

Summary
Little is known about regulatory networks that control metabolic flux in plant cells. Detailed understanding of regulation is crucial for synthetic biology. The difficulty of measuring metabolites with cellular and subcellular precision is a major roadblock. New tools have been developed for monitoring extracellular, cytosolic, organellar and vacuolar ion and metabolite concentrations with a time resolution of milliseconds to hours. Genetically encoded sensors allow quantitative measurement of steady-state concentrations of ions, signaling molecules and metabolites and their respective changes over time. Fluorescence resonance energy transfer (FRET) sensors exploit conformational changes in polypeptides as a proxy for analyte concentrations. Subtle effects of analyte binding on the conformation of the recognition element are translated into a FRET change between two fused green fluorescent protein (GFP) variants, enabling simple monitoring of analyte concentrations using fluorimetry or fluorescence microscopy. Fluorimetry provides information averaged over cell populations, while microscopy detects differences between cells or populations of cells. The genetically encoded sensors can be targeted to subcellular compartments or the cell surface. Confocal microscopy ultimately permits observation of gradients or local differences within a compartment. The FRET assays can be adapted to high-throughput analysis to screen mutant populations in order to systematically identify signaling networks that control individual steps in metabolic flux.
Keywords: fluorescence resonance energy transfer (FRET), fluxomics, metabolomics, metabolic signaling, metabolic pathways, sugar signaling
I. Introduction
Plants play a crucial role in all aspects of our lives. Plants produce oxygen and fix carbon, are the basis of all of our food, feed and clothes, and serve our aesthetic and recreational needs. In today’s world, understanding the factors that limit plant growth has gained extraordinary relevance (Long et al., 2006). Soft commodity prices are on the rise, and the per capita yield increases achieved by breeders do not keep up with the growing population. In addition, massive new demands for increased productivity are emerging, specifically with regard to feedstocks for biofuels (Rothstein, 2007). To address these urgent needs, major goals for the future of plant engineering will be to increasing productivity by expanding the growing season, and to increase above-ground biomass without increasing the need for fertilizer and water (Karp & Shield, 2008). Given the long delays between fundamental research, the development of new technologies that can boost yield, and their introduction into the market, urgent action is required at all levels.
In recent years, the scale of plant research has changed, and we can now begin to use systems biology to accelerate discovery and to create predictive models of plant function. In combination with synthetic biology (Benner & Sismour, 2005), a new scale of engineering will be possible that may help to rationally design plants with increased productivity. The introduction of methods for the synthesis and addition of complete chromosomes is expected to revolutionize biotechnology (Gibson et al., 2008). However, before rational design of crops becomes efficient, a detailed understanding of both cellular and whole-plant physiology will be required.
In this review we provide an overview of the emerging field of flux analysis using fluorescence resonance energy transfer (FRET) sensors, provide a base for data interpretation, and highlight the potential, the possible pitfalls and the need for future developments of this new technology.
II. From individual enzymes to a complete parts list in 100 yr
One hundred years ago, Eduard Buchner gave his Nobel lecture, having obtained the Nobel Prize in recognition of his work, in which he had shown that cell-free extracts can carry out fermentation (Buchner, 1907). He thereby refuted Louis Pasteur’s conclusion that fermentation was a vital physiological act of living yeast cells, and demonstrated instead that fermentation is mediated by a ‘chemical agent’, termed ‘zymase’. Nobel Prizes marked further important milestones in research on metabolism, including those for Otto Meyerhof and Archibald Hill in 1922 for the discovery of glycolysis and its relevance for respiration, Hans Krebs in 1953 for the identification of the citric acid cycle and Melvin Calvin in 1961 for uncovering the dark cycle in photosynthesis. Most work during that time focused on enzymatic reactions, but it became apparent that compartmentation was essential and that transport processes played crucial roles in cellular processes, as recognized by Peter Mitchell who obtained the Nobel Prize for the chemiosmotic hypothesis in 1978. In almost all cases, biochemistry was the centerpiece of the discovery. Using a combination of cell fractionation techniques and biochemical metabolite and protein analysis, in some cases combined with time-dependent quantitative analysis of metabolites, new pathways were uncovered. Today it is possible to view metabolic pathway maps, as exemplified by the Expasy biochemical pathways (www.expasy.ch/cgi-bin/search-biochem-index) or, more recently, databases for the display of organism-specific processes such as in KEGG (www.genome.jp/kegg/), Ecocyc (ecocyc.org) or Aracyc (www.arabidopsis.org/tools/aracyc/), which shows plant pathways. The expectation is that the development of genome-scale metabolic models will in the future help to engineer plant productivity as demonstrated for microorganisms (Feist et al., 2006; Becker et al., 2007; Feist et al., 2007).
Despite the extensive study of the metabolism of plants, we still can be surprised by novel pathways that involve novel enzymes, even in areas that were thought to be well understood; for example, the recent identification of the nonmevalonate pathway (Lichtenthaler, 1999) or the still only partially understood starch degradation pathway in leaves (Zeeman et al., 2007). For many pathways, a major remaining gap relates to inter- and intracellular transport; a prominent example is photorespiration. Most genes for primary metabolic pathways have been cloned, but many of the transporters exchanging intermediates, cofactors and products are unknown at the molecular level (Linka & Weber, 2005). The ability to measure transport or flux across intracellular membrane in vivo could help to identify the underlying processes and their regulation. While the overall network and many of the reactions have been established, one of the major missing elements in our understanding of the functioning of the metabolic pathways is the regulatory layer controlling flux though the pathways. We have probably revealed only a small fraction of the level of complexity that exists.
III. Pathways and flux
Metabolism of a given compound is mediated by a network of enzymatic reactions. The abundance and the properties of the contributing enzymes as well as the concentration of the intermediates determine the flux through the pathway and thus the rates of consumption of the initial compound, for example glucose fed to the cell, and the rate of production of the end products, for example starch and cellulose. Textbooks often suggest that the first step in a metabolic pathway is critical and considered to be highly regulated, thus exerting control over flux. The first enzyme in a pathway is considered to be the first step. However, in many cases the first step is the import into the respective compartment. It apparently makes sense that control is exerted at the transport steps as they are located in strategic positions. In reality, flux control is distributed over the pathway and the contribution of individual steps may vary depending on the conditions (Fernie et al., 2005; Carrari et al., 2006). A detailed discussion of the current state of the art of experimental and mathematical approaches to the modeling of metabolic networks from plants has been published recently (Rios-Estepa & Lange, 2007).
In many cases, pathways have multiple inputs and outputs. Soluble carbohydrates such as glucose and sucrose may be synthesized by photosynthesis, may derive from storage or may be imported from other cells and organs. All of these reactions contribute to accumulation inside the cell or compartment (Fig. 1a). The soluble carbohydrates can then be diverted into many different directions such as cell wall synthesis and storage, or they can serve as precursors for other metabolites. Flux is not a constant, but may change depending on environmental cues or on the status of the organism (Fig. 1b). Changes in light, temperature or pathogen attack will lead to redirection of resources to allow acclimation to the new situation. Regulatory circuits must control the relative fluxes, and thus cells and organisms have to be able to measure cellular parameters such as steady-state concentrations as well as environmental parameters, compute deviations from the set-point and adjust fluxes accordingly. Biological systems make use of every possible mechanism for regulation (Fig. 2). While today transcription is certainly the area that attracts most attention, RNA stability, the new paradigm of regulation by small RNAs, differential splicing, initiation and elongation of translation and of course a wide spectrum of posttranslational mechanisms, including regulation in protein homo- or heterooligomers (Loque et al., 2007), as well as a wide range of covalent modifications, play important roles in regulating flux (Peck, 2006; Niittyla et al., 2007).
Fig. 1.
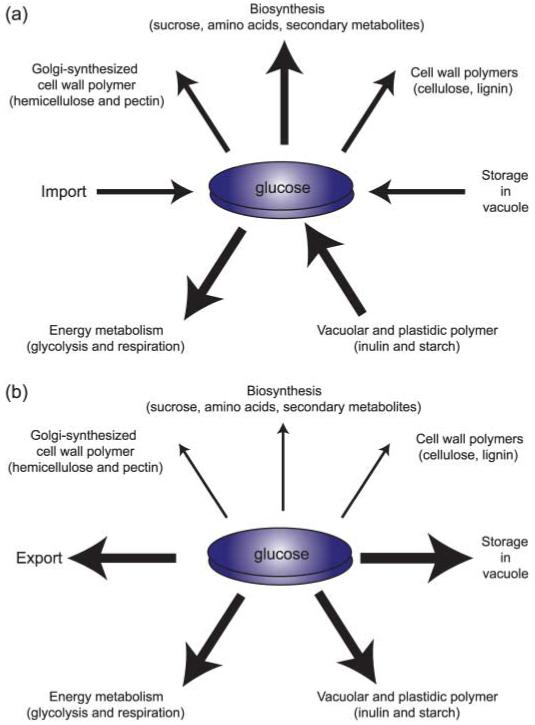
Fluxes affecting steady-state cytosolic glucose concentrations in plant cells. Glucose and its activated derivates can be imported into the cytosol, and stored in the vacuole or plastids. They then serve as precursors for the biosynthesis of primary and secondary metabolites as well as cell wall biosynthesis, or they can be used to produce energy. Cells control the individual fluxes depending on the developmental stage and the environmental conditions. (a) Degradation of storage compounds and import are used to allow rapid biosynthesis; (b) fluxes favor storage and export. A key question is thus: how are the individual fluxes controlled?
Fig. 2.
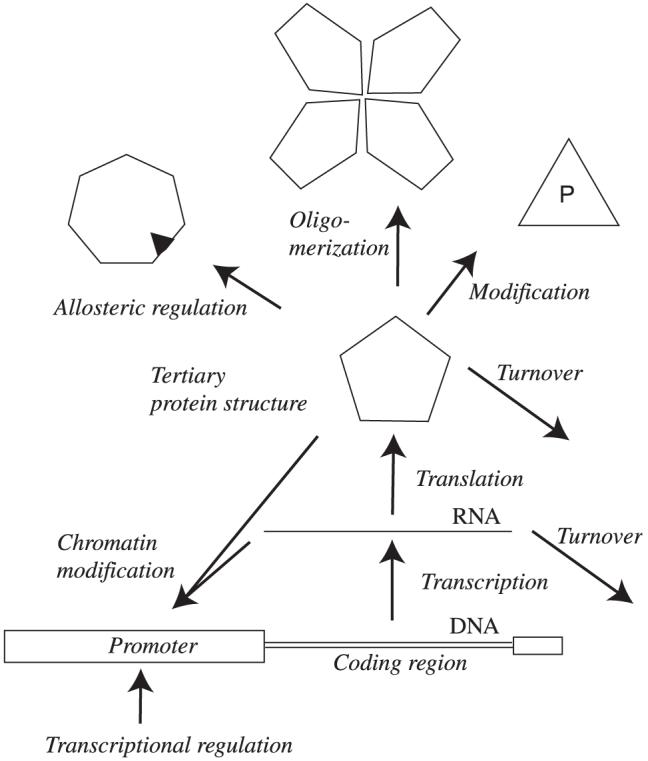
Potential levels of regulation. Organisms use all possible options for regulation, including transcription, splicing, RNA turnover, translation and post-translational mechanisms such as protein modification, oligomerization/protein interactions, turnover and allosteric interactions.
One other poorly studied area is regulation of enzyme or transporter activity by small molecules, a subtype of allostery (the term ‘allostery’ is derived from the Greek allos, ‘other’ and stereos, ‘space’). The most prominent examples of allosteric regulation are Monod’s lactose system and, in plants, the finding that fructose-2,6-phosphate is a key regulator of gluco(sucro)-neogenesis. It is conceivable that allostery is far more extensive than currently thought (Gunasekaran et al., 2004). Novel approaches will be required to reassess the prevalence of allosteric regulation.
Finally, an area that is just beginning to emerge suggests that regulation of enzyme activity can occur at the level of cellular localization, which can be achieved by intracellular RNA transport and localized translation, or by localized control of activity without apparent compartmentation (Pertz & Hahn, 2004; Xu et al., 2005). One of the most illuminating findings in the area of metabolism suggested that UDP-glucose pyro-phosphorylase changes its cellular localization to support cell wall synthesis at the expense of glycogen storage depending on nutrient and stress conditions, a switch controlled by protein phosphorylation through Per-Arnt-Sim (PAS) kinases (Grose et al., 2007).
Thus, above the level of the metabolic pathways, a regulatory network is predicted to control flux though the pathway and the distribution of intermediates; for example, whether sugars are used for cell wall synthesis or respiration (Figs 1, 3). We can infer from findings in other organisms and the extreme efficiency of plants in responding to changes in the environment that a large number of sensors must exist for monitoring parameters such as light, temperature, supply and demand, pathogen attack, and probably a wide range of other parameters as part of the control machinery that adjust fluxes by targeting individual enzymes (Fig. 3). In a simplified cell, the steady-state concentration of a metabolite in the cytosol, for example glucose, is determined by a sum of positive and negative fluxes (Fig. 4). The flux components are potential targets for control. The determination of steady-state concentrations has been complicated by the existence of potential differences in concentrations of metabolites between cells, as well as compartments. Classical methods for metabolite analysis and metabolomics take neither differences between cells nor compartmentation into account. Advanced methods such as single-cell sap sampling provide cellular resolution, but will average over various compartments (Roy et al., 2003). Nonaqueous fractionation techniques can provide data for subcellular compartment; however, they average over many cells in the harvested organ (Farre et al., 2001). Moreover, these techniques are destructive and do not provide the potential for dynamic analysis. Because of the large contribution of the vacuole to the volume of a cell, even a small contamination of the other fractions can lead to artifacts. Thus a key question is how we can measure metabolite concentrations and flux in a multicellular organism, in which the cells are highly compartmentalized. This is especially important in plant cells, where the vacuole typically constitutes the major volume of a cell. To identify the control networks, we need to be able to monitor the metabolites with cellular and even subcellular resolution and we need a high temporal resolution to be able to follow changes of flux.
Fig. 3.
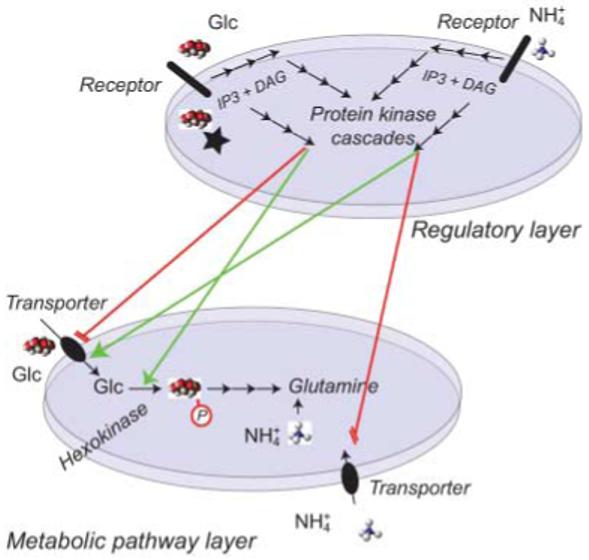
Layers of signaling networks and metabolic pathways. A hypothetical regulatory layer that uses both extracellular and intracellular receptors uses a complex three-dimensional network to transduce, amplify and modulate signals (here for example a glucose (glc) sensor and an ammonium sensor at the cell surface, and potential second messengers such as inositol-triphosphate (IP3) and diacylglycerol (DAG) as well as protein phosphorylation cascades) in order to control the metabolic flux mediated by a network of hypothetical transporters for glucose and ammonium and downstream metabolic pathways.
Fig. 4.
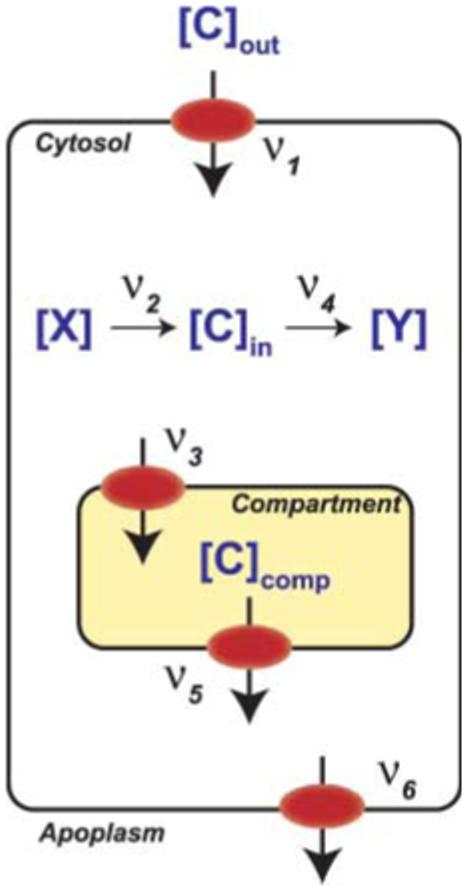
Flux diagram for a metabolite. The intracellular concentration of carbon ([C]in) is determined by the external concentration [C]out, the plasma membrane transport rates (ν1 for influx; ν6 for efflux), the rate of biosynthesis (ν2) and catabolism (ν4) in the cytosol, and the rates of import (ν3) and release (ν5) from/to one or several intracellular compartments. Regulatory effects on any of the parameters will affect steady-state concentrations. Fluorescence resonance energy transfer (FRET) sensors can determine both steady states as well as the rates of accumulation and elimination when external concentrations are changed or when any of the parameters is altered by genetic or chemical modification. X, precursor; Y, synthesized compound.
IV. Quantitative imaging for flux analysis
Microscopy is a classical tool originally developed for structural and anatomical studies. The development of fluorescent dyes has significantly changed the potential of microscopy, especially since the introduction of fluorescent small-molecule dyes and genetically encoded fluorophores. Fluorescence intensity is readily quantifiable using a fluorimeter or microscope. Confocal technologies, high quantum yield cameras, pulsed lasers, novel devices for spectral imaging, fluorescence lifetime imaging (FLIM), polarization microcopy and anisotropy decay imaging have revolutionized microscopy, providing us with a wide array of tools for quantification of microscopic data (Lalonde et al., 2008). These tools combined with the different types of fluorescent sensors now available have made it possible to measure metabolite and ion concentrations as well as cellular processes at the single-cell level with subcellular resolution (Lalonde et al., 2005). Some of the most fascinating tools developed for cell biology are sensors for the state of cell division (Sakaue-Sawano et al., 2008).
In their original implementation, small-molecule ratiometric dyes had a major impact on ions in the transport field (O’Connor & Silver, 2007). Ratiometric fluorescent dyes typically have two emission peaks whose relative intensities change upon ligand binding. The ligand concentration is typically reported as a ratio between the two emission peaks, and as both emission intensities are linear functions of the dye concentration, the ratio between two peaks is relatively independent of the concentration of the dye in the cell. However, these dyes have to be membrane-permeable in order to be able to monitor intracellular ion concentrations, or have to be mechanically introduced by, for example, microinjection. A major advance was thus the development of genetically encoded fluorescent sensors. These sensors can be introduced into any cell that can be transfected or transformed. Moreover, genetically encoded sensors can be targeted to subcellular compartments by fusion to suitable targeting sequences. In its simplest implementation, such a sensor makes use of intrinsic properties of fluorescent proteins, such as their pH sensitivity. While the original green fluorescent protein (GFP) showed limited pH sensitivity, variants of GFP such as pHluorins or the Ptilosarcus gurneyi Pt-GFP, can be used as sensitive pH sensors in vivo (Schulte et al., 2006). A more advanced concept was the development of FRET sensors. These sensors exploit resonance energy transfer as a means of monitoring the conformation of a protein. FRET has been used for a many decades; for example, the resonance energy transfer between tryptophanes in a protein can be used for structural analyses (Moens et al., 2004). If the conformation of the protein fused to FRET reporters changes depending on the presence or absence of the ligand, this principle can be used for monitoring steady-state concentrations of the ligand in vivo. The key advantages of FRET-based metabolite and signaling flux analysis include high time resolution (> 50 Hz), high sensitivity, providing the capability to measure in the nano- and micromolar range, and subcellular resolution as a result of the option to target the sensors to compartments and even subcompartments such as organelle surfaces. Most of these sensors make use of variants of GFP, but recently small-molecule dyes that bind to a specific protein sequence, or are enzymatically coupled to it, have also been exploited for in vivo measurements (Hoffmann et al., 2005; Gassensmith et al., 2007). However, stoichiometric dye coupling and high coupling efficiencies can only be obtained using a genetic approach. Sensors can be characterized in vitro after extraction of the fusion proteins from Escherichia coli (cf. e.g. Fehr et al., 2002).
V. Förster resonance energy transfer (FRET) as a tool
Before entering into a detailed description of the sensors and their use for flux analysis, this chapter provides an introduction to the principle of FRET. FRET is defined as a ‘long-range’ dipole interaction between molecules through resonance dipole-dipole coupling occurring in the pico- to nanometer range and thus at a scale relevant to changes in protein conformation. FRET is based on a quantum mechanical effect between a given pair of fluorophores, that is, a fluorescent donor and an acceptor. Upon excitation of the donor dipole, energy is transferred to the acceptor dipole in a nonradiative manner (Förster, 1948; Jares-Erijman & Jovin, 2003). Consequently, a fraction of the energy absorbed by the donor is emitted in the spectral window of the acceptor. In quantitative terms, the FRET efficiency E is defined as the fraction of the photons absorbed by the donor and transferred to the acceptor. E is a function of the inverse of the distance (r) to the sixth power between the donor and acceptor . The Förster distance (R0) is characteristic for a given donor acceptor pair and includes a number of other parameters as constants. R0 is defined as the distance at which 50% of the energy is transferred. R0 depends on the quantum yield of the donor (QD), the overlap integral of the spectra of donor emission and acceptor absorption (Jλ), the refractive index (n) of the medium and, importantly, the relative orientation of the dipole moment (κ2) of donor and acceptor (Lakowicz, 1999; Jares-Erijman & Jovin, 2003). The orientation factor κ2 can range from 0 to 4 and is set to 2/3 for unrestricted isotropic motion. Because most FRET sensor measurements are not carried out in single-molecule mode, they integrate over many molecules and over periods of time, thus using information from many conformational states of the sensors (Fig. 5). In these cases, FRET measures ensemble behavior, thus increasing the sensitivity of the assay down to the picometer scale.
Fig. 5.
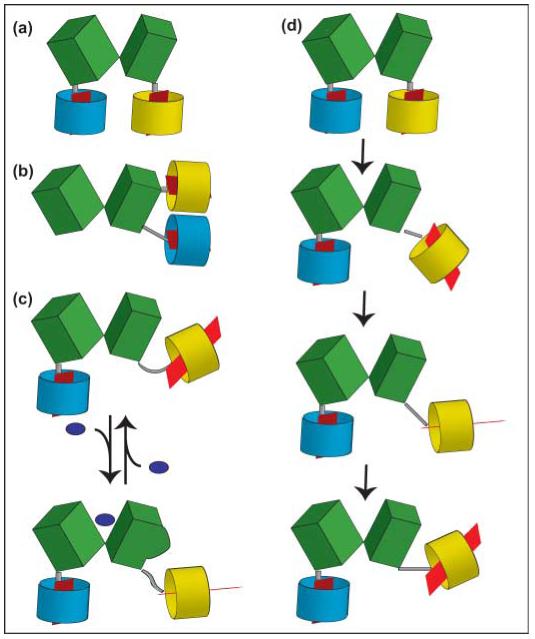
Schematic models of fluorescence resonance energy transfer (FRET) sensors for metabolites. A recognition element, for example a periplasmic binding protein, here consisting of two lobes (green), is coupled to a cyan version of green fluorescent protein (GFP) (cyan barrel) at its N-terminus and to a yellow version of GFP (yellow barrel) at its C-terminus. The red plane inside the barrel indicates the orientation of the dipole (note: dipole orientation is crucial; see k2 in Förster equation); the gray bar represents the linker. (a) C- and N-termini are separated and are located at the bottom of the two lobes of the recognition element (e.g. FRET glucose sensors). (b) C- and N-termini are located on the same lobe of the recognition element (e.g. FRET phosphate sensors). (c) Binding of the ligand to the recognition element leads to a conformational change on the surface of the recognition element, deflecting yellow fluorophore. (d) If not carried out in single-molecule mode, FRET analysis averages over many molecules and many positions of the molecules over time (here only shown for the yellow fluorophore). Thus the analysis averages over many dipole orientations (note positions of red dipole), and the flexible linker and 3D structure of the recognition element determine the freedom of the fluorophore to take different positions and to rotate. Thus, if binding affects the surface of the recognition element as in (c), the number of productive interactions between the dipoles will change, leading to a FRET change. This model may explain the mechanism behind how the sensors, including the one shown in (b), can be functional.
Energy transfer efficiency can be estimated fairly easily and can be calibrated (Vogel et al., 2006). The observed values should be compared to a theoretical value for the donor-acceptor pair to evaluate the potential of random collisions. In addition, independent methods may have to be applied to verify interactions suggested by positive FRET results. The need for appropriate controls also applies to negative FRET results. Low transfer efficiency may be caused by the absence of a molecular interaction, by a stoichiometry of donor-to-acceptor other than 1 : 1 (Vogel et al., 2006), or by excitation of the acceptor in the donor excitation channel (bleed-through).
VI. Methods for FRET determination and analysis of FRET changes
Lalonde et al. (2008) have published a detailed discussion of the different methods that can be used for FRET analysis, focusing on their use for analysis of protein interactions. In its simplest implementation, energy transfer efficiency is determined as the relative fluorescence intensity of the donor fluorescent protein (FP) in the presence or absence of the acceptor FP. Methods employed include (1) filter-based FRET analysis (ratio-imaging; sensitized emission), (2) processed spectral FRET (psFRET), (3) acceptor photobleaching FRET (apFRET), (4) lifetime measurements (FLIM), and (5) analysis of anisotropy decay. The methods have been described in many reviews (Jares-Erijman & Jovin, 2003; Jares-Erijman & Jovin, 2006). Here, we will discuss the methods typically used in combination with FRET metabolite sensors: filter-based FRET and psFRET.
For filter-based FRET quantification, the fluorescence intensities of the donor (donor excitation and emission specific filters) and acceptor (acceptor excitation and emission specific filters) and acceptor-sensitized emission (excitation of the donor and capture of acceptor emission) are acquired using two- or three-filter set configurations. In many studies, only the relative intensity of sensitized emission (emission of the acceptor after excitation of the donor) to the donor emission (emission of the donor after excitation of the donor) is measured using a two-filter configuration. While this type of ratiometric measurement is considered to be sufficient for many applications, it may be prone to artifacts. Sensitized emission is contaminated with spectral bleed-through (SBT) (donor emission passed through the acceptor emission filter (donor spectral bleed-through (DSBT)) and direct excitation of the acceptor by the donor excitation filter (acceptor spectral bleed-through (ASBT))), as well as other components resulting from autofluorescence and detector and optical noise. In addition, the concentration of the FPs influences the ratio between sensitized emission and donor emission. When the two FPs are not physically linked, the method also requires knowledge of the stoichiometory of the donor and acceptor. Filter-based FRET measurements can be significantly improved using appropriate controls in the three-filter set configuration: such methods involve acquisition of three images per sample using the following excitation-emission filter combinations:(1) donor excitation - donor emission, (2) acceptor excitation - acceptor emission, and (3) donor excitation - acceptor emission. Images from cells expressing donor or acceptor alone are also collected in the same configuration in order to calculate DSBT and ASBT. This normalization provides stringent correction for SBT (Vanderklish et al., 2000; Berney & Danuser, 2003; Takanaga & Frommer, 2008). It also allows estimation of donor/acceptor stoichiometry and for the presence of FRET and non-FRET signals in each acquired image (Gordon et al., 1998; Berney & Danuser, 2003).
The method relies on the assumption that the DSBT and ASBT are both proportional to the fluorescence intensity of the donor and acceptor in the sample, respectively. While this assumption is largely applicable when the fluorescence intensities detected in both donor and acceptor channels fall in the linear range of the detector, it could become inaccurate when observing samples with weak fluorescence intensity. Some algorithms take this into account, and correct DBT and SBT according to the fluorescence intensity range of the sample (Chen & Periasamy, 2006).
An alternative to filter-based FRET measurements is psFRET: spectral imaging followed by linear unmixing (Zimmermann et al., 2003). Emission spectra for each pixel are acquired (e.g. using a confocal microscope equipped with spectral detector or using a slit-scanning spectral system such as the SpectralDV) and deconvoluted by spectral unmixing to obtain the ‘pure’ emission for each fluorophore. This system has the additional advantage that it can calculate the contribution of autofluorescence in each pixel. When FRET happens between two independent fluorophores in the sample, however, linear unmixing algorithms tend to underestimate the contribution of the donor and overestimate that of the acceptor (Thaler et al., 2005). To solve this problem, Vogel and colleagues have developed a method in which the emission spectra are captured at two different excitation wavelengths in order to calculate the contribution of energy transfer (Thaler et al., 2005). Moreover, they have implemented a FRET calibration system that can be applied to normalize data and make them comparable between different imaging systems (Koushik et al., 2006).
VII. FRET sensors for determination of steady-state concentrations and composite flux, as well as their change
As outlined above, FRET is an extremely sensitive tool that can detect both changes in the distance and changes in the orientation of the dipoles. FRET can therefore report subtle changes in protein conformation. Thus, when a FRET reporter system consisting of two fluorophores is coupled with a polypeptide that specifically binds a ligand, FRET efficiency will be able to report the conformational change induced by ligand binding. In essence, the ratio between two conformational states serves as a proxy for the ligand concentration. If the polypeptide can exist in two conformations depending on the presence or absence of the ligand, the proportion of the binding protein in either form provides a measure of the ligand concentration.
The first genetically encoded FRET sensor was generated for measuring calcium (Persechini et al., 1997; Romoser et al., 1997). The rationale was that the dramatic conformational change of calmodulin to calcium binding would lead to a change in the distance between two GFP variants, which in turn would lead to a calcium concentration-dependent acceptor/donor intensity ratio change. In an alternative version, a recognition element was constructed by the fusion of calmodulin to a calmodulin-binding domain, which served as a conformational actuator to magnify the allosteric effect (Miyawaki et al., 1997). Today, more than a dozen calcium sensor variants are available (Griesbeck, 2004), some FRET-based, others exploiting the change in the spectral property of a single fluorophore induced by the conformational change of the calcium-binding domain fused to the fluorophore (Heim & Griesbeck, 2004). Systematic comparison of the dynamic properties of these sensors suggests that they differ regarding their ability to function efficiently in vivo (Hasan et al., 2004; Pologruto et al., 2004; Reiff et al., 2005). The reason for the difference in in vivo performance is not fully understood, but may be related to effects of the intracellular milieu on the sensors or the association with endogenous proteins. The calmodulin-based GCaMP2, which is composed of a single circular permuted GFP fused to calmodulin and its binding peptide, as well as the troponin-based TN-XL provide a high signal-to-noise ratio (SNR) in vivo and thus appear to be the best sensors for in vivo use (Garaschuk et al., 2007; Mao et al., 2008). While not classical FRET sensors, pHluorins and the Ptilosarcus GFP can serve as ratiometric dyes for measuring pH in vivo (Miesenbock et al., 1998; Schulte et al., 2006). Because certain versions of the yellow fluorescent protein (YFP) are sensitive to both halides and pH, a tandem fusion of enhanced YFP (eYFP) and enhanced cyan fluorescent protein (eCFP) named ‘clomelion’ can be used to monitor for chloride (Jose et al., 2007; Markova et al., 2008).
In addition to calcium, a rich source of FRET indicators has been engineered for other signaling molecules such as G protein-binding receptors (GPCRs) (Hoffmann et al., 2005), cyclic AMP (cAMP) (Zaccolo, 2004), cyclic GMP (cGMP) (Honda et al., 2001) and phosphatidylinositol phosphate (PIP) (Cicchetti et al., 2004). Excellent summaries of sensors for signaling processes are available, and thus this field will not be reviewed further here (Miyawaki, 2003; Lalonde et al., 2005; Medintz, 2006; Tsien, 2006).
The same overall principle of using conformational changes to report analytes can be used to construct metabolite sensors. Most FRET metabolite sensors developed to date exploit the conformational change of periplasmic binding proteins (PBPs) upon binding to their substrates. These binding proteins undergo large ‘venus fly-trap’ movements of two globular domains that are connected by a hinge region (Fig. 5). As a prototype, the best-studied family member, the maltose-binding protein (MBP), was fused to eCFP and eYFP at its termini to generate a sensor for maltose. Interestingly, the initial version did not show a change in energy transfer upon addition of maltose. Engineering of the domain connecting MBP to the fluorophores yielded a functional maltose sensor that was successfully deployed for a first proof of concept by imaging maltose accumulation in yeast cells (Fehr et al., 2002). MBP is a member of a large family of binding proteins for ions such as nitrate, sulfate and phosphate and for carboydrates including diverse pentoses, hexoses and oligosaccharides as well as amino acids and peptides (Tam & Saier, 1993). Functional sensors built from PBPs include variants for detecting phosphate, arabinose, ribose, glucose, galactose, maltose, glutamate and arginine (Fehr et al., 2003; Lager et al., 2003; Ye & Schultz, 2003; Okumoto et al., 2005; Gu et al., 2006; Bogner & Ludewig, 2007; Ha et al., 2007; Hires et al., 2008; Kaper et al., 2008) (Table 1). The same strategy was also used to verify the function of an unknown PBP from Agrobacterium tumefaciens as a sucrose/maltose/glucose-binding protein (Lager et al., 2006). Subsequent mutations in the binding pocket made this sensor significantly more selective for sucrose. Of various PBPs tested as recognition elements several were found to be nonfunctional; for example, only one of several phosphate-binding proteins from various bacteria was successfully converted into a phosphate FRET sensor. However, in general, most attempts to generate sensors from PBPs were successful. This is surprising, as the primary sequence conservation between the PBPs is limited, and the termini of the proteins are circularly permuted relative to each other. The PBP family is large, with members recognizing a wide spectrum of metabolites; therefore literally hundreds of different sensors can be created by exploiting the full spectrum of available scaffolds in this family. To facilitate this, a Gateway™-based cloning system was developed by which candidate polypeptides can easily be sandwiched between two fluorophores (Kaper et al., 2008).
Table 1.
Fluorescence resonance energy transfer (FRET) sensors for ions and metabolites
| Class | Analyte | Sensor | Selected references |
|---|---|---|---|
| Pentose | Arabinose | FLIPara | Kaper et al. (2008) |
| Ribose | FLIPrib | Lager et al. (2003) | |
| Hexose | Glucose | FLIPglu | Deuschle et al. (2005b); Takanaga et al. (2008) |
| Galactose | FLIPglu | Deuschle et al. (2005b); Takanaga et al. (2008) | |
| Disaccharide | Maltose | FLIPmal | Kaper et al. (2008) |
| Sucrose | FLIPsuc | Lager et al. (2006); Chaudhuri et al. (2008) | |
| Amino acid | Glutamate | FLIPe | Deuschle et al. (2005b); Okumoto et al. (2005); Hires et al. (2008) |
| Arginine | Bogner & Ludewig (2007) | ||
| Tryptophan | FLIPW | Kaper et al. (2007) | |
| Nucleobase | Purine | FLIPpur | A. Schmidt et al. (unpublished) |
| Polyamine | Polyamines | FLIPpa | S. Lalonde & W. B. Frommer (unpublished) |
| Ions | Calcium | Cameleon, TN-XL | Miyawaki et al. (1997); Romoser et al. (1997); Garaschuk et al. (2007) |
| Phosphate | FLIPphos | Gu et al. (2006) | |
| Protons | pHluorin, Ptsilosarcus GFP | Miesenbock et al. (1998); Schulte et al. (2006) | |
| Halides | Clomelion | Jose et al. (2007); Markova et al. (2008) |
Our initial naïve assumption was that FRET would depend mainly on the extent of the distance change. This hypothesis led to the choice of PBPs that show large distance changes between the two lobes when binding their ligands (Shilton et al., 1996). However, the absolute ratio changes of almost all of the initial PBP sensors were relatively similar (c. 25%) even when proteins with smaller distance changes between the N- and C-termini were used as scaffolds (Deuschle et al., 2005a), suggesting that factors other than distance between fluorophores contribute significantly to the FRET efficiency change. Even more compelling was the finding that a phosphate-binding protein, which carries both N- and C-termini attached to the same lobe (Ledvina et al., 1998), produced a functional phosphate sensor (Gu et al., 2006); similarly, the core of the fluorescent indicator protein (FLIP) E glutamate sensor probably also carries the fluorophores attached to same lobe (Okumoto et al., 2005), and several of the functional FRET glucose sensors that have one of the fluorophores inserted into the glucose-binding domain also carry both fluorophores on only one of the two lobes (Deuschle et al., 2005a,b; Takanaga et al., 2008). As it is not conceivable that a large distance change occurs for cases where the fluorophores are attached to the same lobe, different mechanisms must play a central role in signal generation. This conclusion is supported by the observation that deletion of five amino acids from the end of the maltose-binding protein converted a nonfunctional fusion protein into a functional maltose sensor as the distance change caused by the removal of five amino acid is expected to be minimal (Fehr et al., 2002). These unexpected results suggest that a different factor, namely the relative dipole orientation of the fluorophores, plays at least as important a role as the distance change. In all our constructs a similar strategy was used, namely, the PBP was fused via flexible linkers to the GFP variants. The peptide bonds afford free rotation of the fluorescent proteins, and the flexible linkers provide many potential relative positions of the fluorophores relative to each other (Fig. 5d). If we consider ensemble behavior, the dipole orientation will be affected by surface changes of the binding proteins upon interaction with the ligand (Deuschle et al., 2005b; Shimozono & Miyawaki, 2008).
The small initial ratio change proved sufficient to obtain reliable data in vitro and in vivo; however, it was expected that a higher ratio change would improve the sensitivity as a result of increased SNR (Fig. 8c). In many cases cellular metabolites do not fluctuate extensively in given experimental conditions, and therefore the background noise becomes manageable even with sensors with relatively small SNR, provided that a large number of data points can be collected from each sample. However, quantitative analysis for modeling as well as high-throughput screens critically depends on high SNR, for example as defined by the Z factor for high-throughput screens (Zhang et al., 1999).
Fig. 8.
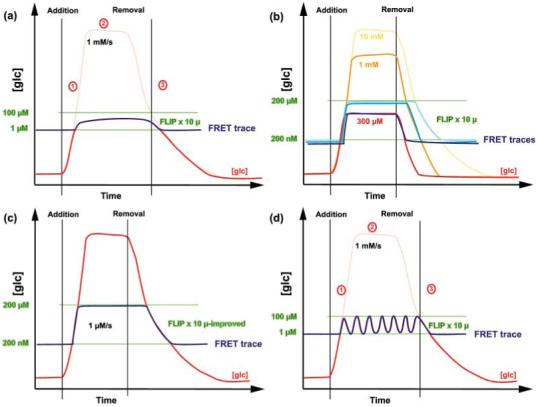
Flux analysis using fluorescence resonance energy transfer (FRET) sensors (Part II). (a) If the external concentration of glucose (glc) is reduced so that the intracellular concentrations fall into the detection interval of the sensor, a concentration-dependent response of a given sensor, here FLIP x 10 μ, can be observed. (b) Red, orange and yellow traces correspond to different hypothetical accumulation, steady-state and elimination rates as the external supply varies. Traces for each of the different accumulation scenarios are shown in three shades of blue. (c) Improvements in the dynamic range of the sensor response (larger FRET change compared with (a) provides a larger detection range). (d) Provided that adequate acquisition frequencies are applied, the sensor can detect potential oscillations of the glucose concentration.
Based on the observations described above, a reduction in the degrees of rotational freedom and a rigidification of the fusions may be a means to increase the ratio change and thus the SNR. A combination of rational and empirical engineering approaches has led to enhanced allosteric linkage of ligand binding and fluorophore rearrangement, and thus improved sensitivity (Deuschle et al., 2005b). The two major approaches used were deletions of the linkers for rigidification (Deuschle et al., 2005b; Takanaga et al., 2008) and insertion of one fluorophore into the backbone of the PBP to decrease the rotational freedom (Deuschle et al., 2005b; Dulla et al., 2008). Both strategies reproducibly lead to an up to tenfold increase in the ratio change. These improved sensors have been introduced into plants (Chaudhuri et al., 2008) and used to develop high-throughput systems for analysis of the metabolo-regulome (Looger et al., 2005). Many of these FRET metabolite sensors are available via http://carnegiedpb.stanford.edu/research/frommer/nanosensors/index.html. A combination of both strategies, however, did not improve the SNR further; in contrast to FLIPglu, in which sequential deletion of the linker sequence either had no effect or increased SNR, short deletions in the intramolecular sensor reduced SNR (Takanaga et al., 2008). Similar observations regarding the effect of linker deletions were also made by other laboratories (Lissandron et al., 2005; Hires et al., 2008). A systematic deletion series, which cannot map all potential linker compositions (2020 possible linkers for a 20 amino acid long linker), suggests a steep energy landscape for optimal linker composition and length (Hires et al., 2008; Takanaga et al., 2008).
These results support the hypothesis that the maximal observed FRET efficiency change depends not only on fluorophore distance but also on the relative orientation and rotational freedom of the reporters. The finding that a high ratio change can be obtained when the fluorophores are attached to the same lobe suggests that changes in the surface properties of the binding protein are responsible for the extent of the ratio change. If this hypothesis is correct, the sensors would have picometer sensitivity for conformational changes. This would have wide-ranging implications, as it would suggest that any protein that undergoes a small conformational change can in principle be converted into a FRET sensor. This hypothesis led to tests for other scaffolds, that is, that of the unrelated tryptophan repressor TrpR (Kaper et al., 2007). It also suggests that both the success in construction of a functional sensor and optimization would largely remain empirical, although with a relatively high success rate.
Before we will be able to use more rational design approaches for generating sensors, additional mechanistic and structural information will be required. A careful analysis of the sensors using anisotropy decay may help in testing the hypothesis. A huge number of FRET sensors with many variants have been generated, providing a scaffold with which to reciprocally develop and examine algorithms for rational optimization of FRET sensors. Thus, modeling approaches may become a means of creating and optimizing FRET sensors in future (Pham et al., 2007).
While CFP and YFP are frequently used as FRET reporter elements (Nagai et al., 2002; Evanko & Haydon, 2005), Citrine or Venus variants of YFP are becoming preferred because of their reduced pH and halide sensitivity (Nagai et al., 2002; Rekas et al., 2002). A new FP pair for FRET generated by DNA shuffling and error-prone PCR, cyan fluorescent protein for energy transfer (CyPet) and yellow fluorescent protein for energy transfer (Ypet), yield higher energy transfer with improved dynamic range (Nguyen & Daugherty, 2005). A pair of coral proteins, Midorishi Cyan (miCy) and monomeric Kusabira Orange (mKO), with larger spectral separation has the advantage of reduced DBT (Karasawa et al., 2004). Other alternatives include, for example, a GFP/Discosoma red fluorescent protein (DsRed) (TDimer2) pair (Yang et al., 2005). One of the limitations of the technology at present is that no two FRET pairs can be used simultaneously because of the spectral overlap. Recently, however, Campbell’s laboratory developed a compatible pair of fluorophores, namely monomeric teal fluorescent protein (mTFP1)/Citrine, which is spectrally similar to eCFP/eYFP and which can be used together with mAmetrine and tandem Tomato (tdTomato) (Ai et al., 2008). This system will enable the parallel analysis of two analytes or two affinity mutants in live cells. Targeting of one of the variants to the nucleus can further improve the data analysis (Ai et al., 2008).
A point that one should keep in mind, however, is that because of the importance of the dipole orientation any change in the chromophore positioning in the barrel or differences in the relative positions of the termini in circular permutated FPs may affect the ratio change, thus potentially requiring additional steps of optimization when any change is made in one of the FPs. Even subtle changes such as switching from eYFP to Venus (in our case a codon modified version named Aphrodite) can affect the ratio change (Deuschle et al., 2006). More dramatic changes to miCy and mKO without further optimization lead to a dramatic reduction in the observed ratio change for the glucose FRET sensors (Deuschle et al., 2006).
VIII. A ‘Gedankenexperiment’ illustrating the potential for flux analysis using FRET sensors
Metabolic flux is defined as the passage of molecules (moles of a particular metabolite) through a metabolic or transport step per unit cell mass per unit time. Flux is determined by the concentrations of compounds participating in a reaction, as well as the amount and properties of the enzyme/transporter (Wiechert et al., 2007). Classically, isotoptomer analysis is used for studying the flux through a pathway. FRET sensors can measure steady-state concentrations, but also the rate of accumulation (or elimination) of a compound in a cellular compartment. As opposed to isoptomer analysis, FRET sensors measure the ‘local’ flux at a single point in a metabolic pathway. For metabolites such as glucose and sucrose, this point is at the interface between extracellular medium and downstream metabolic reactions in the cytoplasm.
In order to be able to quantify metabolite concentrations or flux in vivo, it is necessary to verify that the sensors respond to binding of the metabolite and to determine the binding isotherm, and the affinity, the selectivity and the detection range of the sensors in vitro. As the sensors can be expressed in E. coli and fused to an affinity tag, the sensor proteins or candidate proteins can be affinity-purified by standard methods and titrated with the metabolite or its analogs using a fluorimeter to quantify metabolite-induced ratio changes in vitro (Fig. 6a). A ratio change can either be caused by FRET or by differential sensitivity of the fluorophore to changes in its environment such as ionic conditions or pH. It is thus important to carefully analyze the emission spectra or at least the fluorescence intensity for the two fluorescent proteins. A FRET efficiency change is characterized by a corresponding increase in donor emission and decrease in acceptor emission, or vice versa (Fig. 6b). The total energy in the system, that is, the emission of donor and acceptor, should remain constant. The analysis of the ratio and ratio change and thus the actual energy transfer efficiency is complicated by the presence of two emission peaks for eCFP. Although not the absolute value of energy transfer efficiency, the ratio of peak eYFP emission intensity over eCFP emission intensity for the shorter wavelength peak (485 nm) of eCFP is commonly used as a proxy of FRET efficiency (Fig. 6b). The detection range of the sensors is determined by the SNR, and thus the higher the ratio change, the larger the detection range. For simplicity, the detection range may be approximated as 10 × below and above the dissociation constant (Kd) of the sensor (Takanaga et al., 2008).
Fig. 6.
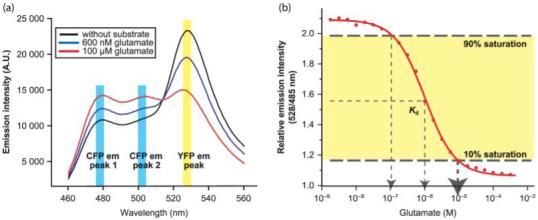
In vitro analysis of fluorescence resonance energy transfer (FRET) sensors. (a) In vitro binding isotherms for the glutamate FRET sensor FLII81PE-1 μ purified from Escherichia coli in the presence of glutamate. (b) Emission spectrum of FLII81PE-1 μ in the presence of 0, 600, and 100 μM glutamate (excitation of eCFP at 433 nm). Note the two emission peaks for eCFP at 477 and 501 nm. The figures are modified with permission from Deuschle et al. (2005b), courtesy of Protein Science. em, emission; Kd, dissociation constant.
The purified FRET metabolite sensors can be used to determine the concentration of analytes in vitro by adding the solution of interest to purified sensor protein samples, as exemplified by their use for quantitative analysis of maltooligosaccharide content in beer, or glutamate and phosphate content in cell extracts, culture media or soy sauce (Fehr et al., 2002; Okumoto et al., 2005; Gu et al., 2006). The affinity of the sensor used for in vitro analysis is less important because the sample of interest can be diluted so that it corresponds to the detection range of the sensor. The purified sensors can also be used for more sophisticated analysis, for example for simultaneous recording of glutamate release and electrical activity in brain slices (Dulla et al., 2008). However, for this purpose as well as for other in vivo applications, the detection range of the sensors is crucial. If the base concentrations in the tissue exceed the concentration at which the sensor is saturated, or if the tissue concentrations do not reach concentrations that can be detected by the sensor, no response will be observed (Fig. 7). A major advantage of using the PBPs as a scaffold for sensor construction is their intrinsic high affinity for their ligands, which is frequently in the nanomolar range. Moreover, the availability of crystal structures for many members of this family helps to guide site-directed mutagenesis of positions that either directly contribute to binding or stabilize the ligand-bound, closed form; mutation of these positions is expected to reduce the affinity and thus will shift the detection range towards the low affinity range. Using this approach, a set of five FRET glucose sensors was constructed (with Kd values of 170 nM, 2 μM, 30 μM, 600 μM and 3.2 mM) that covers the low nanomolar to high millimolar range completely (Deuschle et al., 2006; Takanaga et al., 2008). Similar sets of affinity mutants have also been constructed for the other metabolite sensors (Lager et al., 2003; Okumoto et al., 2005; Gu et al., 2006). The affinity mutants also serve as excellent controls for exclusion of artifacts caused by the effects of parameters other than metabolite binding on the sensors. The affinity mutants are expected to be similar with respect to their sensitivity to other parameters, as they differ in only one or two residues, and therefore should show in vivo response isotherms that correspond to their Kd values (Takanaga & Frommer, 2008).
Fig. 7.
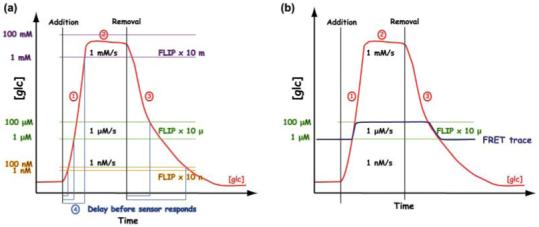
Flux analysis using fluorescence resonance energy transfer (FRET) sensors (Part I). (a) A hypothetical response curve for accumulation and elimination of metabolite x (here glucose; the red line tracks x; the addition and removal of glucose are indicated by black vertical bars) in the cytosol of a cell is shown over time for a defined external glucose concentration. The accumulation rate is dominated by the rate of uptake, and the rate of metabolic conversion. As a result of saturation of the transport system, a steady state (2) is reached at which uptake and metabolism are balanced. The accumulation rate can be obtained from the slope (1). The elimination rate (2) is dominated by the rate of efflux plus the rate of metabolism. Three sensors are shown; each has a defined detection range (FLIP x 10 m, with an affinity of 10 mM and a detection range of 1-100 mM, purple horizontal bars; FLIP x 10 μ, with an affinity of 10 μM and a detection range of 1-100 μM, green horizontal bars; FLIP x 10 n, with an affinity of 10 nM and a detection range of 1-100 nM, yellow horizontal bars). Lower affinity sensors have narrower detection ranges and thus faster reaction times to the external substrate. The actual FLIPglu13 sensor set (Deuschle et al., 2006) has overlapping detection ranges. (b) A given sensor tracks metabolite x within the detection range (blue trace), and thus sensor saturation does not necessarily indicate saturation of the biological system. Note also that the endogenous concentrations of x may be below the detection range of a sensor. If endogenous concentrations approach or exceed the upper limit of the detection range, no sensor response will be observed. If the rate of accumulation is lower than the rate of elimination, no response will be measurable. Quantitative data that can be derived from the measurements include the steady state at a given external concentration, the delay in response, which depends on the accumulation/elimination rate, and the actual rate of accumulation and/or elimination.
Nanosensors can provide a wealth of quantitative information when used for in vivo experiments. The analysis provides information on the steady state, and also on the sum of fluxes during the accumulation phase as well as the sum of fluxes during the removal phase. This is illustrated in Fig. 7. Typically, a cell or tissue expressing the sensor is mounted under a fluorescence imaging system and subjected to perfusion protocols in which the concentration of the metabolite of interest, for example glucose, is changed. When glucose is added, it enters the cell by a transporter with defined kinetic properties and is subsequently phosphorylated by hexokinase in the cytosol, followed by further glycolysis. When glucose enters the cell, intracellular glucose concentrations will rise depending on the relative flux rates across the plasma membrane and the rate of conversion to glucose-6-phosphate (Fig. 7a). If the conversion rate is faster than the uptake, we will not observe an increase in the cytosolic glucose concentration. For example, in the case of the phosphate sensor, no response to perfusion with phosphate was observed, unless the cells were starved (Gu et al., 2006). This suggested that phosphate uptake rates were potentially limiting. When the Na+-dependent phosphate transporter NaPi was overexpressed in these cells, the response of the sensor FLIPPi-30m to phosphate was readily measurable (Gu et al., 2006). In the case of glucose, responses of sensors with Kd c. 600 μM were observed for most cell types analyzed, indicating that uptake was faster than metabolism under the conditions tested (Fehr et al., 2003; Deuschle et al., 2006). Uptake was limiting only in HEK293T cells, in which the cytosolic concentration did not reach the detection range of sensors with Kd c. 600 μM, as demonstrated by overexpression of various glucose transporters and by using sensors with higher affinity (Takanaga & Frommer, 2008). Thus a FRET sensor can be used to determine the relative (composite) flux rates in different cell types. The detection range of the sensor has to match the in vivo concentration range. Typically the detection range of the sensor is smaller than the range of substrate concentration in vivo. Thus, as pointed out above, it is useful to have a set of sensors with different affinities that cover the entire range of the substrate in vivo. Estimating the expected range is difficult because no other methods are currently available that allow measurement of cytosolic concentrations, at least not in cells in which other compartments exist. Thus the best strategy is simply to introduce a wide spectrum of sensors with varied affinities into the organism or cell type of interest, as in the case of FRET glucose sensors covering the low nanomolar to high millimolar range (Deuschle et al., 2006; Takanaga et al., 2008). A Gedankenexperiment helps to illustrate the use of the FRET sensors (Figs 7, 8). If we add a large amount of external glucose to a cell, sensors that are not already saturated or sensors and have a response range below the point of biological saturation will respond. Under conditions in which sensor and acquisition kinetics are not limiting, the slope provides a quantitative measure of the fluxes. The accumulation slope (slope 1 in Fig. 7a) corresponds to a composite of flux rates, namely the uptake rates minus the rate of metabolism (minus flux into compartments) (Fig. 4). It is not easy to resolve the components of the corrective flux rates measured by FRET sensors. Nevertheless, simple mathematical models, for example using Michaelis-Menten equations for the individual reactions and known parameters for transport and metabolism, describe the cellular responses of human cells extraordinarily well (Fehr et al., 2005). Intracellular glucose concentrations will saturate as a consequence of the kinetic properties of the transporters and the relative rates of uptake and metabolism. When glucose is removed from the medium, cytosolic glucose will be eliminated by a combination of efflux, metabolism and release from compartments (slope 3 in Fig. 7a). Thus the slopes for accumulation and elimination will probably be different (Fig. 7a,b). This difference in the composition of accumulation and elimination rates can be used to model the process and to derive individual fluxes. Figure 7b shows the portion of the actual glucose accumulation and elimination curve that is covered by an individual sensor. A high-affinity sensor will respond earlier and will show a steeper slope (slope 4 in Fig. 7a), as the detection range of the sensor becomes exponentially smaller with the increase of the affinity of the sensors. Low-affinity sensors cover a larger range. Note that the set of FLIPglu sensors developed to date covers the range from low nanomolar to high millimolar with overlapping detection ranges (Takanaga et al., 2008). If the saturation of the sensor with the lowest affinity is higher than the biological saturation (Fig. 7a, label 2), the ratio change of the sensor will be smaller, reflecting partial saturation. Apparently, perfusion with different concentrations will yield different degrees of saturation of the sensor that covers the respective cytosolic concentration range (Fig. 8a,b). Perfusion protocols using square pulses with increasing glucose concentrations yield in vivo response curves (Fehr et al., 2005; Deuschle et al., 2006; Takanaga et al., 2008). With an automated perfusion protocol with very accurate control of solution exchange, the delay time between the events, for example change of medium, and the response represents another piece of information; high-affinity sensors are expected to show a short delay after addition of glucose, while low-affinity sensors are expected to show longer delay times. The exact parameters will depend on the initial glucose concentration in the cytosol and the slope of the endogenous change at different concentrations. When uptake is mediated by an obligatory exchange mechanism and under conditions in which metabolic rates are low, as is the case for tryptophan uptake in oral cancer cells (KB cells), removal of tryptophan from medium will not lead to a change (Kaper et al., 2007). The steady-state tryptophan concentrations will return to baseline only after addition of an appropriate exchange substrate, such as histidine or kynurenine. If appropriate acquisition protocols are used, the sensors also allow detection of metabolite oscillation (Fig. 8d). Sensors can also be targeted to the cellular compartments; then, by analyzing cells expressing the glucose sensor in the cytosol and in the compartment (the nucleus or endoplasmic reticulum (ER)), one can extract information about intracellular fluxes between the cytosol and the compartments. In the future the use of multiple sensors covering several steps of a metabolic or signaling pathway, for example glucose and glucose-6-phosphate, will be desirable. Moreover, in addition to the kinetics of a metabolite, any regulatory process acting on individual flux components that leads to a change in steady state can be studied using metabolite sensors, unless the regulation happens in such a way that it affects two antagonizing fluxes and the interference cancels out the individual contributions.
One of the exciting results from the use of metabolic FRET sensors is that sugar concentrations in plants can vary over several orders of magnitude (at least low nanomolar to medium millimolar) depending on supply levels. It has been speculated previously that cellular concentrations may be kept stable by homeostatic control networks, or that free metabolites will not become available in the cell as a result of substrate channeling in transporter/enzyme complexes. The studies summarized here demonstrate that significant concentrations of free metabolites accumulate in the cytosol, at least transiently. It seems that, if channeling occurs, the sensors will underestimate the actual composite fluxes.
In summary, the sensors thus provide rich information for modeling metabolic flux (Wiechert et al., 2007). Theoretically, the cytosolic concentrations and the flux rates can be calculated from the FRET data under the assumption that the affinity of the sensors is identical in vitro and in vivo. Because the in vivo ratio is affected by background fluorescence and SNR, the expression level has an effect on the observed ratio. Thus in most cases in vivo response curves are required for calculations of steady state. The question of whether saturation is a result of the biological system or the sensor appears to be important for the calculation of the cytosolic glucose concentrations. In vivo calibration and clamping of the concentration in the compartment of interest would be advantageous. The use of detergents for permeabilization may be an option; however, their use is often problematic, as cells may lyze, and permeabilization will affect the intracellular ionic environment significantly (Fehr et al., 2005). A careful comparison with data obtained using other techniques may help address these questions.
IX. The in vivo measurement set-up
Live cells expressing FRET metabolite sensors are analyzed by quantitative analysis of the properties of the two fluorophores, specifically FPs for genetically encoded sensors. The fluorescent properties can be measured either in a fluorimeter (Kaper et al., 2008) or under a fluorescence microscope (Niittyla et al., in press; Takanaga et al., 2008). The set-up currently used for FRET analysis in our laboratory is based on an inverted fluorescence microscope equipped with high numerical aperture fluorescence-corrected ×20, ×40, or×63 immersion objectives and either a sensitive CCD camera or, for cases where high-sensitivity measurements are required, an on-chip multiplication gain camera.
The major advantages of fluorimetric imaging analysis over bulk analysis in fluorimeters (e.g. microplate reader) include the ability to observe the response of individual cells even when only a limited number of cells express the sensors, resulting in reduced loss of information relative to the averaging that occurs in bulk analysis. Therefore, the cellular concentration of metabolite in the cells expressing sensors in a heterologous population (e.g. specific cell types in an intact organ) can be monitored with appropriate imaging settings (e.g. multi-photon confocal microscopy) (Lissandron et al., 2007). Most importantly, imaging allows multiple rounds of perfusion of the cells with different media, which is typically not feasible with the use of fluorimeters. The resolution of the system is of less concern because, even when subcellular concentrations need to be analyzed, the localization of the sensors can be genetically controlled. Thus, if the subcellular localization of the sensors has been confirmed independently, for example by confocal microscopy, the fluorescence signal will derive only from the compartment to which the sensor was targeted. In fluorescent microscopes, dual emission intensity ratios can be recorded by motorized switching between emission filters using a filter wheel, by simultaneous recording of both wavelengths on two halves of the CCD chip using an image splitter, or by recording spectra for each pixel with the help of gratings combined with the use of spectral unmixing to resolve the individual components. While psFRET offers more accurate calculation of FRET efficiency, as discussed above, the acquisition time required is longer (typically < 1 Hz) compared with filter-based FRET (typically > 5 Hz). Therefore, when observing dynamic metabolite change or oscillations of signaling molecules (Fig. 8d), filter-based methods have considerable advantages. Here we will discuss the important components of filter-based FRET imaging settings for the CFP/YFP pair and its variants. When required, for example, for signaling studies or to observe metabolite oscillations (Fig. 8d), high-speed acquisition is possible. The association rate constants of wild-type periplasmic binding for sugar binding are c. 107 M-1 s-1; dissociation rate constants are between 1 and 100 s-1 (Miller et al., 1983). As most metabolic analyses use low-affinity mutants of the sensors, one may expect that the measurements are typically not limited by the response kinetics of the sensors.
Excitation for CFP and YFP (required for the bleed-through correction) is provided by light sources equipped with appropriate excitation filters. Filters are chosen depending on the fluorophores used, and require optimization of bandwidth to obtain maximal excitation while minimizing the bleed-through. Specifically, the donor excitation filters should maximize the CFP excitation while minimizing the direct excitation of YFP (ASBT), and the emission filters should maximize the signal while minimizing the cross-talk between the two fluorophores and unwanted autofluorescence. The sensitivity of the system (therefore the SNR) is highly dependent on the choice of the optimal filter sets. Standard filter sets suitable for measurements in plants are available from a variety of companies. The selection requires a compromise between optimal excitation of the donor and minimal cross-excitation of the acceptor, and minimal excitation of endogenous fluorescent compounds. A broader bandwidth for excitation and emission yields higher fluorescence intensities. Thus, for maximal sensitivity, high-transmission modified Magnetron sputter-coated filter sets may be advantageous. These filters are characterized by increased transmission of 93-97%, stricter spectral tolerance and higher pass-band steepness. This higher steepness allows a selection of broader bandwidth combined with reduced bleed-through.
Light sources used for fluorophore excitation include mercury vapor lamps (peak at 436 nm suitable for CFP excitation), xenon arc lamps, lasers and light-emitting diodes (LEDs) (Nishigaki et al., 2006). Laser excitation provides the highest intensity, but peaks available are not optimal for all fluorophores. Especially for CFP, the 458-nm argon laser line typically available in confocal settings is not optimal for CFP excitation (excitation peak 433 nm), and also causes some excitation of YFP (i.e. ASBT). The 407-nm krypton laser line reduces ASBT, but krypton laser sources are relatively expensive. Mercury lamps have several peaks with high excitation, while xenon lamps show a more continuous spectrum, and thus are highly flexible. Typically, the amount of excitation light is limiting, especially when high-speed imaging is required. Thus, high-intensity light sources, in combination with high-transmission filters and high-sensitivity cameras, have proved useful. Obviously too much excitation can lead to photobleaching and phototoxicity of the cells; thus, once a satisfactory level of signal intensity is reached, experimental settings must be optimized to reduce such adverse effects. A reduction of excitation light intensity using neutral density filters, a reduction in the frequency of acquisition, or a compromise between excitation intensity and duration is typically necessary for such optimization.
Images for FRET metabolite sensors are typically acquired at 5-20-s intervals within the linear detection range of the camera. When transport kinetics linearity is studied, it is very important to acquire an adequate number of images to establish a tight time-concentration correlation, but a compromise has to be made between tight linearity and photobleaching. Quantitative data are derived by pixel-by-pixel integration of regions of interest defined in the ratiometric images using software. Typically, such software allows simultaneous image acquisition and ratiometric analyses, therefore providing real-time feedback while the experiment is running. The fluorescence intensity for the CFP and YFP emission channels is monitored sequentially with CFP and YFP excitation in order to normalize the ASBT relative to YFP intensity. The FRET index Fc/D for sensitized emission is then calculated from the peak intensity ratios (YFP/CFP) using background and bleed-through corrections (Vanderklish et al., 2000). For bleed-through correction, images for sensor versions carrying either a defective CFP (FLIPx-†CFP) or a defective YFP (FLIPx-†YFP) can be used for correction (Vanderklish et al., 2000; Takanaga & Frommer, 2008). The FRET index is defined as Fc = Ff-Df × (Fd/Dd) - Af × (Fa/Aa). The uppercase letter represents the respective filter set: D, CFPexCFPem; F, CFPexYFPem; A, YFPexYFPem. The subscript lowercase letter represents the type of fluorophore present in the sample: d, CFP; f, CFP + YFP (i.e. the FRET sensor); a, YFP. Ff is the apparent YFP emission from the FRET sensor, Df is the CFP emission from the FRET sensor, Fd/Dd is the DSBT correction factor normalized to the CFP emission intensity, Af corresponds to YFP emission from the FRET sensor, and Fa/Aa is the ASBT correction factor normalized to the YFP emission intensity. Fd/Dd and Fa/Aa can be measured separately from FLIPx-†CFP and FLIPx-†YFP, respectively. Depending on the expression level of the different nanosensors, exposure times are varied (typically 150-200 ms), and the on-chip multiplication gain of electron multiplying CCD (EMCCD) camera is adjusted as required to obtain a satisfactory signal intensity. Faster acquisitions are possible, provided that the emission intensity does not become limiting. The requirement for the resolution depends on the experimental settings and aims. When analyzing a single cell expressing a FRET sensor in a single compartment, typically the signal from the entire cell is pooled as one region of interest for the analysis. In such settings spatial resolution is not relevant; essentially a single pixel per cell suffices (here the microscope is used analogously to a fluorimeter). Because the resolution of the images is of no major concern, it is possible to use software binning of pixels to increase the sensitivity, minimize the required excitation intensity (and therefore minimize phototoxicity), and reduce noise levels. Typically measurements can be carried out over many hours without causing obvious damaging effects on the cells.
Perfusion is used to manipulate the external concentration of the substrate. It is therefore essential that the cells or tissues to be analyzed remain fixed in all three planes. Some mammalian cell lines are ideal because many of them grow attached to glass surfaces. Pretreatment of the slides with an extracellular matrix such as collagen and poly-L/D-lysine enhances the surface interaction, permitting analysis of the same cells over many hours (Takanaga et al., 2008). Roots of Arabidopsis can be attached to the glass surface with a surgical glue (Chaudhuri et al., 2008), while yeast cells require more sophisticated technologies. The choice of appropriate perfusion chambers is important. Closed laminar-flow chambers with minimal solution exchange time are ideal; however, some applications(i.e. whole-plant measurement) may require the use of an open chamber. When studying the kinetics of metabolite flux, it is very important to determine the rates of solution exchange in the chamber used for the experiment and to ensure that the perfusion rate is not limiting.
X. Spatial resolution of FRET metabolite analyses
High spatial resolution of fluorescent markers and sensors can be achieved by confocal microscopy. If combined with high temporal resolution, spatial differences such as calcium waves can be observed (Weijer, 2003). In the case of FRET sensors, spatial resolution can be achieved by genetic means. Genetically encoded FRET sensors can be expressed in a defined set of cells in an organism using specific promoters. Even more importantly, fusion of the sensor to a signal sequence permits targeting to defined subcellular compartments. Measurement at the cell surface or in the intracellular space can be achieved by extracellular addition of the purified sensor protein (Dulla et al., 2008), by addition of immobilized sensor protein (Sun et al., 2008), or by expressing the sensor in the cells of interest and targeting it to the cell surface using a secretion signal sequence with or without addition of a membrane anchor (Gao et al., 2004; Okumoto et al., 2005). Flux across the ER membrane can be monitored if an ER signal sequence and an ER retention sequence are fused to the sensor, as demonstrated for the glucose sensor, or if the sensor can be targeted to the nucleus to monitor nuclear glucose flux (Fehr et al., 2004; Fehr et al., 2005).
XI. Potential effect of other parameters on the sensor response
As described above, the sensors exploit protein conformation as a proxy for the concentration of the metabolite. Protein conformation is sensitive to a variety of factors, for example ionic concentrations, pH, redox potential and binding to other cellular factors. The sensor will almost certainly respond to a change in the cellular environment even in the absence of a change in metabolic flux. It is necessary to consider the sensitivity of the three major components: the conformation of the recognition element; the structure of the linkers between the core structure of the recognition element and the core structure of the fluorophores; and the fluorophores themselves.
Thus, especially if measurements are carried out in compartments that are known to rapidly change their pH, such as vesicles or the vacuole, additional controls may be required. In this case, it may be useful to determine the in vitro pH dependence of the sensor. However, pH changes will have different kinetics; the effect of the pH change may affect the ratio in the opposite direction relative to the ligand-induced ratio change, in which case it is easy to evaluate the concentration of the analyte even if pH changes affect the response. For the metabolite sensors tested to date, the fluorophores appear to be most sensitive to pH (Chaudhuri et al., 2008); the ratio change can be induced by pH because of the differential sensitivity of two fluorophores to the pH and not because of the change in FRET efficiency. It is thus recommended to use GFP variants such as Citrine or Venus in which pH and halide sensitivities have been minimized (Rekas et al., 2002). While even the improved GFP variants are not free of pH and halide sensitivity, careful analysis of the fluorescence intensities of the two fluorophores can be used to identify pH-induced ratio changes (Chaudhuri et al., 2008). Moreover, the affinity mutants can serve as controls to exclude artifacts (Lager et al., 2003; Takanaga & Frommer, 2008).
However, the sensitivity of the GFP variants to pH and halides has been exploited to develop pH sensors such as pHluorins. pHluorins and the Ptilosarcus GFP can serve as ratiometric dyes for measuring pH in vivo (Miesenbock et al., 1998; Schulte et al., 2006). As certain versions of the YFP are sensitive to both halides and pH, a tandem fusion of eYFP and eCFP named ‘clomelion’ can be used to monitor chloride (Jose et al., 2007; Markova et al., 2008). Variants of GFP with enhanced redox sensitivity (roGFP) are used to visualize the oxidation state of the indicator and thus the redox potential (Dooley et al., 2004). Recently, alternatives using redox-sensitive peptide conformations have also been developed (Kolossov et al., 2008). These sets of sensors for pH, halides and redox status may serve as additional controls to exclude artifacts and to calibrate the in vivo responses to the in vitro behavior.
Another potential drawback is the potential effect of sensor expression on intracellular metabolite pools, which may affect metabolic flux or signaling. Typically, FRET sensors are expressed from strong promoters in order to obtain satisfactory signal intensity; for example, the Cauliflower Mosaic Virus (CaMV) 35S promoter can lead to accumulation of proteins in the range of 1% to several % of total protein (Guenoune et al., 2002). These sensors create a new buffer for the ligand they recognize and thus will necessarily affect metabolism. This may not be an issue when the metabolite concentrations exceed the sensor concentration significantly, but may become relevant when metabolite concentrations are far below the sensor concentration. Moreover, this may be less relevant in the case of major metabolites as opposed to the case of secondary messengers. Careful phenotypic analysis, growth curves and more detailed physiological analyses such as isoptomer-based flux analysis can be used to exclude potential artifacts. In cases of problems, optimization of SNR will allow the use of weaker promoters to reduce interference. Also, the sensors can be expressed under inducible promoters to avoid a potential effect on growth and development. Such artifacts can be excluded if the recognition element is derived from the organism to be analyzed and if the sensor is used to replace the endogenous protein. 13C isoptomer analysis can be used to determine potential effects of the sensors on metabolic flux (Niittyla et al., in press).
XII. Alternatives to FRET sensors for monitoring metabolite concentrations
Promoter-reporter constructs have been used in many systems as biosensors for metabolites (Leveau & Lindow, 2002). One of the most frequently used biosensors is the DR5-Gus construct for measuring auxin (for a critical evaluation see Bai & DeMason, 2006). The activity of enzymes as well as metabolite concentrations can be measured using protein fusions that change subcellular location in response to a signal, that is, relocate from the cytosol to the plasma membrane. Such translocation sensors can be generated by fusion of a translocating peptide to a fluorescent protein. One of the best studied translocation sensors, GFP-AktPH, is based on a GFP fusion to a pleckstrin homology (PH) domain derived from phospholipase Cδ. GFP-AktPH was used for measuring phosphatidylinositol-triphosphate (PIP3) and phospholipase activity (Nahorski et al., 2003). Similarly, the C1 domain of phosphokinase C γ (PKCγ) has been deployed to detect diacylglycerol (DAG) (Liou et al., 2005). These translocation sensors have been used to dissect the timing of the activation of phosphatidylinositol (PI) 3-kinase by insulin and how PIP3 can activate secretion of the glucose transporter GLUT4 (Tengholm & Meyer, 2002). Activity sensors for rho GTPases and heterotimeric G proteins were generated using the same principle (Honda et al., 2001; Pertz & Hahn, 2004; Zaccolo, 2004). Such sensors have successfully been used in plants, for example phospholipase C 3 (PLC3) for measuring accumulation of diacylglycerol or the PH domain of PLC∂1 for measuring phosphatidylinositol 4,5-bisphosphate (Helling et al., 2006; van Leeuwen et al., 2007).
An alternative to the use of polypeptides as recognition elements for FRET sensors is the use of RNA to detect metabolites. Riboswitches occur naturally as genetic control elements which make use of untranslated mRNA sequences that form a secondary structure that can serve as a specific binding pocket for a metabolite and that can regulate gene expression. Riboswitches also undergo a conformational change upon ligand binding and communicate binding to an analyte by affecting transcription. Natural riboswitches exist for coenzyme B12, thiamine pyrophosphate, fravin mononucletotide (FMN), S-adenosylmethionine, guanine, adenine and lysine (Barrick & Breaker, 2007; Kim & Breaker, 2008). The spectrum of sensors can be expanded by engineering of the RNAs, which is possible by selection of cells expressing a mutant collection (Win & Smolke, 2007).
Improvements in biophysical imaging technologies such as mass spectrometry imaging, nuclear magnetic resonance (NMR) imaging, Raman spectroscopy and secondary ion mass spectrometry (SIMS) imaging will lead to significant advances in the metabolomics of tissues and organs (Li et al., 2007). These technologies, as well as the set of sensors described in this section, are complementary to the FRET sensors and will be useful to calibrate the in vivo responses of the FRET sensors and vice versa.
XIII. In vivo analysis of metabolite flux in microorganisms
The synthetic biology of microorganisms requires tailoring of metabolism to optimal productivity (Keasling, 2008). Flux through primary metabolism, especially, needs to be adjusted (Zeng & Biebl, 2002; Causey et al., 2004). To identify and eliminate constraints, extensive efforts have been made to better understand metabolic flux. Most approaches have focused on the use of 13C-isoptomer analysis. FRET sensors can be deployed as a complementary tool. With the help of the sensors, the accumulation of analytes in the cell can be monitored using simple fluorimetric assays in microplates (Kaper et al., 2008). The sensors can replace radiotracer studies, also providing information on steady-state concentrations in the cell. The sensors also provide information on the accumulation rate inside the cell (Kaper et al., 2008). Furthermore, the technology circumvents the problem that metabolites adsorb to the cell wall as a result of nonspecific binding, thus complicating the interpretation of data obtained for intracellular concentrations with metabolomic methods (Teusink et al., 1998). Importantly, when combined with microfluidics, FRET sensors can provide important information on differences in behavior between individual cells or cell populations (Yan et al., 2008). The sensors can also be used to screen mutant collections or chemical libraries for effects on the accumulation of the analyte of interest to help identify the signaling networks that control flux. Immobilized sensors (Sun et al., 2008) or cells expressing the sensors (Kaper et al., 2008) can serve as biosensors, for example for monitoring industrial fermentation processes. Similar to bacterial or fungal bioreporters, which can be used for monitoring chemical, physical or biological environments, both the immobilized sensors and bacterial cells expressing the sensors can be used for monitoring release of metabolites from plants (Leveau & Lindow, 2002; Lindow & Leveau, 2002).
XIV. In vivo analysis of metabolite flux in mammalian cells
Some mammalian model cell lines are easy to grow in culture, adhere to coated coverslips, and have less endogenous fluorescence background compared with other organisms, and are therefore ideal platforms for the study of metabolite flux using FRET sensors. The metabolite concentrations of these cells can be monitored using FRET sensors over hours while external medium is changed by perfusion.
The cellular concentrations of many biologically important molecules change very rapidly. Transport and metabolism of primary metabolites such as sugars and amino acids are known to be especially fast. Therefore, it is very challenging to achieve sufficient time resolution to detect transient concentration changes with classical biochemical approaches. In cases where the cell population consists of more than one cell type, metabolite concentration change in underrepresented cell types could be masked by surrounding cells if the metabolite concentration is measured at the whole-population level.
As genetically encoded sensors allow measurement of rapid, transient concentration change at a single-cell level, FRET and single-fluorophore fluorescent sensors for molecules that work as secondary messengers have received the attention of cell biologists. Sensors for calcium (reviewed in Demaurex & Frieden, 2003), cAMP (reviewed in Mongillo et al., 2005), cGMP (Honda et al., 2001) and inositol 1,4,5-trisphosphate (Sugimoto et al., 2004; Tanimura et al., 2004; Remus et al., 2006) were successfully used to analyze secondary messenger concentration changes in response to various stimuli. In some cases, cellular microdomains with elevated secondary messenger concentrations were discovered, demonstrating the exquisite resolution of this technique (Honda et al., 2001; Remus et al., 2006).
Fluorescent sensors are also useful in tracking rapid metabolite import, export and metabolism in living cells. Fehr et al. (2003) showed that, in COS-7 cells, the glucose concentration can increase from base concentrations by at least two orders of magnitude, indicating very active transport. Removal of external glucose leads to rapid depletion of glucose from the cytosol (typically < 1 min), indicating that glucose metabolism and/or export in this cell type is extremely fast. Similarly, a large change in the cytosolic concentration of ribose could be measured using FRET sensors, although the metabolism/export rate was much slower than that of glucose (Lager et al., 2003). For several metabolites and ions, however, no ratio change was detectable in response to perfusion when the sensors were expressed in the cytosol, specifically for phosphate (Gu et al., 2006) and glutamate (Okumoto et al., 2005) in various cell lines, and surprisingly for glucose in HEK293T cells (Takanaga & Frommer, 2008). This finding suggested that, in these situations, either the rate of uptake is limiting and significantly slower than metabolism/efflux and therefore the cytosolic concentration is kept constant, and/or the concentration is outside of the dynamic range of the sensor. This hypothesis was confirmed by overexpressing an NaPi phosphate transporter for the case of phosphate flux (Gu et al., 2006), and various plasma membrane glucose transporters (GLUTs) for glucose flux in HEK293T cells (Takanaga & Frommer, 2008).
In contrast to the studies described above in which transport across the cell membrane was generally bidirectional, in a study by Kaper et al. (2007) the tryptophan sensor detected accumulation of tryptophan in the cytosol of mammalian cells, and the amino acid was found to be trapped in the cells; after removal of tryptophan from the perfusion medium, the amino acid remained inside the cell. Because of the predominance of amino acid exchangers in the cell lines used, trypotophan effluxed from the cells only after addition of an exchange substrate such as histidine or the degradation products of tryptophan, kynurenines. This example highlights the potential of FRET sensors to detect cellular efflux, a process that is usually difficult to measure, except by preloading of cells with radiotracer.
All of these studies were conducted in cell lines, but these sensors could also be used to visualize the metabolite flux in heterogeneous, physiological populations of cells (i.e. in tissue slices or mixed culture) or in transgenic organisms (Mank et al., 2006) in order to analyze metabolite flux in situ.
Another advantage of genetically encoded sensors is that they can be targeted to different cellular compartments, making them useful for measurement of intercompartmental fluxes. For example, Ca2+ in the ER is kept at a much higher concentration than in the cytosol and the release of Ca2+ into the cytosol triggers cellular responses such as muscle contraction and neurotransmitter release. FRET-based calcium sensors made it possible to study the strict link between the Ca2+ concentration in the ER and store-operated calcium influx (Arnaudeau et al., 2002). The export of glucose synthesized in the ER of hepatocytes is important to maintain blood glucose concentration stability. Glucose flux measurement into and out of the ER using FRET glucose sensors revealed that glucose transport across the ER membrane was rapid and bidirectional (Fehr et al., 2005) Further experiments with improved time resolution and glucose sensors with higher affinity revealed that the kinetics of import and export rate across the ER membrane are asymmetric at lower cellular glucose concentrations (Takanaga & Frommer, 2008). The expression of GLUT plasma membrane glucose transporters in HEK293T cells that have weak endogenous ER glucose transporter activity revealed that the GLUT transporters en route to the plasma membrane can mediate glucose uptake in the ER (Takanaga & Frommer, 2008).
FRET sensors can also be used to detect intercellular metabolite concentration changes. FRET-based glutamate sensors were used to detect glutamate release from neuronal cells when expressed on the outside of the plasma membrane of hippocampal neuronal cell culture, indicating that they can be used to visualize neuronal activities at the cellular level, or even at the single-synapse level if the sensor is targeted to the postsynaptic density (Okumoto et al., 2005). Recently, an improved version of the glutamate sensor was used to show that significant spillover to neighboring neurons persists during burst firing for hundreds of milliseconds. The observed glutamate spillover concentrations appear to be sufficient to prime NMDA receptors, potentially leading to nonsynaptic neuronal communication (Hires et al., 2008).
Calibration of genetic sensors in subcellular compartments is more difficult because the kinetics of plasma membrane transport have to be taken into consideration, and possible variations in ionic conditions between compartments can affect the read-out of the FRET ratio. Drugs that permeate the plasma membrane or equilibrate the pH across organelle membranes can be used to calibrate sensors in organelles of interest (reviewed in Demaurex & Frieden, 2003), but the results have to be interpreted with caution because the intracellular environment will be altered by permeabilization. Alternatively, targeted substrate release using ‘caged’ compounds may allow in situ calibration of sensors. Caging refers to a covalent modification of a compound that renders it inactive in a way in which irradiation with a defined light pulse can lead to quantitative release of the active compound (Ellis-Davies, 2008).
XV. In vivo analysis of metabolite flux in plants
Originally, FRET sensors were developed to study a specific question in plant biology. However, initial problems delayed progress. Specifically, despite the generation of multiple constructs, no stable Arabidopsis line with a high level of sensor-derived fluorescence in leaves was found. This result was in seeming contrast to the successful use of the cameleon calcium sensors for visualizing changes in calcium oscillation patterns in guard cells (Allen et al., 1999, 2000; Young et al., 2006). While the calcium FRET sensors were found to be highly expressed in Arabidopsis guard cells, < 10% of wild-type (Columbia (Col-0)) Arabidopsis plants transformed with various FLIP sensors had levels of fluorescence too low for reliable FRET analysis, and even those plants lost fluorescence during development (Deuschle et al., 2006). We speculated that, whereas guard cells are symplasmically isolated and are not subject to post-transcriptional gene silencing (PTGS) (Voinnet et al., 1998), the expression of sensor proteins in other cell types might be suppressed by PTGS. The problem was overcome by using Arabidopsis plants that carry mutations in SUPPRESSOR OF GENE SILENCING 3 (SGS3) or RNA-DEPENDENT POLYMERASE 6 (RDR6), genes that are required for PTGS (Deuschle et al., 2006). In these mutant backgrounds, > 70% of transformants showed significant fluorescence, strongly indicating that the expression of sensor proteins was indeed suppressed by PTGS. While we cannot rule out the possibility that the regulation of metabolites (i.e. glucose) we are looking at is altered in gene-silencing mutants, this approach enables us to create plants that express sensor proteins at a satisfactory level (Fig. 9).
Fig. 9.
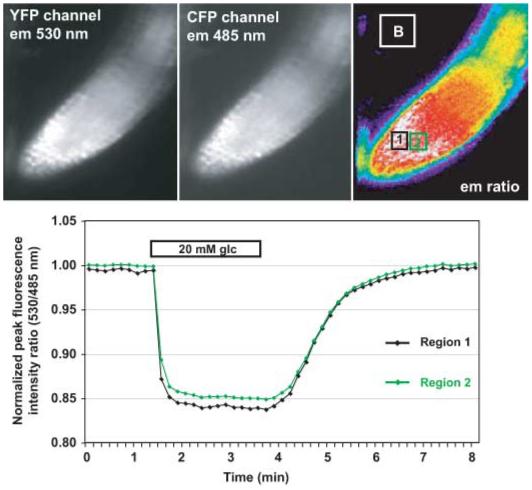
Glucose-induced fluorescence resonance energy transfer (FRET) changes in the cytosol of intact Arabidopsis roots. The FRET sensor FLIPglu-600μΔ13, with an affinity for glucose of 600 μM in stably transformed gene silencing mutant rdr6-11 Arabidopsis plants (Deuschle et al., 2006; Chaudhuri et al., 2008), responds to perfusion with 20 mM glucose. Top panel: Images of the root tip for the yellow fluorescent protein (YFP) and the cyan fluorescent protein (CFP) channels as well as the ratiometric image are shown in pseudocolor (at time 0). The regions from which the quantitative data are calculated are shown on the ratiometric image as black (region 1) and green (region 2) boxes. Data from the white box (B) were used for the background correction. Quantitative data were derived by pixel-by-pixel integration of the regions in the ratiometric image. Bottom panel: The graph shows the ratio of eYFP intensity divided by eCFP intensity (normalized to the starting ratio) for the two regions (the green trace corresponds to the green box (region 2) after background subtraction and normalization; the black trace (region 1) corresponds to the black box at different time-points (here 10-s intervals) measured over 8 min). The bars on top of the trace give the concentration and the duration of the glucose perfusion. The accumulation of glucose is fully reversible. Note that accumulation and elimination phases show different rate constants. Also note the low noise as apparent from the smooth traces. The image has been modified and reproduced in color, with permission, from Niittyla et al. (in press). em, emission.
The in vivo measurement of glucose flux in Arabidopsis cells suggested that cytosolic glucose concentrations are not subject to tight homeostatic control. Moreover, imaging experiments on both root and leaf cells revealed different metabolisms in these two cell types; without an external supply of glucose, cytosolic glucose concentration in root cells drops to < 90 nM whereas the cytosolic glucose concentration in epidermal cells did not decrease far below 300 μM, creating a steeper concentration gradient between these two cell types than previously expected. In the meantime, improved imaging technology has yielded high-quality data for both the glucose and sucrose sensor series in root tips of axenically grown Arabidopsis plants (Chaudhuri et al., 2008). Surprisingly, a careful titration of the responses at different sugar concentrations, and the determination of accumulation and elimination rates suggested that a transport mechanism independent of the previously analysed proton-sugar transporters is responsible for the uptake of sugar from roots. Supporting the hypothesis that uptake is not mediated by proton cotransport, the uptake of sugars was insensitive to the external pH and to protonophores (Chaudhuri et al., 2008). These results underscore the potential of metabolite sensors in detecting unidentified metabolite flux that escaped classical biochemical approaches.
The flux measurements using FRET sensors in plants have so far been largely limited to cellular calcium and sugars. However, with the increasing number of newly available FRET sensors, in combination with the technique allowing circumvention of the problem with PTGS, we expect that flux analyses of other biologically important molecules will be made possible. Recently, FRET sensors for arginine were created based on a glutamine-binding protein from E. coli (the insertion of the fluorophore into the binding domain presumably changed the affinity of the binding domain). The uptake of arginine into plant protoplasts and root cells was visualized using these sensors (Bogner & Ludewig, 2007).
In addition to the functions of transporters per se, chemical and electrical potential gradients across the membrane are an important factor determining the metabolite flux. Traditionally the detection of chemical and electrical gradients across the membrane required the use of electrophysiological techniques such as the use of a voltage or patch clamping. Recently, fluorescent probes including FRET sensors that detect such gradients have been developed. A set of chemical dyes have been synthesized (Gonzalez & Tsien, 1995), which have recently also been deployed in plants (Konrad & Hedrich, 2008). The first protein-based membrane voltage sensors exploit the sensitivity of the fluorophore to being brought close to the membrane. The FlaSh sensor (not to be confused with the biarsenical dye FlAsH), created by fusion of a truncated GFP ‘reporter domain’ to a voltage-gated K+ channel ‘detector domain,’ allows imaging of action potentials (Guerrero et al., 2002). Improved FRET voltage sensors that appears to be more robust were developed on the basis of a voltage sensor-containing phosphatase from a sea squirt (Dimitrov et al., 2007; Tsutsui et al., 2008). In addition, chemical voltage sensors have also been developed (Cacciatore et al., 1999). The FRET sensors, together with sensors for pH and halides and membrane potential sensors, will be used to identify the nature of the unidentified mechanism of metabolite transport into the cells.
It should also be noted that subcellular metabolite imaging, successfully performed for mammalian cells, will also be possible for plant cells. Subcellular metabolite imaging will be especially useful in studying pathways that require transport of molecules between more than two subcellular compartments. Furthermore, we will be able to study metabolite flux in mutants that are affected in the transport and/or metabolism of the molecule of interest, thereby integrating the gene functions into metabolite regulation.
The analysis of mutants in transport, metabolism or signaling is hampered by the necessity to combine homozygous mutants with the FRET sensor in the homozygous silencing background. However, as gene silencing is low in young seedlings and increases during development, it is possible to monitor glucose flux with FRET sensors in the root tips of wild-type Arabidopsis seedlings (Chaudhuri et al., 2008). This observation together with the establishment of a simple root perfusion system will help advance the analysis of small mutant collections with the help of FRET sensors.
XVI. Application of FRET sensors for high-throughput analyses
Over the past decade, many research groups have focused on the analysis of specific genes and their functions. Given that at present only a small number of plant proteins/genes have been studied experimentally, novel approaches are required to increase the speed of the discovery process. At present only 5936 genes out of a total of 28 523 genes found in the Arabidopsis genome in TAIR8 (The Arabidopsis Information Resource, http://www.arabidopsis.org) have been annotated with experimental data (21%). This means that experimental information is lacking for 79% of the Arabidopsis genes (E. Huala, TAIR, Carnegie Institution, unpublished). Therefore, without a large-scale identification of gene functions the possibilities for synthetic biology will be extremely limited in plant biotechnology. Examples of the usefulness of such data sets include TAIR, which can be used to find clones and mutants for the majority of the Arabidopsis genes (http://signal.salk.edu; http://rarge.gsc.riken.jp/), databases such as Atgenexpress (http://jsp.weigelworld.org/expviz/expviz.jsp) and Genevestigator (www.genevestigator.ethz.ch), and software such as eFP browser (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi) which allows fast in silico expression analysis as well as analysis of coexpression as a discovery tool (http://bar.utoronto.ca/ntools/cgi-bin/ntools_expression_angler.cgi).
The flux measurement of metabolites using FRET sensors can be reproduced with unprecedented time and spatial resolution once the automated protocol for detecting metabolite flux is established, making the sensors attractive as a novel platform of high-throughput screens. A proof-of-principle study using HEPG2 cells demonstrated that the decrease in plasma membrane glucose transporter activity, caused by the knockdown of GLUTs, can be detected using a new generation of FRET glucose sensors with larger dynamic range (Takanaga et al., 2008).
FRET sensors can thus be used for fluxomics, defined as the analysis of the contribution of the genome either to all fluxes in an organism as done using isoptomer analysis, or to the control of flux through a single node, as detected by FRET sensors (Wiechert et al., 2007).
High-throughput screening using FRET sensors requires an automated imaging platform suitable for high throughput, and a population of genetically varied samples (e.g. cells expressing a cDNA or siRNA library) compatible with the high-throughput format. Various robotic systems using either photomultipliers or automatic microscopes are available as detection systems. Baker’s yeast is at present one of the most advanced systems in eukaryotes for high-throughput analysis of gene function. A complete knock-out collection is available in microtiter plate format; in addition, currently double mutants are systematically created to analyze synthetic interactions as a tool for rapid functional assignment of gene function (Boone et al., 2007). The yeast knock-out collection can be used to screen for genes that affect metabolic flux. For the human genome, complete siRNA collections are available commercially. Screens of human siRNA collections have successfully been implemented to identify signaling pathways in cell cultures (Liou et al., 2005; Brandman et al., 2007; Galvez et al., 2007). A systematic screening of these collections using FRET metabolite sensors will identify novel players in signaling pathways. For plants, it will be necessary to develop single-cell systems that are compatible with high-throughput format transformation, and to construct a library of genetically varied samples that can be expressed in plant cells (Ogawa et al., 2008).
FRET sensors provide a new tool set suitable for analyzing metabolic flux at defined points of pathways in any organism that is amenable to transfection or transformation. A set of sensors for pentoses, hexoses, disaccharides, amino acids and various ions is available. We expect that the list of substrates that can be measured with FRET sensors will expand rapidly as a result of the use of a wealth of recognition elements in the PBP family, the use of alternative scaffolds and computational design of novel binding proteins. Efficient strategies have been developed for generating affinity series and for improving SNR. In addition, computational design on the scaffold of natural binding proteins would allow the construction of sensors for important biological molecules for which natural binding proteins are either not available or not suitable as sensor scaffolds (e.g. for hormones) (Dwyer & Hellinga, 2004). A major application of these FRET sensors in the future will be in high-throughput screens to identify metabolic signaling. In conjunction with progress in the area of systems biology such as the development of physical protein interaction networks and quantitative phosphoproteomics, the potential for metabolite flux engineering in plants will increase significantly over the next 5 yr.
Acknowledgements
We would like to dedicate this review to Ulf-Ingo Flügge, University of Köln, Widmar Tanner, University of Regensburg and Winslow Briggs, former Director of the Department of Plant Biology, Carnegie Institution for Science, Stanford, on the occasion of their 60th, 70th, and 80th birthdays this year, respectively. This work was made possible by grants from the Department of Energy (DE-FG02-04ER15542) and the National Institute for Health (NIDDK; 1RO1DK079109-01).
References
- Ai HW, Hazelwood KL, Davidson MW, Campbell RE. Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nature Methods. 2008;5:401–403. doi: 10.1038/nmeth.1207. [DOI] [PubMed] [Google Scholar]
- Allen GJ, Chu SP, Schumacher K, Shimazaki CT, Vafeados D, Kemper A, Hawke SD, Tallman G, Tsien RY, Harper JF, et al. Alteration of stimulus-specific guard cell calcium oscillations and stomatal closing in Arabidopsis det3 mutant. Science. 2000;289:2338–2342. doi: 10.1126/science.289.5488.2338. [DOI] [PubMed] [Google Scholar]
- Allen GJ, Kwak JM, Chu SP, Llopis J, Tsien RY, Harper JF, Schroeder JI. Cameleon calcium indicator reports cytoplasmic calcium dynamics in Arabidopsis guard cells. Plant Journal. 1999;19:735–747. doi: 10.1046/j.1365-313x.1999.00574.x. [DOI] [PubMed] [Google Scholar]
- Arnaudeau S, Frieden M, Nakamura K, Castelbou C, Michalak M, Demaurex N. Calreticulin differentially modulates calcium uptake and release in the endoplasmic reticulum and mitochondria. The Journal of Biological Chemistry. 2002;277:46696–46705. doi: 10.1074/jbc.M202395200. [DOI] [PubMed] [Google Scholar]
- Bai F, DeMason DA. Hormone interactions and regulation of Unifoliata, PsPK2, PsPIN1 and LE gene expression in pea (Pisum sativum) shoot tips. Plant & Cell Physiology. 2006;47:935–948. doi: 10.1093/pcp/pcj066. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biology. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nature Protocols. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- Benner SA, Sismour AM. Synthetic biology. Nature Reviews Genetics. 2005;6:533–543. doi: 10.1038/nrg1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophysical Journal. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner M, Ludewig U. Visualization of arginine influx into plant cells using a specific FRET-sensor. Journal of Fluorescence. 2007;17:350–360. doi: 10.1007/s10895-007-0192-2. [DOI] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 2007;8:437–449. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner B. Cell-free fermentation. 1907 http://nobelprize.org/nobel_prizes/lists/all/
- Cacciatore TW, Brodfuehrer PD, Gonzalez JE, Jiang T, Adams SR, Tsien RY, Kristan WB, Jr, Kleinfeld D. Identification of neural circuits by imaging coherent electrical activity with FRET-based dyes. Neuron. 1999;23:449–459. doi: 10.1016/s0896-6273(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Carrari F, Schauer N, Willmitzer L, Fernie AR. Systems biology: a renaissance of the top-down approach for plant analysis. In: Nagata T, Lörz H, Widholm JM, editors. Plant metabolomics. Springer; Heidelberg, Germany: 2006. pp. 185–198. [Google Scholar]
- Causey TB, Shanmugam KT, Yomano LP, Ingram LO. Engineering Escherichia coli for efficient conversion of glucose to pyruvate. Proceedings of the National Academy of Sciences, USA. 2004;101:2235–2240. doi: 10.1073/pnas.0308171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri B, Hörmann F, Lalonde S, Brady S, Orlando DA, Benfey P, Frommer WB. Protonophore- and pH-insensitive glucose and sucrose accumulation detected by FRET nanosensors in Arabidopsis root tips. Plant Journal. 2008 doi: 10.1111/j.1365-313X.2008.03652.x. doi: 10.1111/j.1365-313X.2008.03652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Periasamy A. Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei. Journal of Fluorescence. 2006;16:95–104. doi: 10.1007/s10895-005-0024-1. [DOI] [PubMed] [Google Scholar]
- Cicchetti G, Biernacki M, Farquharson J, Allen PG. A ratiometric expressible FRET sensor for phosphoinositides displays a signal change in highly dynamic membrane structures in fibroblasts. Biochemistry. 2004;43:1939–1949. doi: 10.1021/bi035480w. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Frieden M. Measurements of the free luminal ER Ca2+ concentration with targeted ‘cameleon’ fluorescent proteins. Cell Calcium. 2003;34:109–119. doi: 10.1016/s0143-4160(03)00081-2. [DOI] [PubMed] [Google Scholar]
- Deuschle K, Chaudhuri B, Okumoto S, Lager I, Lalonde S, Frommer WB. Rapid metabolism of glucose detected with FRET glucose nanosensors in epidermal cells and intact roots of Arabidopsis RNA-silencing mutants. Plant Cell. 2006;18:2314–2325. doi: 10.1105/tpc.106.044073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle K, Fehr M, Hilpert M, Lager I, Lalonde S, Looger LL, Okumoto S, Persson J, Schmidt A, Frommer WB. Genetically encoded sensors for metabolites. Cytometry. Part A. 2005a;64:3–9. doi: 10.1002/cyto.a.20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle K, Okumoto S, Fehr M, Looger LL, Kozhukh L, Frommer WB. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Science. 2005b;14:2304–2314. doi: 10.1110/ps.051508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov D, He Y, Mutoh H, Baker BJ, Cohen L, Akemann W, Knopfel T. Engineering and characterization of an enhanced fluorescent protein voltage sensor. PLoS ONE. 2007;2:e440. doi: 10.1371/journal.pone.0000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. The Journal of Biological Chemistry. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Dulla C, Tani H, Okumoto S, Frommer WB, Reimer RJ, Huguenard JR. Imaging of glutamate in brain slices using FRET sensors. Journal of Neuroscience Methods. 2008;168:306–319. doi: 10.1016/j.jneumeth.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer MA, Hellinga HW. Periplasmic binding proteins: a versatile superfamily for protein engineering. Current Opinion in Structural Biology. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Ellis-Davies GC. Neurobiology with caged calcium. Chemical Reviews. 2008;108:1603–1613. doi: 10.1021/cr078210i. [DOI] [PubMed] [Google Scholar]
- Evanko DS, Haydon PG. Elimination of environmental sensitivity in a cameleon FRET-based calcium sensor via replacement of the acceptor with Venus. Cell Calcium. 2005;37:341–348. doi: 10.1016/j.ceca.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Farre EM, Tiessen A, Roessner U, Geigenberger P, Trethewey RN, Willmitzer L. Analysis of the compartmentation of glycolytic intermediates, nucleotides, sugars, organic acids, amino acids, and sugar alcohols in potato tubers using a nonaqueous fractionation method. Plant Physiology. 2001;127:685–700. doi: 10.1104/pp.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proceedings of the National Academy of Sciences, USA. 2002;99:9846–9851. doi: 10.1073/pnas.142089199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr M, Lalonde S, Ehrhardt DW, Frommer WB. Live imaging of glucose homeostasis in nuclei of COS-7 cells. Journal of Fluorescence. 2004;14:603–609. doi: 10.1023/b:jofl.0000039347.94943.99. [DOI] [PubMed] [Google Scholar]
- Fehr M, Lalonde S, Lager I, Wolff MW, Frommer WB. In vivo imaging of the dynamics of glucose uptake in the cytosol of COS-7 cells by fluorescent nanosensors. The Journal of Biological Chemistry. 2003;278:19127–19133. doi: 10.1074/jbc.M301333200. [DOI] [PubMed] [Google Scholar]
- Fehr M, Takanaga H, Ehrhardt DW, Frommer WB. Evidence for high-capacity bidirectional glucose transport across the endoplasmic reticulum membrane by genetically encoded fluorescence resonance energy transfer nanosensors. Molecular and Cellular Biology. 2005;25:11102–11112. doi: 10.1128/MCB.25.24.11102-11112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Molecular Systems Biology. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Scholten JC, Palsson BO, Brockman FJ, Ideker T. Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Molecular Systems Biology. 2006;2 doi: 10.1038/msb4100046. 2006 0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernie AR, Geigenberger P, Stitt M. Flux an important, but neglected, component of functional genomics. Current Opinion in Plant Biology. 2005;8:174–182. doi: 10.1016/j.pbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Förster T. Intermolecular energy migration and fluorescence. Annalen der Physik. 1948;2:55–75. [Google Scholar]
- Galvez T, Teruel MN, Heo WD, Jones JT, Kim ML, Liou J, Myers JW, Meyer T. siRNA screen of the human signaling proteome identifies the PtdIns(3,4,5)P3-mTOR signaling pathway as a primary regulator of transferrin uptake. Genome Biology. 2007;8:R142. doi: 10.1186/gb-2007-8-7-r142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Knight MR, Trewavas AJ, Sattelmacher B, Plieth C. Self-reporting Arabidopsis expressing pH and Ca2+ indicators unveil ion dynamics in the cytoplasm and in the apoplast under abiotic stress. Plant Physiology. 2004;134:898–908. doi: 10.1104/pp.103.032508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Griesbeck O, Konnerth A. Troponin C-based biosensors: a new family of genetically encoded indicators for in vivo calcium imaging in the nervous system. Cell Calcium. 2007;42:351–361. doi: 10.1016/j.ceca.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Gassensmith JJ, Arunkumar E, Barr L, Baumes JM, DiVittorio KM, Johnson JR, Noll BC, Smith BD. Self-assembly of fluorescent inclusion complexes in competitive media including the interior of living cells. Journal of the American Chemical Society. 2007;129:15054–15059. doi: 10.1021/ja075567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Gonzalez JE, Tsien RY. Voltage sensing by fluorescence resonance energy transfer in single cells. Biophysical Journal. 1995;69:1272–1280. doi: 10.1016/S0006-3495(95)80029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophysical Journal. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O. Fluorescent proteins as sensors for cellular functions. Current Opinion in Neurobiology. 2004;14:636–641. doi: 10.1016/j.conb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Grose JH, Smith TL, Sabic H, Rutter J. Yeast PAS kinase coordinates glucose partitioning in response to metabolic and cell integrity signaling. The EMBO Journal. 2007;26:4824–4830. doi: 10.1038/sj.emboj.7601914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Lalonde S, Okumoto S, Looger LL, Scharff-Poulsen AM, Grossman AR, Kossmann J, Jakobsen I, Frommer WB. A novel analytical method for in vivo phosphate tracking. FEBS Letters. 2006;580:5885–5893. doi: 10.1016/j.febslet.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenoune D, Amir R, Badani H, Wolf S, Galili S. Combined expression of S-VSPalpha in two different organelles enhances its accumulation and total lysine production in leaves of transgenic tobacco plants. Journal of Experimental Botany. 2002;53:1867–1870. doi: 10.1093/jxb/erf046. [DOI] [PubMed] [Google Scholar]
- Guerrero G, Siegel MS, Roska B, Loots E, Isacoff EY. Tuning FlaSh: redesign of the dynamics, voltage range, and color of the genetically encoded optical sensor of membrane potential. Biophysical Journal. 2002;83:3607–3618. doi: 10.1016/S0006-3495(02)75361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Ha JS, Song JJ, Lee YM, Kim SJ, Sohn JH, Shin CS, Lee SG. Design and application of highly responsive fluorescence resonance energy transfer biosensors for detection of sugar in living Saccharomyces cerevisiae cells. Applied and Environmental Microbiology. 2007;73:7408–7414. doi: 10.1128/AEM.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese G, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, et al. Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biology. 2004;2:e163. doi: 10.1371/journal.pbio.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim N, Griesbeck O. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. The Journal of Biological Chemistry. 2004;279:14280–14286. doi: 10.1074/jbc.M312751200. [DOI] [PubMed] [Google Scholar]
- Helling D, Possart A, Cottier S, Klahre U, Kost B. Pollen tube tip growth depends on plasma membrane polarization mediated by tobacco PLC3 activity and endocytic membrane recycling. Plant Cell. 2006;18:3519–3534. doi: 10.1105/tpc.106.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires SA, Zhu Y, Tsien RY. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proceedings of the National Academy of Sciences, USA. 2008;105:4411–4416. doi: 10.1073/pnas.0712008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJ. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nature Methods. 2005;2:171–176. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- Honda A, Adams SR, Sawyer CL, Lev-Ram V, Tsien RY, Dostmann WR. Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proceedings of the National Academy of Sciences, USA. 2001;98:2437–2442. doi: 10.1073/pnas.051631298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jares-Erijman EA, Jovin TM. FRET imaging. Nature Biotechnology. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Current Opinion in Chemical Biology. 2006;10:409–416. doi: 10.1016/j.cbpa.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Jose M, Nair DK, Reissner C, Hartig R, Zuschratter W. Photophysics of Clomeleon by FLIM: discriminating excited state reactions along neuronal development. Biophysical Journal. 2007;92:2237–2254. doi: 10.1529/biophysj.106.092841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper T, Lager I, Looger LL, Chermak D, Frommer WB. Fluorescence resonance energy transfer sensors for quantitative monitoring of pentose and disaccharide accumulation in bacteria. Biotechnology for Biofuels. 2008;1:11. doi: 10.1186/1754-6834-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper T, Looger LL, Takanaga H, Platten M, Steinman L, Frommer WB. Nanosensor detection of an immunoregulatory tryptophan influx/kynurenine efflux cycle. PLoS Biology. 2007;5:e257. doi: 10.1371/journal.pbio.0050257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. The Biochemical Journal. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp A, Shield I. Bioenergy from plants and the sustainable yield challenge. New Phytologist. 2008;179:15–32. doi: 10.1111/j.1469-8137.2008.02432.x. [DOI] [PubMed] [Google Scholar]
- Keasling JD. Synthetic biology for synthetic chemistry. ACS Chemical Biology. 2008;3:64–76. doi: 10.1021/cb7002434. [DOI] [PubMed] [Google Scholar]
- Kim JN, Breaker RR. Purine sensing by riboswitches. Biologie Cell. 2008;100:1–11. doi: 10.1042/BC20070088. [DOI] [PubMed] [Google Scholar]
- Kolossov VL, Spring BQ, Sokolowski A, Conour JE, Clegg RM, Kenis PJ, Gaskins HR. Engineering redox-sensitive linkers for genetically encoded FRET-based biosensors. Experimental Biology and Medicine (Maywood, N.J.) 2008;233:238–248. doi: 10.3181/0707-RM-192. [DOI] [PubMed] [Google Scholar]
- Konrad KR, Hedrich R. The use of voltage-sensitive dyes to monitor signal-induced changes in membrane potential-ABA triggered membrane depolarization in guard cells. Plant Journal. 2008;55:161–173. doi: 10.1111/j.1365-313X.2008.03498.x. [DOI] [PubMed] [Google Scholar]
- Koushik SV, Chen H, Thaler C, Puhl HL, 3rd, Vogel SS. Cerulean, Venus, and VenusY67C FRET reference standards. Biophysical Journal. 2006;91:L99–L101. doi: 10.1529/biophysj.106.096206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager I, Fehr M, Frommer WB, Lalonde S. Development of a fluorescent nanosensor for ribose. FEBS Letters. 2003;553:85–89. doi: 10.1016/s0014-5793(03)00976-1. [DOI] [PubMed] [Google Scholar]
- Lager I, Looger LL, Hilpert M, Lalonde S, Frommer WB. Conversion of a putative Agrobacterium sugar-binding protein into a FRET sensor with high selectivity for sucrose. The Journal of Biological Chemistry. 2006;281:30875–30883. doi: 10.1074/jbc.M605257200. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of fluorescence spectroscopy. Kluwer Academic/Plenum Publishers; New York, NY, USA: 1999. [Google Scholar]
- Lalonde S, Ehrhardt DW, Frommer WB. Shining light on signaling and metabolic networks by genetically encoded biosensors. Current Opinion in Plant Biology. 2005;8:574–581. doi: 10.1016/j.pbi.2005.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde S, Ehrhardt DW, Loque D, Chen J, Rhee SY, Frommer WB. Molecular and cellular approaches for the detection of protein-protein interactions: latest techniques and current limitations. Plant Journal. 2008;53:610–635. doi: 10.1111/j.1365-313X.2007.03332.x. [DOI] [PubMed] [Google Scholar]
- Ledvina PS, Tsai AL, Wang Z, Koehl E, Quiocho FA. Dominant role of local dipolar interactions in phosphate binding to a receptor cleft with an electronegative charge surface: equilibrium, kinetic, and crystallographic studies. Protein Science. 1998;7:2550–2559. doi: 10.1002/pro.5560071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen W, Vermeer JE, Gadella TW, Jr, Munnik T. Visualization of phosphatidylinositol 4,5-bisphosphate in the plasma membrane of suspension-cultured tobacco BY-2 cells and whole Arabidopsis seedlings. Plant Journal. 2007;52:1014–1026. doi: 10.1111/j.1365-313X.2007.03292.x. [DOI] [PubMed] [Google Scholar]
- Leveau JH, Lindow SE. Bioreporters in microbial ecology. Current Opinion in Microbiology. 2002;5:259–265. doi: 10.1016/s1369-5274(02)00321-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Shrestha B, Vertes A. Atmospheric pressure molecular imaging by infrared MALDI mass spectrometry. Analytical Chemistry. 2007;79:523–532. doi: 10.1021/ac061577n. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler HK. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annual Review of Plant Physiology and Plant Molecular Biology. 1999;50:47–65. doi: 10.1146/annurev.arplant.50.1.47. [DOI] [PubMed] [Google Scholar]
- Lindow SE, Leveau JH. Phyllosphere microbiology. Current Opinion in Biotechnology. 2002;13:238–243. doi: 10.1016/s0958-1669(02)00313-0. [DOI] [PubMed] [Google Scholar]
- Linka M, Weber AP. Shuffling ammonia between mitochondria and plastids during photorespiration. Trends in Plant Science. 2005;10:461–465. doi: 10.1016/j.tplants.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Current Biology. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissandron V, Rossetto MG, Erbguth K, Fiala A, Daga A, Zaccolo M. Transgenic fruit-flies expressing a FRET-based sensor for in vivo imaging of cAMP dynamics. Cellular Signalling. 2007;19:2296–2303. doi: 10.1016/j.cellsig.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Lissandron V, Terrin A, Collini M, D’Alfonso L, Chirico G, Pantano S, Zaccolo M. Improvement of a FRET-based indicator for cAMP by linker design and stabilization of donor-acceptor interaction. Journal of Molecular Biology. 2005;354:546–555. doi: 10.1016/j.jmb.2005.09.089. [DOI] [PubMed] [Google Scholar]
- Long SP, Zhu XG, Naidu SL, Ort DR. Can improvement in photosynthesis increase crop yields? Plant, Cell & Environment. 2006;29:315–330. doi: 10.1111/j.1365-3040.2005.01493.x. [DOI] [PubMed] [Google Scholar]
- Looger LL, Lalonde S, Frommer WB. Genetically encoded FRET sensors for visualizing metabolites with subcellular resolution in living cells. Plant Physiology. 2005;138:555–557. doi: 10.1104/pp.104.900151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loque D, Lalonde S, Looger LL, von Wiren N, Frommer WB. A cytosolic trans-activation domain essential for ammonium uptake. Nature. 2007;446:195–198. doi: 10.1038/nature05579. [DOI] [PubMed] [Google Scholar]
- Mank M, Reiff DF, Heim N, Friedrich MW, Borst A, Griesbeck O. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophysical Journal. 2006;90:1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, O’Connor DH, Scheuss V, Nakai J, Svoboda K. Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS ONE. 2008;3:e1796. doi: 10.1371/journal.pone.0001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova O, Mukhtarov M, Real E, Jacob Y, Bregestovski P. Genetically encoded chloride indicator with improved sensitivity. Journal of Neuroscience Methods. 2008;170:67–76. doi: 10.1016/j.jneumeth.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Medintz IL. Recent progress in developing FRET-based intracellular sensors for the detection of small molecule nutrients and ligands. Trends in Biotechnology. 2006;24:539–542. doi: 10.1016/j.tibtech.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Miller DM, 3rd, Olson JS, Pflugrath JW, Quiocho FA. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. The Journal of Biological Chemistry. 1983;258:13665–13672. [PubMed] [Google Scholar]
- Miyawaki A. Visualization of the spatial and temporal dynamics of intracellular signaling. Developmental Cell. 2003;4:295–305. doi: 10.1016/s1534-5807(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Moens PD, Helms MK, Jameson DM. Detection of tryptophan to tryptophan energy transfer in proteins. The Protein Journal. 2004;23:79–83. doi: 10.1023/b:jopc.0000016261.97474.2e. [DOI] [PubMed] [Google Scholar]
- Mongillo M, Terrin A, Evellin S, Lissandron V, Zaccolo M. Study of cyclic adenosine monophosphate microdomains in cells. Methods in Molecular Biology. 2005;307:1–13. doi: 10.1385/1-59259-839-0:001. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Nahorski SR, Young KW, John Challiss RA, Nash MS. Visualizing phosphoinositide signalling in single neurons gets a green light. Trends in Neurosciences. 2003;26:444–452. doi: 10.1016/S0166-2236(03)00178-4. [DOI] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nature Biotechnology. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Niittyla T, Chaudhuri B, Sauer U, Frommer WB. Trends in neurosciences metabolite imaging tools and carbon-13 techniques for fluxomics. In: Belostotsky DA, editor. Methods in molecular biology. Humana Press; New York, NY, USA: in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niittyla T, Fuglsang AT, Palmgren MG, Frommer WB, Schulze WX. Temporal analysis of sucrose-induced phosphorylation changes in plasma membrane proteins of Arabidopsis. Molecular & Cellular Proteomics. 2007;6:1711–1726. doi: 10.1074/mcp.M700164-MCP200. [DOI] [PubMed] [Google Scholar]
- Nishigaki T, Wood CD, Shiba K, Baba SA, Darszon A. Stroboscopic illumination using light emitting diodes reduces phototoxicity in fluorescence cell imaging. Biotechniques. 2006;41:191–197. doi: 10.2144/000112220. [DOI] [PubMed] [Google Scholar]
- O’Connor N, Silver RB. Ratio imaging: practical considerations for measuring intracellular Ca2+ and pH in living cells. Methods in Cell Biology. 2007;81:415–433. doi: 10.1016/S0091-679X(06)81019-8. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Dansako T, Yano K, Sakurai N, Suzuki H, Aoki K, Noji M, Saito K, Shibata D. Efficient and high-throughput vector construction and Agrobacterium-mediated transformation of Arabidopsis thaliana suspension-cultured cells for functional genomics. Plant & Cell Physiology. 2008;49:242–250. doi: 10.1093/pcp/pcm181. [DOI] [PubMed] [Google Scholar]
- Okumoto S, Looger LL, Micheva KD, Reimer RJ, Smith SJ, Frommer WB. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proceedings of the National Academy of Sciences, USA. 2005;102:8740–8745. doi: 10.1073/pnas.0503274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck SC. Phosphoproteomics in Arabidopsis: moving from empirical to predictive science. Journal of Experimental Botany. 2006;57:1523–1527. doi: 10.1093/jxb/erj126. [DOI] [PubMed] [Google Scholar]
- Persechini A, Lynch JA, Romoser VA. Novel fluorescent indicator proteins for monitoring free intracellular Ca2+ Cell Calcium. 1997;22:209–216. doi: 10.1016/s0143-4160(97)90014-2. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hahn KM. Designing biosensors for Rho family proteins-deciphering the dynamics of Rho family GTPase activation in living cells. Journal of Cell Science. 2004;117:1313–1318. doi: 10.1242/jcs.01117. [DOI] [PubMed] [Google Scholar]
- Pham E, Chiang J, Li I, Shum W, Truong K. A computational tool for designing FRET protein biosensors by rigid-body sampling of their conformational space. Structure. 2007;15:515–523. doi: 10.1016/j.str.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Yasuda R, Svoboda K. Monitoring neural activity and [Ca2+] with genetically encoded Ca2+ indicators. The Journal of Neuroscience. 2004;24:9572–9579. doi: 10.1523/JNEUROSCI.2854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Ihring A, Guerrero G, Isacoff EY, Joesch M, Nakai J, Borst A. In vivo performance of genetically encoded indicators of neural activity in flies. The Journal of Neuroscience. 2005;25:4766–4778. doi: 10.1523/JNEUROSCI.4900-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekas A, Alattia JR, Nagai T, Miyawaki A, Ikura M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. The Journal of Biological Chemistry. 2002;277:50573–50578. doi: 10.1074/jbc.M209524200. [DOI] [PubMed] [Google Scholar]
- Remus TP, Zima AV, Bossuyt J, Bare DJ, Martin JL, Blatter LA, Bers DM, Mignery GA. Biosensors to measure inositol 1,4,5-trisphosphate concentration in living cells with spatiotemporal resolution. The Journal of Biological Chemistry. 2006;281:608–616. doi: 10.1074/jbc.M509645200. [DOI] [PubMed] [Google Scholar]
- Rios-Estepa R, Lange BM. Experimental and mathematical approaches to modeling plant metabolic networks. Phytochemical. 2007;68:2351–2374. doi: 10.1016/j.phytochem.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Romoser VA, Hinkle PM, Persechini A. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. The Journal of Biological Chemistry. 1997;272:13270–13274. doi: 10.1074/jbc.272.20.13270. [DOI] [PubMed] [Google Scholar]
- Rothstein SJ. Returning to our roots: making plant biology research relevant to future challenges in agriculture. Plant Cell. 2007;19:2695–2699. doi: 10.1105/tpc.107.053074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SJ, Cuin TA, Leigh RA. Nanolitre-scale assays to determine the activities of enzymes in individual plant cells. Plant Journal. 2003;34:555–564. doi: 10.1046/j.1365-313x.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Schulte A, Lorenzen I, Bottcher M, Plieth C. A novel fluorescent pH probe for expression in plants. Plant Methods. 2006;2:7. doi: 10.1186/1746-4811-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilton BH, Flocco MM, Nilsson M, Mowbray SL. Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: the maltose-, glucose/galactose- and ribose-binding proteins. Journal of Molecular Biology. 1996;264:350–363. doi: 10.1006/jmbi.1996.0645. [DOI] [PubMed] [Google Scholar]
- Shimozono S, Miyawaki A. Engineering FRET constructs using CFP and YFP. Methods in Cell Biology. 2008;85:381–393. doi: 10.1016/S0091-679X(08)85016-9. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Nishida M, Otsuka M, Makino K, Ohkubo K, Mori Y, Morii T. Novel real-time sensors to quantitatively assess in vivo inositol 1,4,5-trisphosphate production in intact cells. Chemistry & Biology. 2004;11:475–485. doi: 10.1016/j.chembiol.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Sun H, Scharff-Poulsen AM, Gu H, Jakobsen I, Kossmann JM, Frommer WB, Almdal K. Phosphate sensing by fluorescent reporter proteins embedded in polyacrylamide nanoparticles. ACS Nano. 2008;2:19–24. doi: 10.1021/nn700166x. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Chaudhuri B, Frommer WB. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochimica et Biophysica Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga H, Frommer WB. Plasma membrane GLUT facilitators mediate glucose transport during ER transit. Molecular and Cellular Biology. 2008 in press. [Google Scholar]
- Tam R, Saier MH., Jr Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiological Reviews. 1993;57:320–346. doi: 10.1128/mr.57.2.320-346.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Nezu A, Morita T, Turner RJ, Tojyo Y. Fluorescent biosensor for quantitative real-time measurements of inositol 1,4,5-trisphosphate in single living cells. The Journal of Biological Chemistry. 2004;279:38095–38098. doi: 10.1074/jbc.C400312200. [DOI] [PubMed] [Google Scholar]
- Tengholm A, Meyer T. A pi3-kinase signaling code for insulin-triggered insertion of glucose transporters into the plasma membrane. Current Biology. 2002;12:1871–1876. doi: 10.1016/s0960-9822(02)01223-x. [DOI] [PubMed] [Google Scholar]
- Teusink B, Diderich JA, Westerhoff HV, van Dam K, Walsh MC. Intracellular glucose concentration in derepressed yeast cells consuming glucose is high enough to reduce the glucose transport rate by 50% Journal of Bacteriology. 1998;180:556–562. doi: 10.1128/jb.180.3.556-562.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler C, Koushik SV, Blank PS, Vogel SS. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophysical Journal. 2005;89:2736–2749. doi: 10.1529/biophysj.105.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. Breeding and building molecules to spy on cells and tumors. The Keio Journal of Medicine. 2006;55:127–140. doi: 10.2302/kjm.55.127. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Karasawa S, Okamura Y, Miyawaki A. Improving membrane voltage measurements using FRET with new fluorescent proteins. Nature Methods. 2008;5:683–685. doi: 10.1038/nmeth.1235. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proceedings of the National Academy of Sciences, USA. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SS, Thaler C, Koushik SV. Fanciful FRET. Science Signaling. 2006;2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- Voinnet O, Vain P, Angell S, Baulcombe DC. Systemic spread of sequence-specific transgene RNA degradation in plants is initiated by localized introduction of ectopic promoterless DNA. Cell. 1998;95:177–187. doi: 10.1016/s0092-8674(00)81749-3. [DOI] [PubMed] [Google Scholar]
- Weijer CJ. Visualizing signals moving in cells. Science. 2003;300:96–100. doi: 10.1126/science.1082830. [DOI] [PubMed] [Google Scholar]
- Wiechert W, Schweissgut O, Takanaga H, Frommer WB. Fluxomics: mass spectrometry versus quantitative imaging. Current Opinion in Plant Biology. 2007;10:323–330. doi: 10.1016/j.pbi.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win MN, Smolke CD. A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proceedings of the National Academy of Sciences, USA. 2007;104:14283–14288. doi: 10.1073/pnas.0703961104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Meier-Schellersheim M, Jiao X, Nelson LE, Jin T. Quantitative imaging of single live cells reveals spatiotemporal dynamics of multistep signaling events of chemoattractant gradient sensing in Dictyostelium. Molecular Biology of the Cell. 2005;16:676–688. doi: 10.1091/mbc.E04-07-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Zhang B, Wu H. Chemical cytometry on microfluidic chips. Electrophoresis. 2008;29:1775–1786. doi: 10.1002/elps.200700561. [DOI] [PubMed] [Google Scholar]
- Yang X, Xu P, Xu T. A new pair for inter- and intra-molecular FRET measurement. Biochemical and Biophysical Research Communications. 2005;330:914–920. doi: 10.1016/j.bbrc.2005.03.054. [DOI] [PubMed] [Google Scholar]
- Ye K, Schultz JS. Genetic engineering of an allosterically based glucose indicator protein for continuous glucose monitoring by fluorescence resonance energy transfer. Analytical Chemistry. 2003;75:3451–3459. doi: 10.1021/ac034022q. [DOI] [PubMed] [Google Scholar]
- Young JJ, Mehta S, Israelsson M, Godoski J, Grill E, Schroeder JI. CO2 signaling in guard cells: calcium sensitivity response modulation, a Ca2+-independent phase, and CO2 insensitivity of the gca2 mutant. Proceedings of the National Academy of Sciences, USA. 2006;103:7506–7511. doi: 10.1073/pnas.0602225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M. Use of chimeric fluorescent proteins and fluorescence resonance energy transfer to monitor cellular responses. Circulation Research. 2004;94:866–873. doi: 10.1161/01.RES.0000123825.83803.CD. [DOI] [PubMed] [Google Scholar]
- Zeeman SC, Smith SM, Smith AM. The diurnal metabolism of leaf starch. The Biochemical Journal. 2007;401:13–28. doi: 10.1042/BJ20061393. [DOI] [PubMed] [Google Scholar]
- Zeng AP, Biebl H. Bulk chemicals from biotechnology: the case of 1,3-propanediol production and the new trends. Advances in Biochemical Engineering/Biotechnology. 2002;74:239–259. doi: 10.1007/3-540-45736-4_11. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zimmermann T, Rietdorf J, Pepperkok R. Spectral imaging and its applications in live cell microscopy. FEBS Letters. 2003;546:87–92. doi: 10.1016/s0014-5793(03)00521-0. [DOI] [PubMed] [Google Scholar]