNA into a pshuttle-CMV transfer vector (Stratagene); GFP-expressing adenovirus was obtained via homologous recombination between pshuttle-CMV-GFP and pAdEasy-1 (Stratagene). Titers were obtained using HEK293 cells [13]. Hepatocytes were infected at a multiplicity of infection = 10.
BrdU L.I., immunofluorescence and Western blots
L.I. measurements were made in cultures exposed 18 h to 10 μM BrdU, followed by washing, overnight fixation in 4% formalin and re-washing. Immunofluorescence staining (BrdU = red, albumin = green) was performed using 1°-rabbit anti-BrdU and goat anti-albumin antibodies (Megabase), and 2°-donkey Alexa 594-conjugated anti-rabbit- and chick Alexa 488-conjugated anti-goat antibodies (Invitrogen), respectively; no staining occurred in the absence of 1°- or 2°-antibodies. To circumvent problems arising from heterogeneously distributed cells, ⩾ 2000 hepatocytes, visualized microscopically using a dual red–green filter (Chroma Technology), were scored over similar evenly-spaced sites/dish. Photomicrographs (Ni-kon DXM1200C camera) were archived using Nikon software. Western blots were performed using anti-PCNA and cyclin D1 (PC10 and M-20, Santa Cruz sc-56 and 718), IKKβ (10AG2, Imgenex IMG129A) and anti-actin (Sigma A4700) antibodies [7].
Statistical analysis
Data were expressed as means ± S.E. Significance (P ≤ 0.05) was judged by 1-way ANOVA or Student’s t-tests. Areas under curves (AUC) were calculated with SigmaPlot v.7 software.
Results
Targeted Ikkβ deletion accelerates PCNA and cyclin D1 expression and shortens hepatocyte G0 → G1 transitions during liver regeneration after PH
G0 → G1 transitions were shortened after 70% PH in IkkβΔhep mice (Fig. 1A), as shown by elevated and sustained levels of PCNA at 24–60 h, and cyclin D1 at 24–48 h. Similar changes, absent after sham hepatectomy, did not occur until 36 h in control IkkβF/F mice, in which cyclin D1 levels oscillated and peaked between 36 and 60 h (Fig. 1A).
Fig. 1.

IkkβΔhep liver regeneration is accelerated after 70% PH. (A) Precocious PCNA and cyclin D1 expression. Liver lysates were gel separated and immunoblotted. (B) Elevated hepatocyte M.I. Hemotoxylin-and-eosin stained liver sections 48 h post-PH (200×): mitotic hepatocytes (→). (C) Apoptotic IkkβF/F and IkkβΔhep hepatocytes quantified by TUNEL assays (N = 4).
Intergenotypic differences (Fig. 1A) agreed with published hepatocyte BrdU L.I. values: S-phase onset times, St [14], were shortened in IkkβΔhep mice, as shown by a 200-fold higher BrdU L.I. (10%) compared to controls (0.05%) at 24 h (P < 0.0001); BrdU L.I. levels peaked normally in both groups [15] by 36 h, and BrdU-staining occurred only in hepatocytes [7]. IkkβΔhep mitotic indexes (M.I.) were higher than IkkβF/F controls by ~40% at peak mitotic times (48 h; Fig. 1B); however, apoptotic hepatocyte fractions were equivalent in both strains at 0–36 h (Fig. 1C). α1-Fetoprotein positive oval-like cells (not shown), inflammatory cells or necrotic hepatocytes were not observed (Fig. 1B). Thus, Ikkβ deletion enhanced hepatocyte proliferation during liver regeneration without affecting apoptosis.
Differences between plating and growth properties of IkkβF/F and Ikkβhep hepatocytes are revealed in primary culture
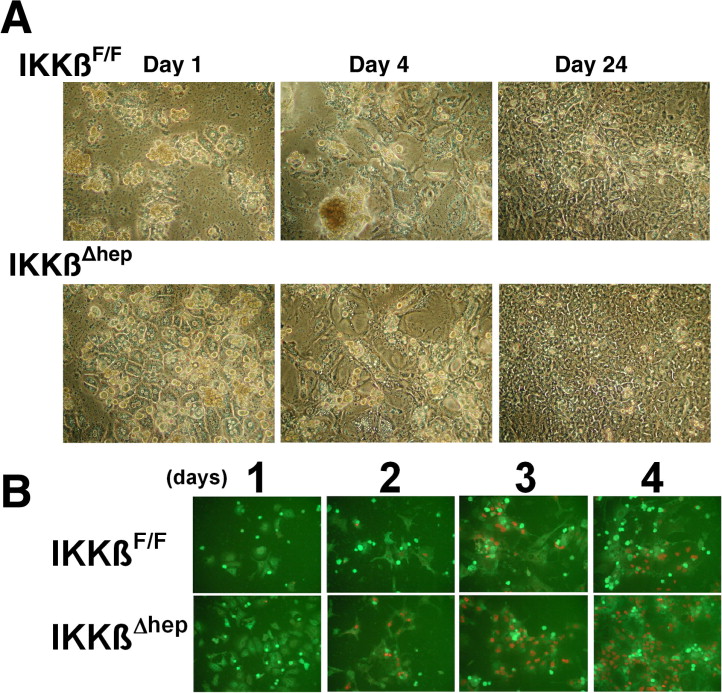
To determine whether Ikkβ deletion directly affects growth, primary cultures were prepared in the absence of non-parenchymal cells. Hepatocytes spread and flattened between d1 and d4, as expected [12] (Suppl. Fig. 1A); consistent with growth (Fig. 2A), more hepatocytes were seen between d4 and d24 and, after ~3 population doublings, large differentiated hepatocyte-aggregates appeared (Suppl. Fig. 1A).
Fig. 2.

(A–C) Cultured IkkβΔhep hepatocyte growth advantages. (A) Growth dependence upon N0. N0 = 1.5 × 105 (N = 2 dishes/point, 1 plating); N0 = 3 × 105 (IkkβF/F [N = 137] or IkkβΔhep [N = 176 dishes/curve]; 6–10 platings); N0 = 6 × 105 (N = 2 dishes/point, 1 plating). (B) Selection against non-hepatocytes. Ikkβ deletion analyses were performed at indicated times (N0 = 3 × 105). (C) Reduced IKKβ mRNA levels. N0 = 3 × 105; N = 2 dishes/point, 2 platings. (D–F) Augmented cultured IkkβΔhep hepatocyte L.I. and cyclin D1 mRNA expression (N0 = 3 × 105). (D) L.I. through mid-log phase: albumin + (top curves) or BrdU + albumin + cells (bottom curves); N = 6 dishes/point, 3 platings. (E) IkkβΔhep S- and M-phase hepatocytes (200×): mitotic cells (←). (F) Augmented cyclin D1 expression. N = 8 dishes/point, 4 platings.
Growth curves (Fig. 2A) showed intergenotypic differences when cells were plated at varying concentrations. At low N0(1.5 ×105 cells/dish), d1 IkkβΔhep hepatocyte R.E. [15] exceeded controls (IkkβΔhep ~ 58–68% > IkkβF/F ~ 36–38%, Suppl. Fig. 1A). Because R.E. curves were linear over a broad N0 range (Suppl. Fig. 2), such differences were unlikely results of differential media conditioning [16]. When plateau phase was attained (d12), IkkβΔhep hepatocyte numbers exceeded controls by ~1.5-fold (P < 0.01). At progressively higher N0 (3 × 105 or 6 × 105), although d2–d8 growth rates and d12–d21 plateau densities were similar, intergenotypic R.E. differences were sustained (IkkβΔhep > IkkβF/F, Suppl. Fig. 2), IkkβΔhep hepatocytes/dish exceeded controls until d8 (P < 0.0001), and IkkβΔhep lag phases relative to controls were attenuated (P < 0.003, Fig. 2A).
Additional findings support cell-autonomous growth differences between IkkβF/F and IkkβΔhep hepatocytes. First, because both kinds of hepatocytes came from age- and weight-matched littermates, and were isolated with similar yields and viabilities (Suppl. Table 2), the results could not be explained by differences in these parameters. Second, non-parenchymal cells could not have contributed measurably to growth because cell isolation and plating conditions select against non-hepatocyte survival [10,12,16]; this was visually evident from uniform d1 hepatocyte morphologies (Suppl. Fig. 1) and findings that >90% of cells expressed hepatocyte-specific albumin in d1 cultures (Fig. 2D [top curves]; Suppl. Fig. 1B). Genotyping analysis failed to reveal significant levels of non-parenchymal Ikkβ DNA (an ~2-kb product generated from intact Ikkβ genomes) in freshly isolated (0-time) or cultured IkkβΔhep hepatocytes on d3–d4 (Fig. 2B) or d8 (not shown). The origins of trace 2-kb bands in d1–d2 cultures is unclear; such bands represented <0.1% of observed PCR products and were absent in 0-time cells (they may have come from rare non-hepatocyte contaminants, undetected at 0-time and lost from cultures as selection-pressure prevailed). Non-hepatocyte-derived IKKβ proteins were also undetectable in d1 IkkβΔhep cultures [6], consistent with ~6-fold intergenotypic differences in Ikkβ mRNA levels in d1–d6 cultures (IkkβF/F > IkkβΔhep, P < 0.00004; Fig. 2C). Thus targeted Ikkβ deletion conferred growth advantages to IkkβΔhep hepatocytes that are independent of non-parenchymal cells.
Targeted Ikkβ deletion causes precocious expression of cyclin D1 and enhanced S-phase entry in primary hepatocytes
To investigate growth-associated cell-cycle parameters, S-phase entry and cyclin D1 expression were measured d1–d8 post-plating. If precocious G0,1 → S transitions were properties of growth-stimulated IkkβΔhep hepatocytes, BrdU L.I. and cyclin D1 expression levels should be augmented compared to controls. Indeed, S-phase entry (visualized by scoring BrdU + albumin + cells, Suppl. Fig. 1B) was specifically 2-fold higher in IkkβΔhep hepatocytes between d1 and d4 (P < 0.009; Fig. 2D, bottom curves). Using 18h labeling intervals to exclude scoring log-phase cells transiting S-phase more than once (given ~72 h population doubling times, Fig. 2A), BrdU labeled mitotic hepatocytes were also detected (Fig. 2E). Cyclin D1 mRNA expression peaked bimodaly d3 and d6, and declined towards baseline on d8 in both groups; however, IkkβΔhep hepatocyte rates and peak amplitudes of cyclin D1 mRNA expression exceeded controls (P < 1.8 ×10−5, Fig. 2F). Albumin expression did not parallel intergenotypic G1 → S transition differences; instead, it changed inversely with growth-state (12): highest initially on d1, with its L.I. falling to 80–82% (Fig. 2D, top curves) as log–growth ensued (Fig. 2A and D [bottom curves]). Thus, constitutive Ikkβ deletion directly enhanced hepatocyte proliferative transitions.
Ex vivo deletion of Ikkβ stimulates hepatocyte proliferation
To circumvent strain-dependent R.E., the effects of abrupt Ikkβ deletion were investigated in equal numbers of IkkβF/F hepatocytes (N0 = 3 × 105) infected with Cre recombinase-expressing (Ad-Cre) or control GFP-expressing adenovirus (Ad-GFP). Ad-Cre infection effectively deleted Ikkβ (Suppl. Fig. 3A) and stimulated proliferation compared to controls (P < 0.0009; Fig. 3A, top panel). Ad-Cre infection also augmented rates and levels of hepatocyte BrdU labeling (P < 0.05; Fig. 3A, bottom panel). Net increases in cells/dish and L.I. were similar to uninfected IkkβΔhep hepatocytes (Figs. 2A and D [bottom curves]). Ad-Cre infection also augmented cyclin D1 (P < 0.05; Fig. 3B, top panel) and reduced IKKβ mRNA expression (P < 0.0001; Fig. 3B, bottom panel) compared to controls. Neither Cre expression artifacts [17] nor cytotoxicity could have caused Ad-Cre-augmented responses because equivalent growth (Suppl. Fig. 3B) and albumin expression curves (not shown) of Ad-Cre, Ad-GFP- or vehicle-treated C57BL/6 hepatocytes were observed.
Fig. 3.

Ikkβ deletion ex vivo stimulates cultured IkkβF/F hepatocyte proliferation. (A) Growth and BrdU L.I. curves (top and bottom panels, respectively; additions, ↓). (B) Ad-Cre infection augments cyclin D1 and reduces IKKβ mRNA expression (top and bottom panels, respectively). Panels (A and B): N = 6 dishes/point, 3 platings.
Ikkβ deletion affects hepatocyte growth-signaling systems
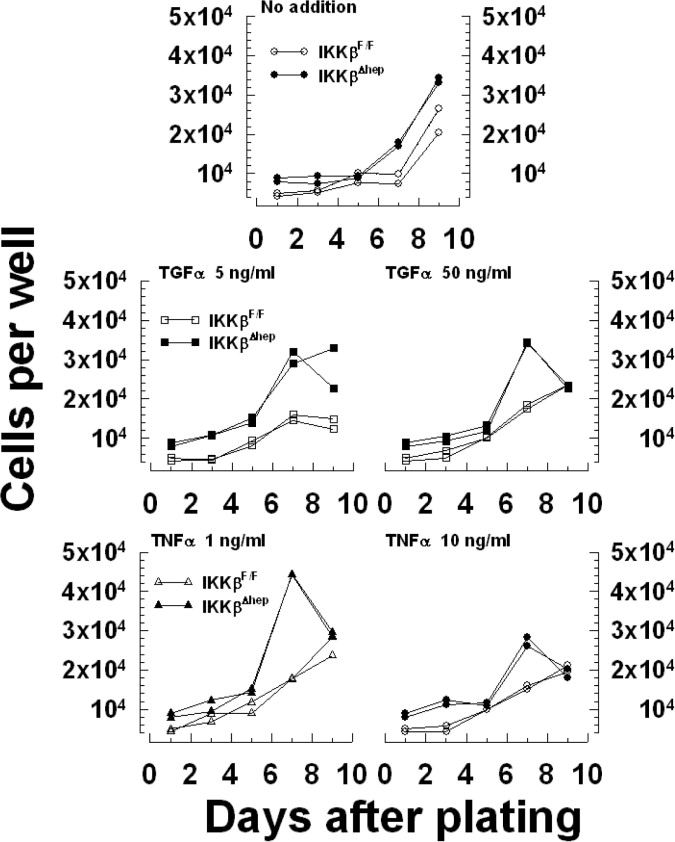
Signaling system components, including mitogens and mitogen receptor/ECM/basement membrane/cytoskeleton/integrin complexes concertedly regulate hepatocyte growth [14,18]. Therefore, selected components were investigated for growth advantage-correlations in IkkβΔhep cells. Firstly, increased receptor sensitivities to mitogens were monitored by quantifying growth-responses to TGFα or TNFα. Mitogens were added after attachment (d1) and replicate cell counts measured d1–d9 (Suppl. Fig. 4 and Table 3). Again, IkkβΔhep hepatocyte R.E. exceeded controls by ~2-fold. Without added mitogens, following a long lag-phase >3–5d (perhaps due to differences between multiwells and dishes), IkkβΔhep hepatocyte growth surged between d5 and d7, ahead of controls d7–d9. With added mitogens, lag phases shortened; by d7, IkkβΔhep hepatocytes responded better than controls to TNFα (P < 0.0005) and TGFα (P < 0.03): Suppl. Fig. 4 shows nearly doubled peak numbers of cells. These results suggested that IkkβΔhep hepatocytes were more sensitive to mitogens than controls, a conclusion more evident when data were plotted as mitogen dose-response curves (Fig. 4A). Left-shifts occurred in both groups. IkkβΔhep responses to TGFα plateaued before controls (attenuated responses were not observed.). In contrast, both groups showed biphasic TNFα responses, heightened in IkkβΔhep cultures in which TNFα-dependent growth was considerably higher at low or blunted at high ligand concentrations (Fig. 4A).
Fig. 4.

Effects of Ikkβ deletion on growth-signaling systems in cultured hepatocytes. (A) Heightened IkkβΔhep sensitivity to mitogens. Duplicate wells of cells plated (N0 = 1.5 × 105) into 24-multiwell plates received serum-free plating medium (0-dose), TGFα (top panel) or TNFα (bottom panel). Curves are differences between the average AUC ± S.E[each treatment] minus the average AUC ± S.E[0-dose] integrated over d1–d9 cell counts. Average AUC values[0-dose] subtracted from each AUC[ligand dose] were 42026 (IkkβF/F) and 61023 (IkkβΔhep; see Suppl. Table 3). Intergenotypic P values between curves: TGFα, P < 0.0005; TNFα, P < 0.03. (B) Augmented IkkβΔhep cytoskeletal, ECM, basement membrane and integrin mRNA expression. Curves are averages, 3 platings (N0 = 3 × 105); N = 6 dishes/primer pair/point (sister culture growth was similar to Fig. 2A). Primer pair results were combined (Suppl. Table 1), except for nidogen (N = 6 dishes/primer pair 1/point; N = 4 dishes, primer pair 2/point). Intergenotypic P values d0–d6: fibronectin (P < 0.07); collagen I (P < 0.003), collagen III (P < 0.0003); nidogen (P < 0.02); β-actin (P < 0.03); integrin B1 (P < 0.0001).
Secondly, correlations with ECM, basement membrane, cytoskeletal and mitogen receptor-associated integrin mRNA expression were investigated [19]. Specifically elevated changes (IkkβΔhep > IkkβF/F) were observed in 6 of 12 candidate mRNAs surveyed between d1 and d6 (Fig. 4B): fibronectin, collagens I and III, nidogen, β-actin, and EGF receptor-associated integrin B1. No differences were seen in laminin, elastin, FAK, SPARC, or mitogen receptor-associated integrin B3 (c-MET) or B4 (insulin) mRNAs, or in freshly isolated hepatocyte, or IkkβΔhep and control liver mRNAs.
Discussion
We have shown that constitutively Ikkβ deleted (IkkβΔhep) or ex vivo Ikkβ deleted IkkβF/F hepatocytes have growth advantages over Ikkβ-expressing hepatocytes. These advantages are associated with attenuated levels of IKKβ mRNA and augmented levels of cy-clin D1 mRNA; cell-cycle expression patterns of IKKβ and cyclin D1 proteins and activities will require further clarification. In contrast, when intrahepatic mitogen mRNA expression is reduced throughout liver by the deletion of Ikkβ in hepatocytes and hematopoietic-derived cells (IkkβΔLIV mice), in vivo hepatocyte growth advantages expected of Ikkβ deleted hepatocytes following 70% PH are absent [7]. Thus, IKKβ is a bifunctional regulator of hepatocyte proliferation and HCC, with different roles played by hepatocytes and liver non-parenchymal cells; enhanced HCC in DEN-treated IkkβΔhep mice requires the effects of IKKβ-dependant Kupffer cell-derived cytokines and the loss of growth-inhibitory hepatocyte IKKβ. Because primary culture studies, and dual investigations with IkkβΔhep and IkkβΔLIV knockout mice [7], were needed to reach both conclusions (otherwise missed by unicellular targeting), the potential regulation of HCC-suppressors including pRb, p53 and p21 [20] by non-parenchymal cells should not be overlooked.
Bifunctional IKKβ implicates cell-specificity. This is supported by studies with embryonic Ikkβ−/− fibroblasts which grow faster but plate with identical efficiency compared to controls [21], and Ikkβ−/− Hela cells which undergo mitotic arrest [22]. Investigations with Ikk2Δhepa mice [23,24] also suggest that hepatocyte IKKβ survival functions depend on IKKα and IKKγ [25]; however, different Ikkβ-targeting strategies may have differentially affected Ikk2Δhepa mice because findings of PH-induced inflammation indirectly shortening hepatocyte St and abnormally delayed post-PH BrdU L.I. peaks [15,24] disagree with findings in IkkβΔhep mice [6,23].
G0 and proliferating IkkβΔhep populations consist only of hepatocytes. Therefore, exceedingly rare hepatocyte progenitors like oval cells, or putative progenitors like stellate cells that differentiate into albumin + cells [26], are unlikely to have undergone Ikkβ deletion in regenerating liver or primary culture. Defined G0-culture conditions, which best simulate complex growth control processes free from confounding in vivo variables [27], should help clarify mechanisms by which Ikkβ deletion confers growth advantages. Although in vitro measurements were consistent with in vivo results [7], which suggested that 70% PH shortened IkkβΔhep hepatocyte St without affecting DNA-synthesis entry rates (SΔ), they were made in cells exposed to pre-plating stresses that induce early gene expression [28]. However, pre-plating artifacts like these or trace mitogens in the serum supplement are unlikely to have caused post-plating IkkβΔhep hepatocyte growth advantages because ex vivo Ikkβ deletion also stimulates IkkβF/F hepatocyte proliferation. Future G0-culture investigations should also clarify whether IkkβΔhep growth advantages result from signal-1 or -2 accelerated G0,1 → G1 transitions with or without increased rates of SΔ [14,29]. These investigations might be feasible with the culture system described here, which may have undergone G0-arrest as evident from stable plateau phases d8–d24, reduced cyclin D1 mRNA expression and near-baseline BrdU L.I. levels d8–d10.
Our results show that increased sensitivities to mitogens, including increased expression of ECM, basement membrane, cytoskeletal and integrin mRNAs accompany increased IkkβΔhep hepatocyte growth. TNFα-attenuated growth in IkkβF/F and IkkβΔhep cultures is consistent with TNFα-stimulated apoptosis at high (0.58 nM) but not low (0.058 nM) TNFα doses [6], suggesting that ligand interactions with TNFR2 may account for attenuated growth in high-dose TNFα-treated cells. In further agreement, biphasic dose-response curves of TNFα-stimulated AP-1 activation were also seen in transfected hepatocytes: AP-1 expression peaked at 0.01 nM and returned to basal levels at 1 nM [30]. Significant changes in the expression of IkkβΔhep fibronectin, collagens I and III, nidogen, β-actin and TGFα/EGF receptor-associated integrin B1 mRNAs were not seen in tissues or cells from which the cells were plated; instead 1–2d were required before IkkβΔhep-associated elevations were observed. This suggests that regulatory effects of Ikkβ deletion occurred post-plating after exposure to serum factors, to factors released by necrotic cells [10], and possibly to culture conditions like uncoated plastic used to preclude cell attachment bias. Further investigations are needed to determine how and if these signaling systems underlie the growth advantages of IkkβΔhep hepatocytes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Supported by the NIH (CA113602, AI067354 [H.L.]), Superfund Basic Research Program (ES10337 [M.K., H.L.]) and Damon Runyon Cancer Fund (G.H.). We thank K. Juson (and partial salary support from NIH CA112481 [S. Sell]) and M. Zhang for technical assistance, M. Kagnoff (UCSD) in whose laboratory Q-PCR was performed, and A. Chang (husbandry). M.K. is an American Cancer Society Research Professor.
Abbreviations
- Ikkβ
IκB kinase β
- IKK
IκB kinase complex
- PH
partial hepatectomy
- DEN
diethylnitrosamine
- HCC
hepatocellular carcinoma
- ECM
extracellular matrix
- TGF
transforming growth factor
- PBS
phosphate-buffered saline
- GFP
green fluorescent protein
- FAK
focal adhesion kinase
- SPARC
secreted protein acidic and rich in cysteine
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi: 10.1016/j.bbrc.2009.01.085.
References
- 1.Ghosh S, Karin M. Missing pieces in the NF-κB puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 2.Heyninck K, Wullaert A, Beyaert R. Nuclear factor-κB plays a central role in tumour necrosis factor-mediated liver disease. Biochem Pharmacol. 2003;66:1409–1415. doi: 10.1016/s0006-2952(03)00491-x. [DOI] [PubMed] [Google Scholar]
- 3.Joyce D, Albanese C, Steer C, Fu J, Bouzahzah M, Pestell RG. NF-κB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001;12:73–90. doi: 10.1016/s1359-6101(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 4.Shishodia S, Aggarwal BB. Nuclear factor-κB: a friend or a foe in cancer? Biochem Pharmacol. 2004;68:1071–1080. doi: 10.1016/j.bcp.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 5.Laurent S, Horsmans Y, Stärkel P, Leclercq I, Sempoux C, Lambotte L. Disrupted NF-κB activation after partial hepatectomy does not impair hepatocyte proliferation in rats. World J Gastroenterol. 2005;11:7345–7350. doi: 10.3748/wjg.v11.i46.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 7.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Aldeguer X, Debonera F, Shaked A, Krasinkas AM, Gelman AE, Que X, Zamir GA, Hiroyasu S, Kovalovich KK, Taub R, Olthoff KM. Interleukin-6 from intrahepatic cells of bone marrow origin is required for normal murine liver regeneration. Hepatology. 2002;35:40–48. doi: 10.1053/jhep.2002.30081. [DOI] [PubMed] [Google Scholar]
- 9.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;17:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 10.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koch KS, Son KH, Maehr R, Pellicciotta I, Ploegh HL, Zanetti M, Sell S, Leffert HL. Immune-privileged embryonic Swiss mouse STO and STO cell-derived progenitor cells: MHC and cell differentiation antigen expression patterns resemble those of human embryonic stem cell lines. Immunology. 2006;119:98–115. doi: 10.1111/j.1365-2567.2006.02412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leffert HL, Koch KS, Skelly H. Primary culture of hepatocytes. In: Sato G, Sirbasku D, Barnes D, editors. Cell Culture Methods for Molecular and Cell Biology. III. Alan R. Liss; N.Y.: 1984. pp. 43–55. [Google Scholar]
- 13.Bewig B, Schmidt WE. Accelerated titering of adenoviruses. Biotechniques. 2000;28:870–873. doi: 10.2144/00285bm08. [DOI] [PubMed] [Google Scholar]
- 14.Leffert HL, Koch KS, Lad PJ, Skelly H, de Hemptinne B. Hepatocyte growth factors. In: Zakim D, Boyer TD, editors. Hepatology. W.B. Saunders; Philadelphia: 1982. pp. 64–75. [Google Scholar]
- 15.Sakamoto T, Liu Z, Murase N, Ezure T, Yokomuro S, Poli V, Demetris AJ. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology. 1999;29:403–411. doi: 10.1002/hep.510290244. [DOI] [PubMed] [Google Scholar]
- 16.Leffert HL, Paul D. Studies on primary cultures of differentiated fetal liver cells. J Cell Biol. 1972;52:559–568. doi: 10.1083/jcb.52.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prost S, Sheahan S, Rannie D, Harrison DJ. Adenovirus-mediated Cre deletion of floxed sequences in primary mouse cells is an efficient alternative for studies of gene deletion. Nucleic Acids Res. 2001;29:E80. doi: 10.1093/nar/29.16.e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch KS, Leffert HL. Hepatic regeneration and gene expression. J Tumour Marker Oncol. 1994;9:35–56. [Google Scholar]
- 19.Martinez-Hernandez A, Amenta PS. The extracellular matrix in hepatic regeneration. FASEB J. 1995;9:1401–1410. doi: 10.1096/fasebj.9.14.7589981. [DOI] [PubMed] [Google Scholar]
- 20.Martin J, Dufour JF. Tumor suppressor and hepatocellular carcinoma. World J Gastroenterol. 2008;14:1720–1733. doi: 10.3748/wjg.14.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen F, Lu Y, Castranova V, Li Z, Karin M. Loss of Ikkβ promotes migration and proliferation of mouse embryo fibroblast cells. J Biol Chem. 2006;281:37142–37149. doi: 10.1074/jbc.M603631200. [DOI] [PubMed] [Google Scholar]
- 22.Irelan JT, Murphy TJ, DeJesus PD, Teo H, Xu D, Gomez-Ferreria MA, Zhou Y, Miraglia LJ, Rines DR, Verma IM, Sharp DJ, Tergaonkar V, Chanda SK. A role for IκB kinase 2 in bipolar spindle assembly. Proc Natl Acad Sci USA. 2007;104:16940–16945. doi: 10.1073/pnas.0706493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luedde T, Assmus U, Wüstefeld T, Vilsendorf AMZ, Roskams T, Schmidt-Supprian M, Rajewsky K, Brenner DA, Manns MP, Pasparakis M, Trautwein C. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest. 2005;115:849–859. doi: 10.1172/JCI23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malato Y, Sander LE, Liedtke C, Al-Masaoudi M, Tacke F, Trautwein C, Beraza N. Hepatocyte-specific inhibitor-of-κB-kinase deletion triggers the innate immune response and promotes earlier cell proliferation during liver regeneration. Hepatology. 2008;47:2036–2050. doi: 10.1002/hep.22264. [DOI] [PubMed] [Google Scholar]
- 25.Luedde T, Heinrichsdorff J, de Lorenzi R, De Vos R, Roskams T, Pasparakis M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc Natl Acad Sci USA. 2008;105:9733–9738. doi: 10.1073/pnas.0800198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Jung Y, Omenetti A, Witek RP, Choi S, Vandongen HM, Huang J, Alpini GD, Diehl AM. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–2113. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koch KS, Leffert HL. Increased sodium ion influx is necessary to initiate rat hepatocyte proliferation. Cell. 1979;18:153–163. doi: 10.1016/0092-8674(79)90364-7. [DOI] [PubMed] [Google Scholar]
- 28.Paine AJ, Andreakos E. Activation of signaling pathways during hepatocyte isolation: relevance to toxicology in vitro. Toxicol In Vitro. 2004;2:187–193. doi: 10.1016/s0887-2333(03)00146-2. [DOI] [PubMed] [Google Scholar]
- 29.Leffert HL, Koch KS. Hepatocyte growth regulation by hormones in chemically-defined medium: a two-signal hypothesis. In: Sato GH, Pardee AB, Sirbasku DA, editors. Growth of Cells in Hormonally Defined Media (Cold Spring Harbor Conferences on Cell Proliferation 9) Cold Spring Harbor Press; Cold Spring Harbor: 1982. pp. 597–613. [Google Scholar]
- 30.Hattori M, Tugores A, Westwick JK, Veloz L, Leffert HL, Karin M, Brenner DA. Activation of AP-1 during the hepatic acute phase response. Amer J Physiol. 1993;264:G95–G103. doi: 10.1152/ajpgi.1993.264.1.G95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.